

1 Introduction
Helicenes represent a fascinating class of polycyclic aromatic molecules in which a regular cylindrical helix is formed through an all-ortho annelation of aromatic or heteroaromatic rings. They have a strong inherent chromophor which exhibits very high specific optical rotations depending on a gross structure, a fine geometry and electronic interactions [1]. These organic molecules present left- and right-handed chiral helical structures of M and P configuration, respectively [2]. Helicene backbones have been used as rotors [3], in molecular recognition research [4] and as self-assembling components of helical structures with high circular dichroism and a specific rotation [5]. Furthermore, enantiomerically enriched helicenes, especially phosphorus- or oxygen-containing species have proved successful as chiral ligands [6] and catalysts [7] in asymmetric synthesis because of their stable polyconjugated helical framework. In these cases, coordination of the heteroatoms with metals is considered to play an essential role in the induction of asymmetry. The first enantiomerically-enriched helical diol with a bis[5]helicene skeleton 1 (called [5]-HELOL) was prepared by Katz in 2000 and used as a chiral ligand in the addition of Et2Zn to aromatic aldehydes (Fig. 1). Reetz and Sostmann reported the synthesis of the first dihydroxy[6]helicene 2 (called HELIXOL) in its optically active form, which was used as an enantioselective fluorescent sensor [8]. Shortly after, Wachsmann et al. [9] prepared the 2,15-dihydroxymethyl[6]helicene 3. The antipode separation of each helicenediol 2 and 3 was achieved by chiral HPLC.
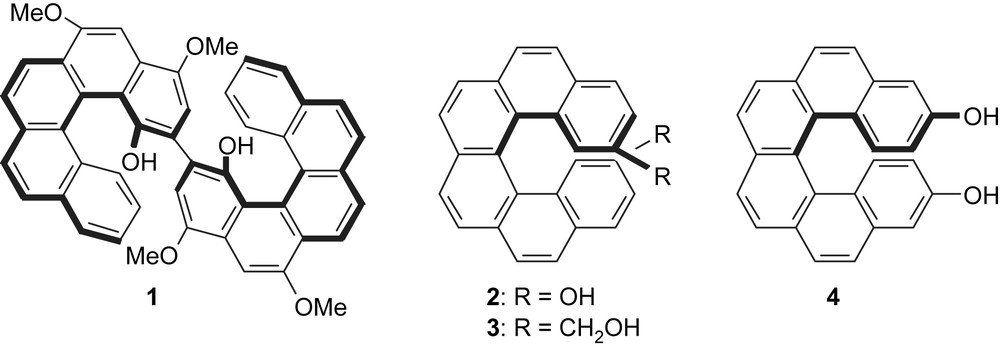
In the present work, we demonstrate that the Mizoroki–Heck coupling reaction between a benzo[c]phenanthrene moiety and a styryl derivative, followed by a photocyclisation step, yields in a regioselective manner the helically chiral hexacyclic framework of 7, which is a suitable precursor for [6]helicenediol 4.
2 Results and discussion
In order to utilize the optically active helicenes as chiral elements in asymmetric synthesis, we have designed a heterohelicene with two hydroxy groups in selected positions at the terminal aromatic nuclei and synthesized 3,14-dihydroxy[6]helicene 4, through a “3 + 2” approach. The synthetic approach makes use of a bromophenanthrene derivative as a starting material [10] for the synthesis of the helicene precursor 5 (Scheme 1). At the last step of the synthesis of the helicene framework, a benzo[c]naphtho[1,2-h]anthracene derivative 6 was produced as a byproduct, in a considerable amount, and was found to be partially separable from the expected helicene 7 by column chromatography.
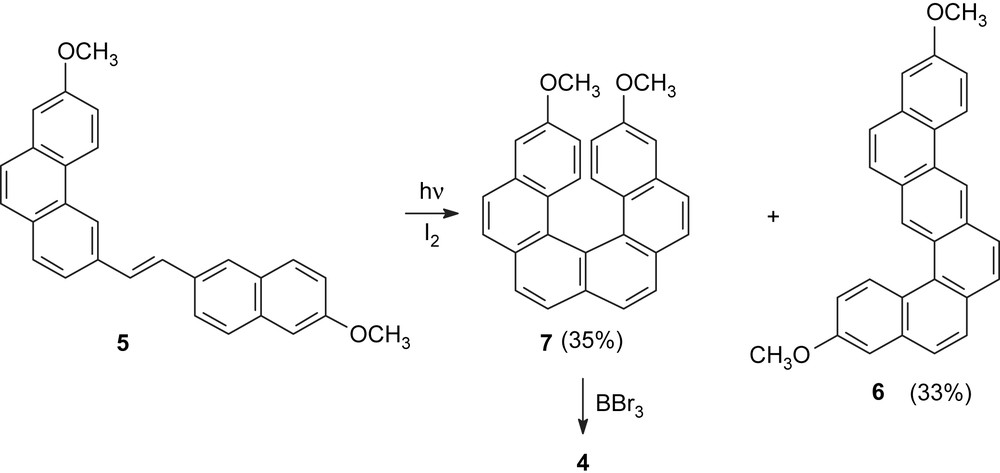
Synthesis of the helicenediol 4 via the “3 + 2” approach.
In order to improve the synthesis of the helicenediol 4, to make it suitable for a synthesis of higher analogues as well, we thought it necessary to prevent the formation of the regioisomer 6. Towards this end, we envisaged a “4 + 1” approach involving a Mizoroki–Heck coupling [11] of the benzo[c]phenanthrene moiety 8 with an excess amount of 3-methoxystyrene using 1% of Hermann's palladacycle [trans-di(μ-acetato)-bis[o-(di-o-tolylphosphino)benzyl]dipalladium] as the catalyst. The starting material 2-bromo-10-methoxybenzo[c]phenanthrene 8 is available in two steps by photocyclisation of 2-(4-bromostyryl)-6-methoxynaphthalene, which is conveniently prepared by a Heck-type reaction [12]. The desired helicene-precursor 9, possessing the (E)-stereochemistry at the double bond, is obtained in 78% yield after a 48-h heating at 140 °C, according to Scheme 2. To complete the synthesis of [6]helicene 7, diarylethene 9 was exposed to photocyclisation in the presence of a stoichiometric amount of iodine as an oxidating agent and an excess of propylene oxide as a hydrogen iodide scavenger [13]. Thus, the photolysis of 9 was performed in cyclohexane, for about 120 min, on a 200-mg scale per run and afforded the expected 3,14-dimethoxy[6]helicene 7 in 66% yield. The treatment of compound 7 with a BBr3 solution in CH2Cl2 at room temperature provided the corresponding 3,14-dihydroxyhexahelicene 4 in 90% yield, with a total 51% yield over three steps starting from the benzo[c]phenanthrene moiety 8 (Scheme 2).
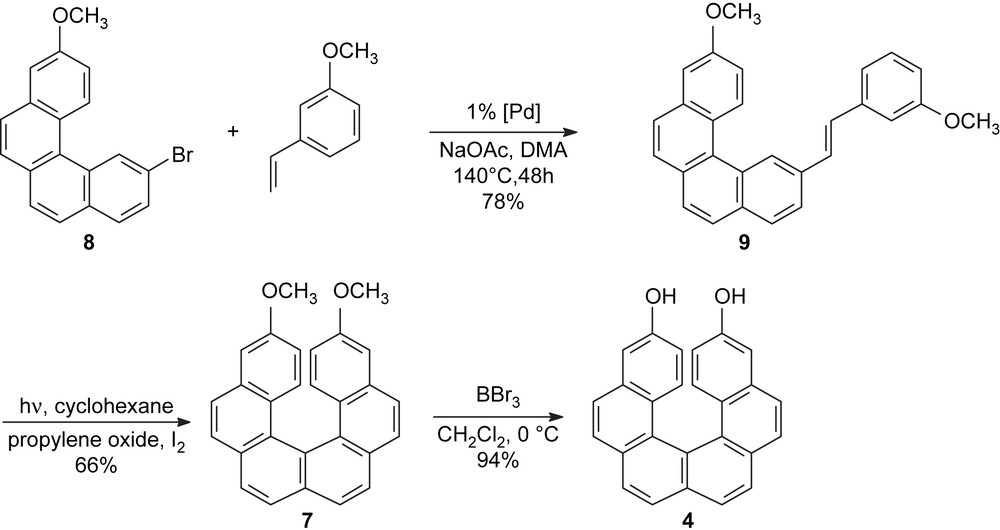
Synthesis of the helicenediol 4 via the “4 + 1” approach.
The ring closure of olefin 9 was not completely regioselective since 1,14-dimethoxyhexahelicene 10 was isolated in 9% yield and identified as a minor, helically chiral compound in the reaction mixture (Fig. 2). Helicenes 7 and 10 were successfully separated by column chromatography on silica gel.
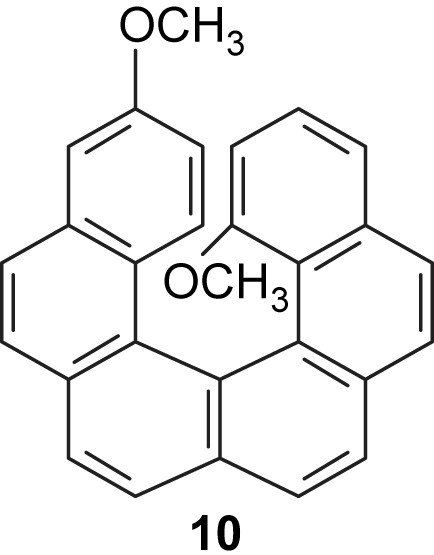
Structure of helically chiral compound 10.
To perform the resolution of racemic 3,14-dihydroxyhexahelicene into its enantiomers, we first investigated the usefulness of (S)-(−)-camphanoyl chloride as a chiral resolving reagent used to convert related alcohols to their corresponding ester camphanates [14]. Thus, the treatment of racemic helicenediol 4 with (S)-(−)-camphanoyl chloride, in the presence of triethylamine as a base and dichloromethane as the solvent at room temperature, led to a 1:1-mixture of two diastereomeric helicene dicamphanates (M,S)-11 and (P,S)-11 in 91% yield (Scheme 3). The first attempt to separate the diastereomers of 11 consisted in the use of chromatographic column over silica gel. Different solvent mixtures were tried, but none of them proved suitable. Subsequently, we attempted fractional crystallisations. Yellow crystals of 11 were obtained from a CH2Cl2/pentane mixture. Their analysis by 1H NMR spectroscopy indicated the presence of a 1:1 mixture of the two diastereomers, revealing no d.e. for the crystallisation process.
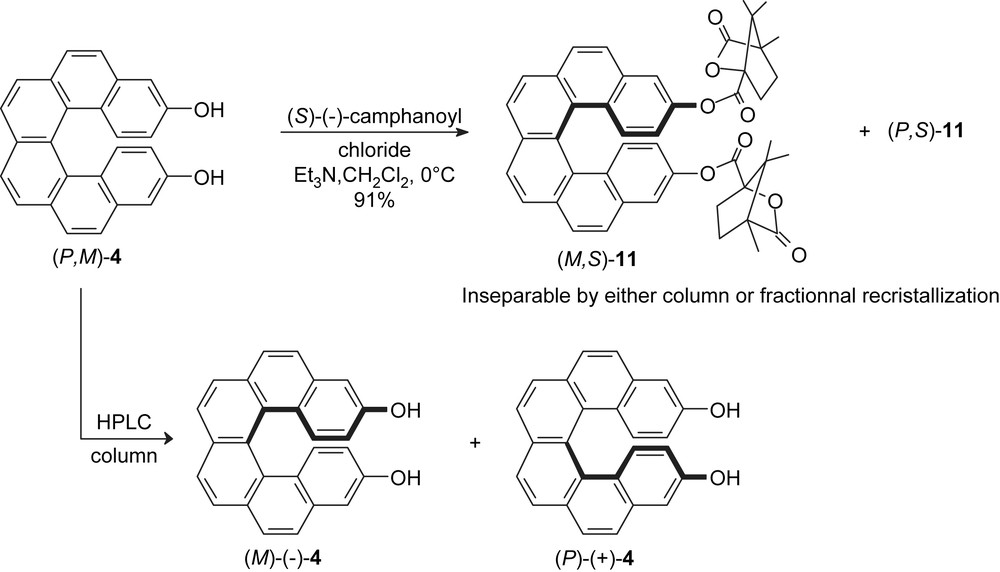
Enantiomeric resolution of the helically chiral alcohol 4.
The alternative resolution method was finally accomplished by chiral HPLC. The direct resolution of 3,14-dihydroxyhexahelicene 4 using a chiral column (250 × 20 mm) packed with cellulose-tris(3,5-dimethylphenylcarbamate) [15] when an n-heptane/2-propanol (80:20) mixture was used, as the mobile phase, was found to be successful. Thus, starting from 100 mg of rac-4, a total of 69 mg of pure product was separated, equivalent to a yield of 69% (Scheme 3). The earlier eluting fractions, of >99% ee, consisted of the enantiomer exhibiting a negative optical rotation (−) which was isolated in 39% yield. Later eluting fractions gave compound (+)-4 in 30% yield with only 97.6% ee. The enantiomeric purity of both enantiomers was established by chiral HPLC using the same stationary phase as indicated above. The specific rotation values obtained for (−)-4 and (+)-4 were found to be [α]D −2287 (c 0.19, acetone) and +2371 (c 0.20, acetone), respectively. The very high specific rotation value is a known feature of helical structures.
Unfortunately neither of the two enantiomers of 4 afforded crystals suitable for the determination of their structure by X-ray diffraction. Nevertheless, we can assign the absolute configuration of both enantiomers on the basis of circular dichroism spectra. The chiroptical properties of 3,14-dihydroxyhexahelicene 4 in the form of the CD spectrum were also measured. Fig. 3 shows the results of this study. The CD spectrum of the dextrorotatory enantiomer (P)-(+)-4 recorded a distinct positive maximum at 337 nm, as depicted in Fig. 3. The CD spectral characteristic is in good agreement with those of known (P)-helicenes [16], so the absolute configuration of (−)- and (+)-helicenediols 4 must be M (left-handed helix) and P (right-handed helix), respectively.

CD spectra of (M)-(−)-4 and (P)-(+)-4 in acetone.
3 Conclusion
We have built in a good overall yield the helically hexacyclic framework of 4 starting from a benzo[c]phenanthrene moiety. The resolution of the helicenediol 4 was successfully achieved and led to the corresponding enantiomers in a high optical purity. The application of these optically active helicenediols as chiral ligands in enantioselective reactions is currently in progress.
4 Experimental section
All reactions were performed under an argon atmosphere and monitored by TLC Merck 60F-254 silica gel plates (layer thickness 0.25 mm). Column chromatography was performed on silica gel (70–230 mesh) using ethyl acetate and cyclohexane mixture as eluents. Melting temperatures were determined on an Electrothermal 9002 apparatus and were reported uncorrected. NMR spectra were recorded on a Bruker AC-300 spectrometer at 300 MHz (1H) and 75 MHz (13C). All chemical shifts were reported as δ values (ppm) relative to internal tetramethylsilane. N,N-Dimethylacetamide and dichloromethane were distilled over CaH2. Photocyclizations were carried out in a 1-L water-cooled quartz photoreactor equipped with a high-pressure mercury immersion lamp [Heraeus TQ 150]. Optical rotations were measured with a JASCO P-1010 polarimeter. Mass spectra were recorded on a Hewlett-Packard HP 5989. High-resolution mass spectra were recorded on a MALDI-TOF Perspective Biosystems Voyager DE-STR. Enantiomeric excess determinations were carried out using a Waters 2695 system equipped with an analytical HPLC column (Chiralcel OD 250 × 4.6 mm). The CD spectra were recorded on a JASCO J-810 instrument flushed with dry nitrogen.
4.1 2-(3-Methoxystyryl)-10-methoxybenzo[c]phenanthrene: 9
A solution of 2-bromo-10-methoxybenzo[c]phenanthrene (1.5 g, 4.4 mmol) and dry sodium acetate (400 mg, 4.9 mmol) in N,N-dimethylacetamide (10 mL) was placed in a double-necked flask fitted with a septum, and repeatedly degassed and purged with argon. To this was then added 0.84 mL of 3-methoxystyrene (6.2 mmol) and the mixture was heated to 100 °C. When this temperature was reached, a solution of the Herrmann's catalyst (41 mg, 1%) in N,N-dimethylacetamide (2 mL) was added and the reaction mixture heated to 140 °C. Heating was maintained for about 48 h. The reaction was quenched by an addition of 5% HCl solution, stirred for 30 min at room temperature and then extracted with CH2Cl2. The combined layers were dried over MgSO4 and evaporated to dryness. After column chromatography with cyclohexane/ethyl acetate 90:10 as the eluent (Rf = 0.37), the final product 9 was obtained in 78% yield (1.3 g) as a light yellow solid. Mp = 82–84 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 3.77 (s, 3H, OCH3), 3.91(s, 3H, OCH3), 3.75 (dd, J = 2.4, 7.5 Hz, 1H), 7.04 (s, 1H), 7.08–7.30 (m, 6H), 7.64–7.71 (m, 4H), 7.74 (dd, J = 1.2, 8.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 8.94 (d, J = 9.6 Hz, 1H), 8.95 (s, 1H, H-1); 13C NMR (75 MHz, CDCl3): δ (ppm): 55.37 (OCH3), 55.50 (OCH3), 108.14 (C–H), 111.82 (C–H), 113.43 (C–H), 117.20 (C–H), 119.38 (C–H), 123.25 (C–H), 125.13 (C), 126.20 (C–H), 126.96 (C–H), 127.03 (C–H), 127.25 (C–H), 127.52 (C–H), 127.70 (C), 128.96 (2C–H), 129.42 (C–H), 129.68 (C–H), 129.77 (C–H), 130.12 (C), 130.41 (C), 133.24 (C), 134.93 (C), 135.20 (C), 138.92 (C), 157.62 (C–O), 160.02 (C–O); MS (EI, 50 eV): C28H22O2: M = 390 g mol−1: m/z = 390 ([M]+, 100%), 375 (3%), 359 (2%), 332 (1%), 300 (4%), 276 (3%), 263 (3%), 239 (5%), 213 (5%), 188 (4%), 151 (8%).
4.2 3,14-Dimethoxyhexahelicene: 7 [10]
2-(3-Methoxystyryl)-10-methoxybenzo[c]phenanthrene 9 was used to give 66% of a light yellow solid; Rf = 0.35 (eluent: cyclohexane/ethyl acetate 90:10). Mp = 245–247 °C; HRMS (MALDI-TOF) calcd. for C28H20O2Na [M + Na]+: 411.1361. Found: 411.1381.
4.3 1,14-Dimethoxyhexahelicene: 10
Helicene 10 was obtained as a colourless solid from 9 in 9% yield. It was purified by column chromatography with cyclohexane/ethyl acetate (90:10) as the eluent (Rf = 0.43). Mp = 191–193 °C; 1H NMR (300 MHz, CDCl3): δ (ppm): 3.72 (s, 6H, 2OCH3), 6.08 (d, J = 9.3 Hz, 1H, H-2 or 15), 6.10 (d, J = 9 Hz, 1H, H-2 or 15), 6.94 (d, J = 9.3 Hz, 1H), 7.05 (d, J = 2.7 Hz, 1H, H-13), 7.15 (d, J = 6.3 Hz, 1H), 7.18 (d, J = 7.8 Hz, 1H), 7,44 (dd, J = 1.2, 8.1 Hz, 1H, H-4), 7.69 (d, J = 9 Hz, 1H), 7.81–7.84 (m, 4H), 7.88 (d, J = 8.1 Hz, 1H), 7.94 (d, J = 8.1 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ (ppm): 54.08 (OCH3), 55.68 (OCH3), 105.98 (C–H), 106.93 (C–H), 114.34 (C–H), 120.41 (C–H), 122.21 (C–H), 123.81 (C), 124.78 (C), 125.55 (C–H), 125.63 (C), 126.67 (C–H), 126.84 (C–H), 127.21 (C–H), 127.26 (C–H), 127.32 (C–H), 127.36 (C–H), 127.71 (C–H), 127.78 (C), 128.23 (C), 128.46 (C–H), 131.30 (C), 132.10 (C), 132.15 (C), 133.75 (C), 133.80 (C), 157.17 (C–O), 155.57 (C–O); HRMS (MALDI-TOF) calcd. for C28H20O2 [M]+: 388.14578. Found: 388.14649.
4.4 Dicamphanate esters: 11
Helicenediol 4 (150 mg, 0.41 mmol) was dissolved in anhydrous dichloromethane (10 mL) under argon. (S)-(−)-Camphanoyl chloride (263 mg, 1.08 mmol) and triethylamine (151 μL, 1.0 mmol) were added successively at 0 °C. The mixture was stirred for 30 min at 0 °C and afterward allowed to warm up to room temperature. The solution was stirred for an additional 12 h, washed with HCl (1 M) and then saturated NaHCO3 and finally dried over MgSO4. After evaporation of the solvent, the final product 11 was obtained in 91% yield (273 mg). Crystallisation from a CH2Cl2/pentane mixture gave yellow crystals. 1H NMR (300 MHz, CDCl3): δ (ppm): 1.08 (s, 6H, 2CH3), 1,15 (s, 12H, 4CH3), 1.75 (m, 2H, 2CH), 2.00 (m, 2H, 2CH), 2.20 (m, 2H, 2CH), 2.57 (m, 2H, 2CH), 6.50 (dd, J = 2.4, 9.3 Hz, 2H, H-2 and H-15), 7.58 (dd, J = 3.3, 9.3 Hz, 2H, H-1 and H-16), 7.62 (d, J = 2.4 Hz, 2H, H-4 and H-13), 7.87 (d, J = 8.4 Hz, 2H), 7.96–8.04 (m, 6H); 13C NMR (75 MHz, CDCl3): δ (ppm): 9.81, 16.98, 29.02, 30.78, 30.86, 54.77, 54.97, 91.01, 118.60, 118.80, 123.80, 127.28, 127.51, 127.89, 129.36, 131.34, 132.51, 133.43, 147.36, 165.91, 178.03; ESI-MS: m/z = 743.2 [M + Na]+; HRMS (MALDI-TOF) calcd. for C46H40O2Na [M + Na]+: 743.2645. Found: 743.2659.
4.5 Resolution of 3,14-dihydroxyhexahelicene: 4
The enantiomeric resolution of the helicenediol 4 was performed by HPLC as follows: PHARMACHIR 314A column (250 × 20 mm); mobile phase: n-C6H14/2-propanol = 80/20 v/v; flow rate 17.0 mL min−1; temperature 289 K; detection: UV, λ = 270 nm. The levorotatory enantiomer (M)-(−)-4 was firstly isolated in 39% yield (>99% ee), followed by the second dextrorotatory enantiomer which was obtained in 30% yield (97.6% ee).
(M)-(−)-3,14-Dihydroxyhexahelicene 4: mp >300 °C (decomposed); Rf = 0.38 (chloroform/ethyl acetate 70:30); [α]D −2287 (c 0.19, acetone); 1H NMR (selected data, 500 MHz, acetone-d6): δ (ppm): 6.38 (dd, J = 2.5, 9 Hz, 2H, H-2 and H-15), 7.27 (d, J = 2.5 Hz, 2H, H-4 and H-13), 7.50 (d, J = 9 Hz, 2H, H-1 and H-16), 7.85 (d, J = 8.5 Hz, 2H, H-5 and H-12), 7.97 (d, J = 8.5 Hz, 2H, H-6 and H-11), 7.99 (d, J = 8.5 Hz, 2H, H-7 and H-10), 8.02 (d, J = 8 Hz, 2H, H-8 and H-9), 8.57 (s, 2H, OH); 13C NMR (selected data, 75 MHz, acetone-d6): δ (ppm): 110.12 (C-4 and C-13), 115.69 (C-2 and C-15), 123.08 (C), 123.85 (2C), 125.61 (C-7 and C-10), 126.58 (C-6 and C-11), 126.95 (C-5 and C-12), 127.19 (C-8 and C-9), 128.37 (2C), 129.11 (C-1 and C-16), 129.89 (2C), 133.42 (C), 133.85 (2C), 155.08 (C-3 and C-14); MS (EI, 50 eV) m/z = 361 ([M + H]+, 100%), 360 (M+, 82%), 316 (9%), 300 (17%), 289 (21%), 274 (4%), 199 (4%), 156 (17%), 150 (21%), 144 (16%), 91 (82%); HRMS (MALDI-TOF) calcd. for C26H16O2 [M+]: 360.11448. Found: 360.11581.
(P)-(+)-3,14-Dihydroxyhexahelicene 4: mp >300 °C (decomposed); Rf = 0.38 (chloroform/ethyl acetate 70:30); [α]D 2371 (c 0.20, acetone); 1H NMR (selected data, 500 MHz, acetone-d6): δ (ppm): 6.38 (dd, J = 2.5, 9 Hz, 2H, H-2 and H-15), 7.27 (d, J = 2.5 Hz, 2H, H-4 and H-13), 7.51 (d, J = 9 Hz, 2H, H-1 and H-16), 7.85 (d, J = 8.5 Hz, 2H, H-5 and H-12), 7.97 (d, J = 9 Hz, 2H, H-6 and H-11), 7.99 (d, J = 8 Hz, 2H, H-7 and H-10), 8.02 (d, J = 8 Hz, 2H, H-8 and H-9), 8.62 (s, 2H, OH); 13C NMR (selected data, 75 MHz, acetone-d6): δ (ppm): 110.12 (C-4 and C-13), 115.70 (C-2 and C-15), 123.08 (C), 123.83 (2C), 125.61 (C-7 and C-10), 126.57 (C-6 and C-11), 126.96 (C-5 and C-12), 127.19 (C-8 and C-9), 128.37 (2C), 129.10 (C-1 and C-16), 129.88 (2C), 133.42 (C), 133.85 (2C), 155.10 (C-3 and C-14); HRMS (MALDI-TOF) calcd. for C26H16O2 [M+]: 360.11448. Found: 360.11624.
The enantiomeric purity of 4 was determined by HPLC as follows: column Chiralcel OD (250 × 4.6 mm); mobile phase: n-C6H14/2-propanol = 80/20 v/v under isocratic conditions; flow rate 1.0 mL min−1; temperature 303 K; detection: UV, λ = 270 nm; retention times of the two enantiomers were 8.019 and 9.993 (the enantiomer exhibiting negative optical rotation (−) was the first to be eluted). The configuration of the helical moiety of helicenediols 4 was assigned on the basis of their CD spectra.
Acknowledgements
The authors are grateful to DGRSRT (Direction générale de la Recherche scientifique et de la Rénovation technologique) of the Tunisian Ministry of Higher Education, Scientific Research and Technology, for financial support.