

1 Introduction
Synthetic dyes, such as acid red 37 (AR-37) and azo dyes, are widely used in the textile, leather, polyamide fiber, cosmetics, and food industries [1]; however, their toxicity, carcinogenic, or genotoxic effects on the environment and humans [2] remain the major drawbacks and limited their wide applications. Moreover, removal of these dyes from the industrial effluents or converting them into useful byproducts before discharge is a great environmental challenge for safe industries [3]. Conventional physicochemical methods including adsorption, floatation, coagulation, incineration, neutralization, reduction, oxidation, electrolysis, and ion-exchange of these dyes are quite expensive, inapplicable for a wide variety of dyes, and may produce a lot of more toxic sludge and byproducts [4]. Therefore, identification of a novel strategy to detoxify these synthetic dyes is of great interest. Motivated by our interest in the development of new pharmacologically relevant architectures for pharmaceutical applications [5,6], we aimed herein to apply subsequent Schiff-base (SB) condensation and palladination strategies for conversion of AR-37 into pharmacologically useful active products.
Our choice for SBs and their organopalladium complexes as target products is because of their unrivaled features such as simplicity of the preparation, remarkable flexibility of their molecular and electronic structure, and their wide range of pharmacological activities including antibacterial, antifungal, antimalarial, antitubercular, antiproliferative, anti-inflammatory, and antiviral [7]. Furthermore, many reported azomethine Pd(II) chelates exhibited significant promising antiviral, fungicidal, bactericidal, and antitumor activities [8]. Moreover, palladium plays a crucial role in the metallotherapeutic drugs and metal-based diagnostic agents [8]. For example, anticancer screening for organopalladium of acetone SBs that bear S-methyldithiocarbazate and S-benzyldithiocarbazate compartments against T-lymphoblastic leukemia cell lines demonstrated excellent cytotoxicity in comparison to the standard anticancer drug, tamoxifen [9].
Inspired by the aforementioned literature outputs and in continuation of our endeavor directed toward designing and development of novel biologically potent materials, we planned herein to apply SB condensation and palladination chemistries for conversion of toxic AR-37 into beneficial pharmacologically (antimicrobial and anticancer) active acid red Schiff bases (ARSBs) and endo-cyclic five/six-membered cyclopalladated complexes.
2 Experimental section
2.1 Reagents and materials
Chemicals were obtained from the following suppliers and used without further purification: AR-37, 4-chlorobenzaldehyde, 4-methoxybenzaldehyde, anthracen-9(10H)-one, 4-nitrobenzaldehyde, and anthracene-9-carbaldehyde (Sigma–Aldrich) and palladium(II) chloride (PdCl2) (Acros).
2.2 Instrumentation
Melting points were measured using a Gallenkamp melting point apparatus; all melting points were measured in open glass capillaries and are uncorrected. Thin layer chromatography was performed with fluorescent silica gel plates HF254 (Merck), and plates were viewed under ultraviolet light at 254 and 265 nm. The elemental analyses for C, H, and N were determined by a Perkin–Elmer Analyzer 2440. Fourier transform infrared (FTIR) spectra were recorded on a Bruker Vector Germany and on a Mattson FTIR spectrophotometer in the range of 400–4000 cm−1 as KBr disc with 2 cm−1 resolution. For signal intensities the following abbreviations were used: br (broad), sh (sharp), w (weak), m (medium), s (strong), vs (very strong). 1H NMR spectra were recorded on a Gemini 200 MHz spectrometer, in dimethyl sulfoxide (DMSO)-d6 solution with calibration to the residual proton solvent signal in DMSO-d6 (1H NMR: 2.52 ppm) against tetramethylsilane (TMS) (δ = 0.00 ppm). Multiplicities of the signals were specified as s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). The mass spectra of the synthesized compounds were measured on GCQ Finnigan MAT. For the mass spectral assignment, peaks are based on 12C with 12.0000 Da.
2.3 Solvent-free microwave-assisted synthesis of ligands (ARSB1–6)
AR-37 SB ligands were synthesized according to the literature procedure [10] assisted with microwave irradiation. Generally, in a microwave flask, the AR-37 (0.25 mmol) and appropriate aromatic aldehydes (0.50 mmol) were introduced and homogenized. The mixture was submitted to microwave irradiation at ambient pressure for 15 min at 200 W (150 °C) without the solvent. The crude product was cooled, washed with CH2Cl2 (5 mL), and crystallized from the suitable solvent. The pure product was filtered off under reduced pressure. The details for the physical, elemental analysis, and spectral data of all ligands (ARSB1–6) are provided in our previous work [10].
2.4 Synthesis of organopalladium complexes (Pd(II)-ARSBs)
Generally, 1 mmol of the parent ligand (ARSB1–6) was added to an ethanolic solution (60 mL) of palladium(II) chloride (1 mmol) containing few drops of concentrated HCl. Then, the mixture was stirred for 4 h under reflux and nitrogen atmosphere, the isolated product was filtered off, washed with cold ethanol (5 mL ×3), ether (5 mL ×3), and finally dried under vacuum. Further crystallization from the methanol affords pure complexes (Pd(II)-ARSBs). Samples of isolated organopalladium complexes were characterized as follow.
[Pd(ARSB1)Cl] (Pd-ARSB1): Obtained as orange crystals in 80.5% yield; mp = 330 °C. FTIR (KBr, cm−1): 3405 (s, br, ν(OH)), 3065 (m, sh, νasym(CH), Ar), 3040 (m, sh, νsym(CH), Ar), 2940 (s, br, ν(CH)), 1644 (s, sh, ν(CO)), 1599 (s, sh, ν(CN), azomethine), 1497, 1421 (s, sh, ), 575 (m, sh, ν(CPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 12.28 (s, 1H), 10.20 (s, 1H), 8.10 (S, 1H), 7.83 (d, J = 6.8 Hz, 1H), 7.78–7.65 (m, 5H), 7.59 (s, 1H), 7.45–7.33 (m, 3H), 7.30–7.19 (m, 2H), 7.11–6.98 (m, 3H), 2.50 (s, br, 2H), 2.02 (s, 2H). EI-MS: m/z (intensity) 883.50 (31.05%) (C32H21Cl3N4O8PdS2 [M]+). Anal. Calcd (%) for C33H21Cl3N4O8PdS2 (M = 866.44 g/mol): C, 44.36; H, 2.44; N, 6.47; S, 7.40. Found (%): C, 44.28; H, 2.56; N, 6.32; S, 7.11.
[Pd(ARSB2)Cl] (Pd-ARSB2): Obtained as red crystals in 85.5% yield; mp = 327 °C. FTIR (KBr, cm−1): 3435 (s, br, ν(OH)), 3195 (s, br, ν(NH)), 3065 (m, sh, νasym(CH), Ar), 3040 (m, sh, νsym(CH), Ar), 2608 (s, br, ν(CH)), 1644 (s, sh, ν(CO)), 1598 (s, sh, ν(CN), azomethine), 1497, 1393 (s, sh, ), 575 (m, sh, ν(CPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 12.33 (s, 1H), 10.28 (s, 1H), 8.56 (s, 1H), 8.21 (d, J = 7.1 Hz, 1H), 7.92 (d, J = 6.9 Hz, 2H), 7.79–7.68 (m, 3H), 7.51 (d, J = 7.2 Hz, 1H), 7.44 (d, J = 1.8 Hz, 1H), 7.38–7.21 (m, 2H), 7.15–7.03 (m, 3H), 3.97 (s, 3H), 3.90 (s, 3H), 2.53 (s, br, 2H). EI-MS: m/z (intensity) 857.50 (5.06%) (C34H27ClN4O10PdS2 [M]+). Anal. Calcd (%) for C34H27ClN4O10PdS2 (M = 857.50 g/mol): C, 47.62; H, 3.17; N, 6.53; S, 7.48. Found (%): C, 47.58; H, 3.25; N, 6.43; S, 7.31.
[Pd(ARSB3)Cl] (Pd-ARSB3): Obtained as brown crystals in 85.3% yield; mp = 319 °C. FTIR (KBr, cm−1): 3397 (s, br, ν(OH)), 3299 (s, sh, NH), 3062 (m, sh, νasym(CH), Ar), 3025 (m, sh, νsym(CH), Ar), 2927 (s, br, ν(CH)), 1658 (s, sh, ν(CO)), 1595 (s, sh, ν(CN), azomethine), 1458, 1403 (s, sh, ), 572 (m, sh, ν(CPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.98 (s, 1H), 10.28 (s, 1H), 8.45 (s, 1H), 8.13 (d, J = 7.3 Hz, 1H), 7.96–7.78 (m, 5H), 7.63–7.49 (m, 4H), 7.37–7.25 (m, 4H), 7.21–7.07 (m, 4H), 6.96 (d, J = 1.8 Hz, 1H), 2.61 (s, br, 2H). EI-MS: m/z (intensity) 956 (0.56%) (C46H30ClN4O7PdS2 [M−OH]+). Anal. Calcd (%) for C46H31ClN4O8PdS2 (M = 973.76 g/mol): C, 56.74; H, 3.21; N, 5.75; S, 6.59. Found (%): C, 56.50; H, 3.25; N, 5.54; S, 6.50.
[Pd(ARSB4)(CH3OH)(H2O)] (Pd-ARSB4): Obtained as brown crystals in 86.0% yield; mp = 341 °C. FTIR (KBr, cm−1): 3436 (s, br, ν(OH)), 3066 (m, sh, νasym(CH), Ar), 2923 (s, br, ν(CH)), 1697 (s, sh, ν(CO)), 1626 (s, sh, ν(CN), azomethine), 1525, 1349 (m, sh, NO2), 1494, 1441 (s, sh, ), 575 (m, sh, ν(CPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.67 (s, 1H), 8.71 (s, 1H), 8.53–8.34 (m, 5H), 8.12–8.01 (m, 3H), 7.77 (d, J = 7.3 Hz, 2H), 7.65 (d, J = 7.0 Hz, 1H), 7.37–7.28 (m, 2H), 2.51 (s, br, 2H), 2.23 (s, 3H), 1.18 (s, 3H). EI-MS: m/z (intensity) 869 (1.03%) (C32H22N6O13PdS2 [M−CH3OH−H2O]+). Anal. Calcd (%) for C33H28N6O15PdS2 (M = 919.16 g/mol) C, 43.12; H, 3.07; N, 9.14; S, 6.98. Found (%): C, 43.00; H, 3.00; N, 9.20; S, 6.90.
[Pd(ARSB5)(CH3OH)(H2O)] (Pd-ARSB5): Obtained as orange crystals in 84.0% yield; mp = 325 °C. FTIR (KBr, cm−1): 3432 (s, br, ν(OH)), 3073 (m, sh, νasym(CH), Ar), 2925 (s, br, ν(CH)), 1664 (s, sh, ν(CO)), 1623 (s, sh, ν(CN), azomethine), 1487, 1445 (s, sh, ), 578 (m, sh, ν(CPd)), 528 (m, sh, ν(OPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 11.98 (s, 1H), 8.75 (s, 1H), 8.58–8.43 (m, 4H), 8.10–7.96 (m, 4H), 7.78–7.63 (m, 5H), 7.59 (d, J = 7.0 Hz, 2H), 7.55 (d, J = 7.1 Hz, 2H), 7.51 (t, J1 = J2 = 7.2 Hz, 2H), 7.48 (d, J = 7.3 Hz, 2H), 7.39–7.25 (m, 5H), 2.51 (s, br, 2H), 2.26 (s, 3H), 1.22 (s, 3H). EI-MS: m/z (intensity) 979 (13.93%) (C48H32N4O9PdS2 [M−CH3OH−H2O]+). Anal. Calcd (%) for C49H38N4O11PdS2 (M = 1029.40 g/mol) C, 57.17; H, 3.72; N, 5.44; S, 6.23. Found (%): C, 57.00; H, 3.82; N, 5.30; S, 6.20.
[Pd(ARSB6)(CH3OH)2]H2O (Pd-ARSB6): Obtained as red crystals in 88.0% yield; mp = 337 °C. FTIR (KBr, cm−1): 3438 (s, br, ν(OH)), 3076 (m, sh, νasym(CH), Ar), 2931 (s, br, ν(CH)), 1658 (s, sh, ν(CO)), 1625 (s, sh, ν(CN), azomethine), 1485, 1448 (s, sh, ), 580 (m, sh, ν(CPd)), 529 (m, sh, ν(OPd)). 1H NMR (200 MHz, DMSO-d6) δ (ppm): 12.27 (s, 1H), 8.78 (s, 1H), 8.61–8.49 (m, 3H), 7.87–7.70 (m, 5H), 7.61–7.51 (m, 2H), 7.22 (d, J = 7.0 Hz, 2H), 7.13 (d, J = 6.9 Hz, 2H), 7.01 (t, J1 = J2 = 7.1 Hz, 2H), 6.88–6.76 (m, 3H), 3.37 (s, 3H), 3.28 (s, 3H), 3.19 (s, 3H), 2.48 (s, br, 2H), 2.35 (s, 2H), 1.16 (s, 6H). EI-MS: m/z (intensity) 1077 (5.52%) (C47H52N7O12PdS2 [M−H]+). Anal. Calcd (%) for C47H53N7O12PdS2 (M = 1078.51 g/mol) C, 52.34; H, 4.95; N, 9.09; S, 5.95. Found (%): C, 52.25; H, 4.80; N, 9.00; S, 5.92.
3 Pharmacological studies
3.1 Antimicrobial activity
3.1.1 Reagents
DMSO, tetracycline (Tetrcy) antibiotic (C22H24N2O8, 444,44 g mol−1), and amphotericin B (Am B) (C47H73NO17, 923.49 g mol−1), antifungal drug were obtained from Sigma Chemical Co (St. Louis, MO).
3.1.2 Microbial cultures
Strains used in this study were from National Organization for Drug Control and Research, Cairo, Egypt. The different strains are Staphylococcus aureus (RCMB 010027) and Streptococcus faecalis as representatives for the G+ bacteria, whereas Escherichia coli (RCMB 010059) and Pseudomonas aeruginosa as the most important G− pathogenic bacteria. Antifungal species are Aspergillus flavus and Candida albicans (RCMB 05038). Stock cultures grown aerobically on nutrient broth agar slants (Hi-Media) at 37 °C were maintained at 4 °C. Precultures containing 105 CFU mL−1, grown aerobically in Mueller Hinton liquid medium (Hi-Media) at 37 °C for 5 h, were used as inoculum for all experiments. Antimicrobial susceptibility of the microbial strains was carried out by agar well diffusion method [11] for the target compounds and standard drugs, Tetrcy, and Am B. The diameter of the zones of inhibition (ZOI, mm) was measured accurately as indicative of antimicrobial activity.
3.1.3 Determination of MIC
As parameters of the antibacterial efficacy, the minimal inhibitory concentrations (MICs) of the new compounds against infection isolates were determined using the macrodilution broth susceptibility test. Freshly prepared Mueller Hinton broth was used as diluent in the macrodilution method. A serial dilution of each compound was prepared within a desired range (0.25–20.00 mM). One milliliter of the stock cultures was then inoculated and tubes were incubated at 37 °C for 24 h, control tubes were assayed simultaneously. MIC was examined visually by checking the turbidity of the tubes.
3.2 In vitro anticancer activity
3.2.1 Cell cultures
Human breast carcinoma (MCF-7) cell lines were obtained from the American type culture collection (Rockville, MD). The cells were grown on RPMI-1640 medium supplemented with 10% inactivated fetal calf serum and 50 mg/mL gentamicin. The cells were maintained at 37 °C in a humidified atmosphere with 5% CO2 and were subcultured two to three times a week.
3.2.2 Antitumor assay
The antitumor activity of new organopalladium complexes was evaluated against MCF-7 cell lines. The cancer cells were grown as monolayers in growth RPMI-1640 medium supplemented with 10% inactivated fetal calf serum and 50 mg/mL gentamicin. The monolayers of 104 cells adhered at the bottom of the wells in 69-well microtiter plates were incubated for 24 h at 37 °C in a humidified incubator with 5% CO2. Fresh medium containing different concentrations of the tested sample in DMSO was added after 24 h of seeding. The monolayers were then washed with sterile phosphate-buffered saline (0.01 M, pH 7.2) with simultaneous treatment of the cells with 100 mL from different dilutions of a tested sample in a fresh maintenance medium and incubated at 37 °C for 48 h. A control of untreated cells was cultured in the absence of a tested sample. A positive control containing doxorubicin (Dox) drug was used for comparison. Six wells were used for each concentration of the examined sample. Every 24 h the observation under the inverted microscope was carried out. The number of the surviving cells was determined by staining the cells with sulforhodamine B stain [12], followed by cell lysing using 33% glacial acetic acid and then recording the absorbance at 590 nm using enzyme-linked immunosorbent assay (ELISA) reader (SunRise, TECAN, Inc,) after well mixing. The absorbance values collected from untreated cells were considered as 100% proliferation. The number of viable cells was determined using ELISA reader as previously mentioned, and the percentage of viability was calculated as {(1 − (ODt/ODc)) × 100}, where ODt is the mean optical density of wells treated with the tested sample and ODc is the mean optical density of untreated cells. The 50% inhibitory concentration (IC50), the concentration required to cause toxic effect in 50% of intact cells, was estimated from graphic plots.
4 Results and discussion
4.1 Chemistry protocol
Microwave irradiation was used to induce a facile simultaneous dual functionalization (chalcone and SB) in AR-37 with aromatic aldehydes (p-chlorobenzaldehyde and p-methoxybenzaldehyde or anthrone) for preparation of a series new chalcone-functionalized ARSB1–3, which undergo further palladination with in situ prepared H2[PdCl4] to give organopalladium complexes (Pd(II)-ARSB1–3) (see Scheme 1).
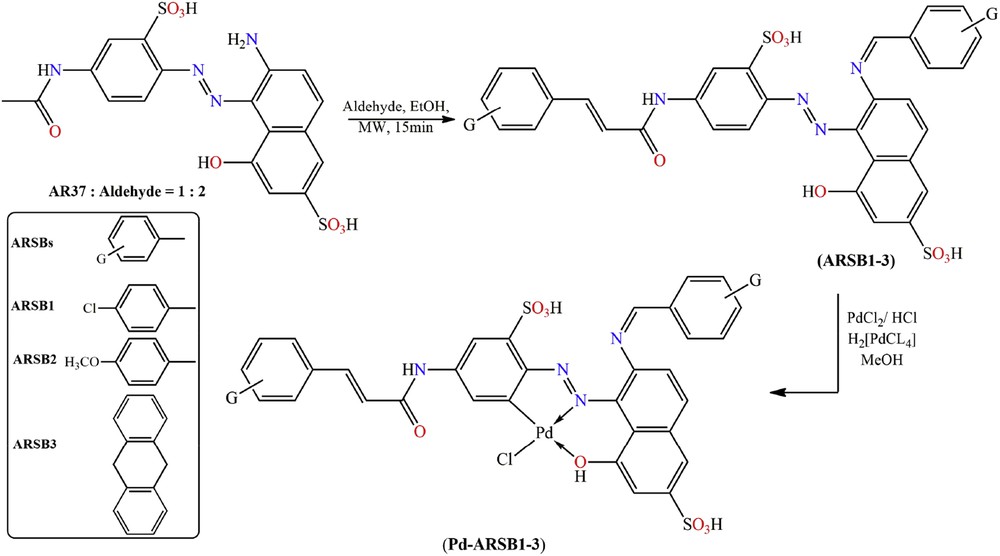
Schematic diagram for the synthesis of ARSB1–3 and their palladination to (Pd(II)-ARSB1–3).
On the other hand, the electron-deficient aromatic aldehyde, p-nitrobenzaldehyde, or 9-anthracenecarbaldehyde reacts with AR-37 by nucleophilic addition along with SB condensation reactions under the same microwave conditions to yield new aminohydrin-functionalized ARSB4,5, which were palladinated with H2[PdCl4] to give organopalladium complexes Pd(II)-ARSB4,5 (see Scheme 2).
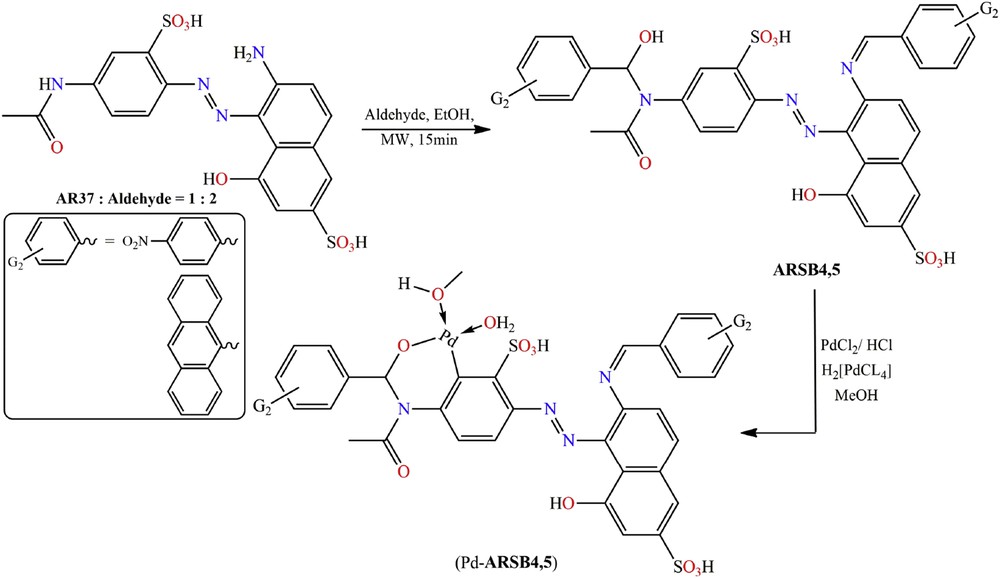
Schematic preparation of ARSB4,5 and their organopalladium compounds, (Pd(II)-ARSB4,5).
However, a highly electron-rich aromatic aldehyde, p-(N,N-dimethylamino)benzaldehyde (DMAB), reacts with AR-37 by an unexpected triple-reactions pathway (condensation, nucleophilic addition, and SB) to give bifunctional chalcone/aminohydrin-functionalized ARSB6, which was further palladinated with H2[PdCl4] affording the corresponding organopalladium complex Pd(II)-ARSB6 (see Scheme 3).
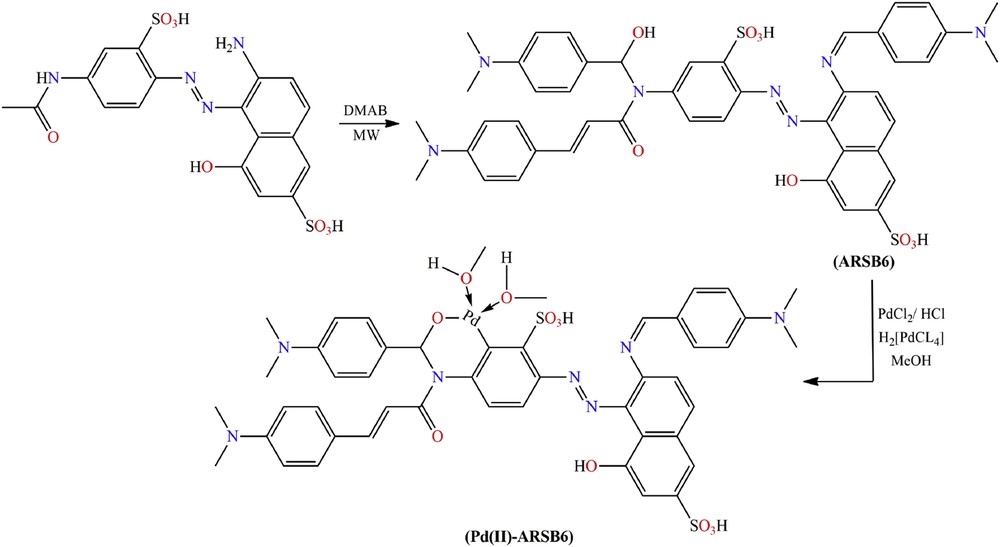
Schematic preparation of ARSB6 and its organopalladium complex.
The structures of a new organopalladium compound were assigned based upon the microanalytical and spectral analysis (IR, 1H NMR, and electron impact mass spectra [EI-MS]).
4.2 Characterizations of the organopalladium compounds
4.2.1 Microanalytical data and mass spectrometry
The new organopalladium compounds (Pd(II)-ARSB1–6) were prepared in convinced yields, and their microanalytical data (C, H, N, and S analyses) are in good agreement with our proposed structural formulas (see Section 2).
The EI-MS of new organopalladium complexes are synonymous with their low stability as revealed by minimum contribution (0.56%–13.93%) of its molecular ion peak [M]+, a molecular mass signature. This reduced stability led to a higher level of fragmentation along with enhanced contribution of produced fragments. The fragmentation pattern of Pd(II)-ARSB5, as a representative example for organopalladium complexes, is depicted in Scheme 4.
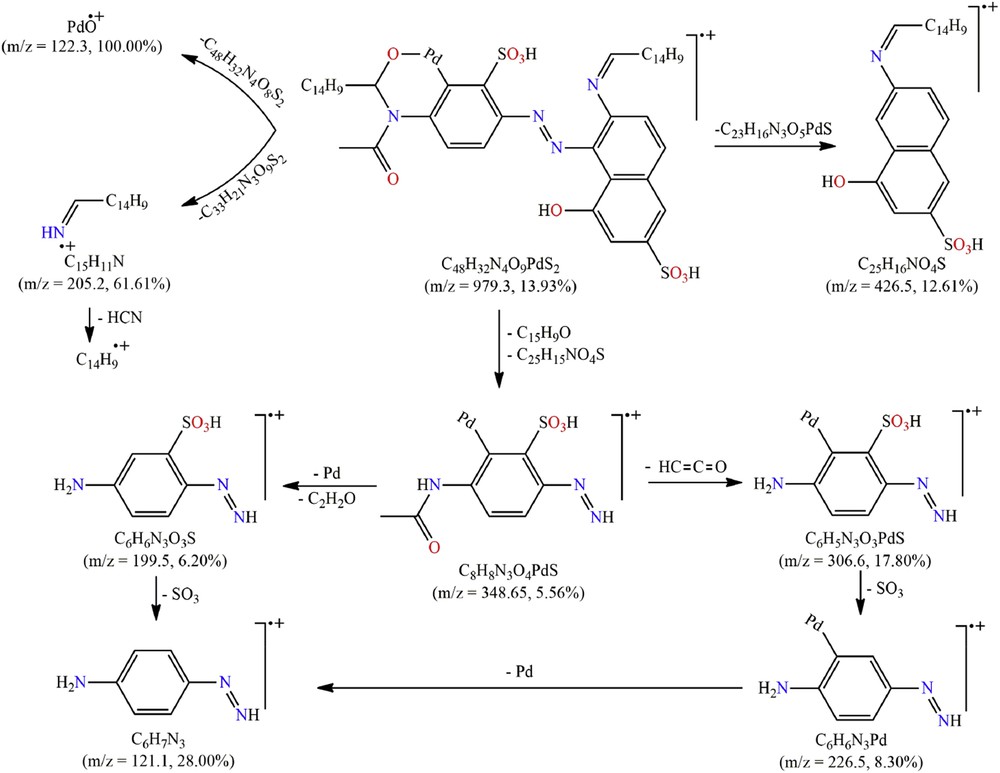
All possible mass fragmentation pathways for (Pd(II)-ARSB5).
4.2.2 IR spectroscopic data
Emergence of new CPd vibrational band along with the remarkable shift in the native bands in the recorded FTIR spectra of organopalladium complexes as compared with those of parent ligands provides strong evidence for the successful palladination of ARSB1–6 and simultaneously assigns the coordination sphere for each Pd(II) ion in its complexes. For instance, the two remarkable displacements of the azo (NN) and phenolic (CO) groups to a lower and higher frequencies by 22–29 and 12–21 cm−1, respectively, in the spectra of Pd(II)-ARSB1–3 confirm the participation of azo nitrogen and phenolic oxygen in the coordination sphere of Pd(II) ion belongs to these complexes. Moreover, consistent with the palladination of the phenyl segment of ARSB is the observation of a new medium stretch around 575 cm−1 assignable to CPd vibrational mode. In conclusion, FTIR data suggested that ARSB1–3 architectures act as tridentate CNO-chelating ligands. On the other hand, the alcoholic-OH stretches, which have been observed in the FTIR spectra of the ARSB4–6 around 2650 cm−1, were missed from the spectra of their organopalladium complexes confirming the deprotonation of the alcoholic oxygen and replacement of alcoholic proton by Pd(II) ion. In addition, the infinitesimal changes in the position of the vibration band characteristics for azo (NN) and phenolic (CO) groups prove the departure of these moieties from the coordination sphere of Pd(II) ion. Finally, the broad band at ca. 3441–3401 cm−1 agrees with the hydrated nature of Pd(II)-ARSB4–6 complexes as suggested by the microanalytical data. Conclusively, the ARSB4–6 act as bidentate CO-chelating ligands.
4.2.3 NMR studies
1H NMR spectra of Pd(II)-ARSBs complexes are dominated by common remarkable features' evidence for their successful preparation: (1) the downfield shift of naphthyl-OH resonance for Pd(II)-ARSB1–3 in comparison to parent ARSB1–3, by δ = +(0.08–0.15) ppm, corroborates the engaging of naphthyl-OH in coordination to Pd(II) ion. (2) The immutability of sulfonic (SO3H), δ = 2.05 ± 0.06 ppm, and azomethine (HCN), δ = 8.10 ± 0.15 ppm, groups in the 1H NMR spectra for all Pd(II)-ARSBs complexes as compared with native ligands suggested nonengagement of these moieties in chelation to Pd(II) ion. (3) The downfield shifts of phenyl-H resonances along with lowering of the proton integral value by one proton provide strong evidence replacement of such aromatic proton with Pd(II) ion (see Fig. 1).
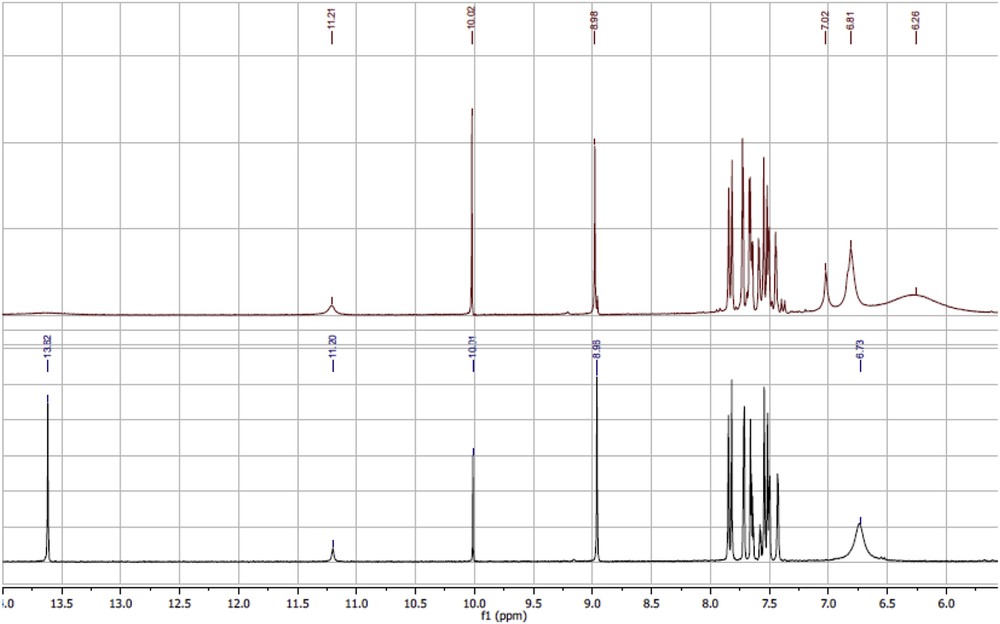
Partial 1H NMR region (6.0–14.0 ppm), for comparison of the phenolic protons' resonance and their splitting patterns in ARSB4 and its Pd(II) complex.
4.3 Pharmacology
4.3.1 Antimicrobial activity
Parent ARSB ligands and Pd(II)-ARSB complexes were in vitro evaluated in comparison with standard antibiotics (antibacterial, Tetrcy; antifungal, Am B) for their efficacy to curb the growth of a common panel of pathogenic microbial species including S. aureus and S. faecalis, G+ bacteria, E. coli and P. aeruginosa, G− bacteria, and A. flavus and C. albicans, fungal pathogens. Generally, palladination of ARSBs synergistically enhanced the antibacterial and antifungal (i.e., antimicrobial) efficacies in comparison to the native ARSBs as revealed from the diameters of ZOI (Fig. 2). Also, noticeable, organopalladium complexes (and Pd(II)-ARSB1–6) exhibited a wide range of antibacterial activities from inactive derivative such as Pd-ARSB3 to an extremely potent one Pd-ARSB1 (MICE. coli/P. aeruginosa = 1.18/2.65 mM and MICS. aureus/S. faecalis = 9.85/11.02 mM). These promoted bactericidal activities of Pd(II) complexes can be explained on the basis of Overtone's concept [13] and Tweedy's chelation theory [14], which attributed the higher potency of complexes against pathogens to their enhanced liposolubility action owing to diminishing of the polarity of the Pd(II) ion through its coordination and partial sharing of its cationic charge with the donor sites of ARSBs. This enhanced lipophilicity facilitates penetration and diffusion of the target complex into the lipid membrane of pathogen along with blocking metal active enzymatic binding sites in the microorganisms.
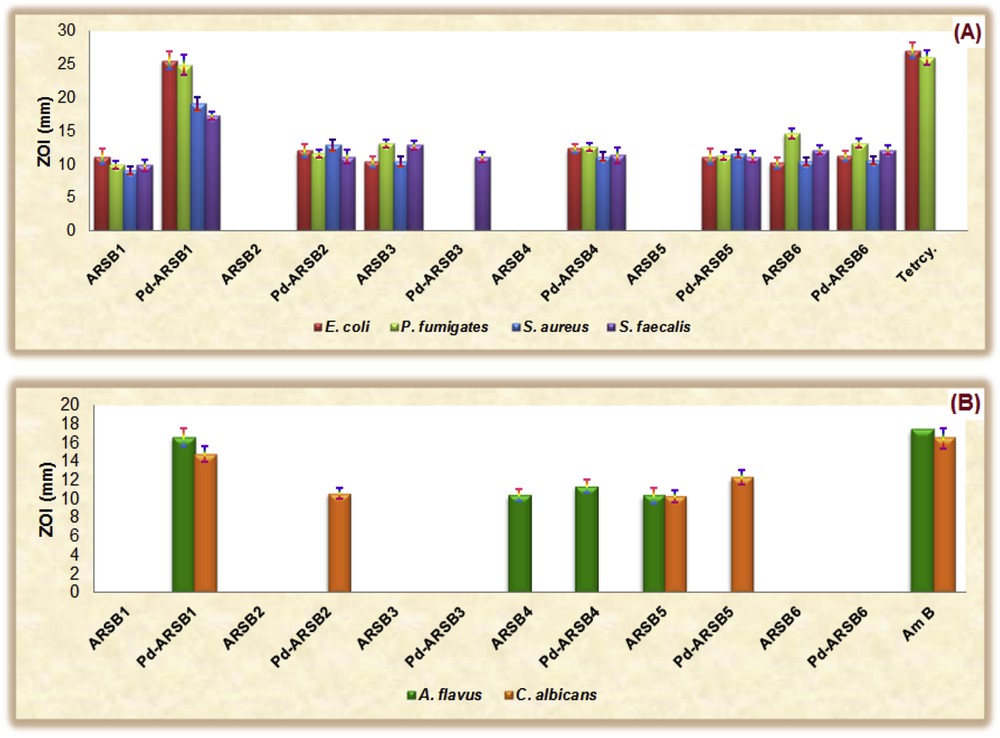
Graph of zone of inhibition (ZOI, mm) for most ARSBs and their organopalladium complexes against (A) bacterial cells and (B) fungal strains.
Noteworthy, most tested compounds are less potent as fungicides where nearly all parent ARSBs are antifungal inactive except ARSB4, which exhibits moderate inhibitory activity against A. flavus, and a more potent antifungal agent, ARSB5, which has the ability to eradicate both fungal pathogens (A. flavus and C. albicans). However, organopalladium complexes demonstrated higher antifungal efficacy than the native ligands with significantly promoted antifungal activity registered for Pd-ARSB1. In conclusion, among assaying compounds, Pd-ARSB1 was signed as the most potent and broad-spectrum antimicrobial agent (MICE. coli/P. aeruginosa = 1.18/2.65 mM and MICS. aureus/S. faecalis = 9.85/11.02 mM) and (MICA. flavus/C. albicans = 12.50/14.10 mM). Inspired with this notice, further structural refinement along with intensive microbiological assessments for this organopalladium complex may open new avenue for generation of new promising antibiotic candidates.
4.3.2 Preliminary in vitro antitumor assay
Preliminary in vitro antitumor survey of new organopalladium complexes was investigated in comparison to a common antitumor drug, doxorubicin (Dox) (C27H29NO11, 543.52 g/mol), against human breast carcinoma (MCF-7) cell lines. Noticeable, all selected complexes exhibit growth inhibitory effects against MCF-7 cell lines within structure–activity relationship profile as revealed from the depicted cell viability values in Fig. 3. Interestingly, refinement of the ARSB skeleton can modulate the anticancer efficacy of the organopalladium complex where the IC50 values for Pd-ARSB5 and 6 are 6.63 and 20.2 μg/mL, respectively; consequently, Pd-ARSB5 is a threefold more cytotoxic against MCF-7 cell lines than Pd-ARSB6. On the contrary, the Pd-ARSB4 is fully inactive against MCF-7 cells. Memorably, the remarkable effectiveness of Pd-ARSB5 as an MCF-7 antitumor agent in comparison to other organopalladium complexes could be attributed to the promoted nonpolarizing effect of two anthranyl side groups, which significantly affected the DNA-organopalladium van der Waals interaction, whereas the hydrophobic nature of anthranyl segments enhances the van der Waals energy of Pd-ARSB5, causing an improvement in DNA-organopalladium association via van der Waals forces, which played a crucial role than hydrogen bonding in the binding of different anticancer agents to DNA [15].
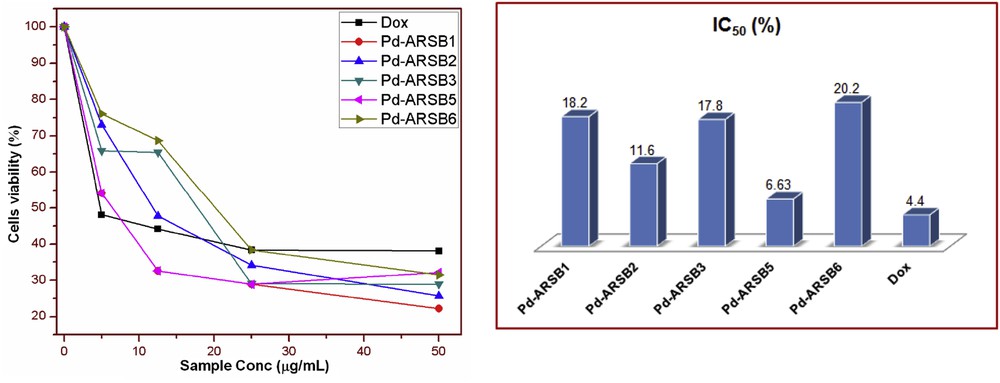
Concentration-dependent antitumor efficacy assessment and IC50 values of new organopalladium complexes against human MCF-7 cell lines.
5 Conclusions
Our protocol has succeeded in the conversion of harmful dye (AR-37) into new pharmacological candidates with synergistic antibacterial, antifungal, and anticancer performance. In this endeavor, the biocidal activities of the newly synthesized endo-cyclic five/six-membered cyclopalladated complexes (Pd(II)-ARSBs) have been investigated against common pathogenic G+ and G− bacterial and fungal strains. Moreover, the antitumor efficacy of these complexes against human breast carcinoma (MCF-7) cell lines has been addressed. The recorded ZOIs and MIC values revealed a moderate to superb broad-spectrum antimicrobial efficacy of cyclopalladated complexes (Pd(II)-ARSBs) in comparison to the parent ARSB standard antibiotic with a preferential efficacy to act as bactericides than fungicides. Structure–activity relationship for new organopalladium complexes against MCF-7 cell lines revealed a correlation between the hydrophobicity of the target compound, as tuned by the substituents, and its antitumor activity.