

1 Introduction
Pyrazolo[3,4-d]pyrimidines are of pharmaceutical interest in a variety of therapeutic areas [1]. They have attracted much attention in drug discovery programs because of their structural resemblance to purine nucleobases. In recent years, researchers have reported the use of purine derivatives of allopurinol (Scheme 1, compound 1) as kinase inhibitors [2], antiviral agents [3], adenosine antagonists [3c,4], glutamate modulators [5], and antitubercular agents [6].

Strategy for the synthesis of C-3 arylated pyrazolo[3,4-d]pyrimidines.
Recently, the direct CH arylation of aromatic and heteroaromatic compounds has proved to be an attractive alternative method to traditional cross-coupling reactions [7]. Contrary to palladium-catalyzed couplings, including Suzuki et al. [8], Stille [9], or Negishi et al. [10] couplings, which require the preparation of organometallic reagents (boronic acids, tin, or zinc derivatives), preliminary functionalization is not carried out in direct CH arylation. In our research program on the stimulation of CH activation [11], we suggest a new approach for the direct and selective C-3 arylation of substituted 1H-pyrazolo[3,4-d]pyrimidines using Pd(OAc)2 as a catalyst, with various ligands and bases. The results obtained with this approach in the present investigation are reported and discussed herein.
2 Results and discussion
The general synthetic pathway for the preparation of a series of 1H-pyrazolo[3,4-d]pyrimidines is shown in Scheme 1.
In the pyrazolopyrimidine ring system, the chloro substituent (as a leaving group) was introduced at C-4, which was the most reactive site for nucleophilic attack. The high-temperature reaction of the commercially available allopurinol 1 with excess phosphorus oxychloride in the presence of NN-dimethylaniline gave an intermediate product 2 [12], which was then allowed to react with methyl iodide so as to afford the corresponding 4-chloro-1-methyl-1H-pyrazolo[3,4-d]pyrimidine [13] 3. The use of 1 equiv of methyl iodide allowed us to selectively obtain N1 methylated isomer. Compound 3 also demonstrated the Suzuki cross-coupling reaction [14]. The 4-chloro substituent of 3 was directly displaced with phenyl to generate compound 4, affording an additional opportunity for the selective functionalization of this molecule.
We started our optimization by investigating the direct arylation of 4-phenyl-1-methyl pyrazolopyrimidine 4 with 4-bromotoluene 5a. The reaction conditions were the same as those already used in our previous study [11f]. Compound 4 (1 equiv) was allowed to react with 5a (2 equiv) in the presence of palladium(II) acetate (10 mol %), triphenylphosphine (20 mol %), and K2CO3 (2 equiv) in toluene at 110 °C for 48 h, but no C-3 arylation was observed (Table 1, entry 1). On replacing the ligand by tricyclohexylphosphine (20%), direct CH arylation then took place at the 3-position of 4 to give product 7 in 28% yield (Table 1, entry 3). Using another base such as cesium carbonate and increasing the amount of Pd(OAc)2 and tricyclohexylphosphine to 20% and 40% mol, respectively, no noticeable improvement in the reaction was detected (Table 1, entries 4 and 5). Using 1,10-phenanthroline instead of tricyclohexylphosphine in the presence of Pd(OAc)2, 4-iodotoluene (2 equiv), and Cs2CO3 (3 equiv) in DMA at 165 °C produced 7 in 65% yield (Table 1, entry 7). With K3PO4 as additive conversion improved to 80% (Table 1, entry 8), whereas using 4-bromotoluene instead of 4-iodotoluene led to a decrease in conversion to 72% (Table 1, entry 9). When decreasing the amount of Pd(OAc)2 and 1,10-phenanthroline to 10 and 20 mol %, respectively, the yield of the desired product 7 diminished (60%, Table 1, entry 10).
Optimization of the C-3 arylation reaction of 4.Image 1
| Entry | Catalyst (mol %) | Ligand (mol %) | Base (equiv) | X (equiv) | Solvent | T (°C) | % Yield |
| 1 | Pd(OAc)2 (10%) | PPh3 (20%) | K2CO3 (2.0) | Br (2.0) | Toluene | 110 | 0 |
| 2 | Pd(OAc)2 (10%) | PPh3 (20%) | K2CO3 (2.0) | I (2.0) | Toluene | 110 | 0 |
| 3 | Pd(OAc)2 (10%) | PCy3 (20%) | K2CO3 (2.0) | I (2.0) | Dioxane | 120 | 28 |
| 4 | Pd(OAc)2 (10%) | PCy3 (20%) | Cs2CO3 (2.0) | I (2.0) | Dioxane | 120 | 35 |
| 5 | Pd(OAc)2 (20%) | PCy3 (40%) | Cs2CO3 (3.0) | I (2.0) | Dioxane | 120 | 40 |
| 6 | Pd(OAc)2 (20%) | Phen (40%) | K2CO3 (3.0) | I (2.0) | DMA | 165 | 48 |
| 7 | Pd(OAc)2 (20%) | Phen (40%) | Cs2CO3 (3.0) | I (2.0) | DMA | 165 | 65 |
| 8 | Pd(OAc)2 (20%) | Phen (40%) | Cs2CO3 (3.0)/K3PO4 (2.5) | I (2.0) | DMA | 165 | 80 |
| 9 | Pd(OAc)2 (20%) | Phen (40%) | Cs2CO3 (3.0)/K3PO4 (2.5) | Br (2.0) | DMA | 165 | 72 |
| 10 | Pd(OAc)2 (10%) | Phen (20%) | Cs2CO3 (3.0)/K3PO4 (2.5) | I (2.0) | DMA | 165 | 60 |
| 11 | Pd(OAc)2 (20%) | – | Cs2CO3 (3.0)/K3PO4 (2.5) | I (2.0) | DMA | 165 | 0 |
We tested the latter conditions without using 1,10-phenanthroline, but no C3 arylation was detected (Table 1, entry 11). This result displays the crucial role of the ligand in CH activation.
Finally the best results were obtained in the presence of palladium (II) acetate (20 mol %), 1,10-phenanthroline (40 mol %), 4-iodotoluene (2 equiv), Cs2CO3 (3 equiv), and K3PO4 (2.5 equiv) in DMA at 165 °C for 48 h (entry 8).
Comparable yields were attained using diverse aryl iodides bearing ortho, meta, and para substituents (Table 2, entries 1–7). This result highlights the great adaptability of our strategy. Indeed, we were pleased to observe that aryl-bearing electron-rich (MeO, Me) or electron-poor (CF3, CN, and so forth) substituents in the C-3 position were well tolerated. The molecular structure of compound 7 was confirmed by X-ray crystallographic analysis and proved that CH arylation occurred regioselectively at position 3 (Fig. 1).
Scope and limitation study of C-3 arylation.Image 2
| Entry | Ar-I | Product | Yield (%)a |
| 1 | Image 3 | Image 4 | 80b |
| 2 | Image 5 | Image 6 | 71b |
| 3 | Image 7 | Image 8 | 65b |
| 4 | Image 9 | Image 10 | 69b |
| 5 | Image 11 | Image 12 | 63b |
| 6 | Image 13 | Image 14 | 49/61c |
| 7 | Image 15 | Image 16 | 43/59c |
a Isolated yield after column chromatography.
b Conditions: Pd(OAc)2 (20 mol %), 1,10-phenanthroline (40 mol %), aryl iodide (2.0 equiv), Cs2CO3 (3.0 equiv), and K3PO4 (2.5 equiv) in DMA.
c Using 4 equiv of ArI.
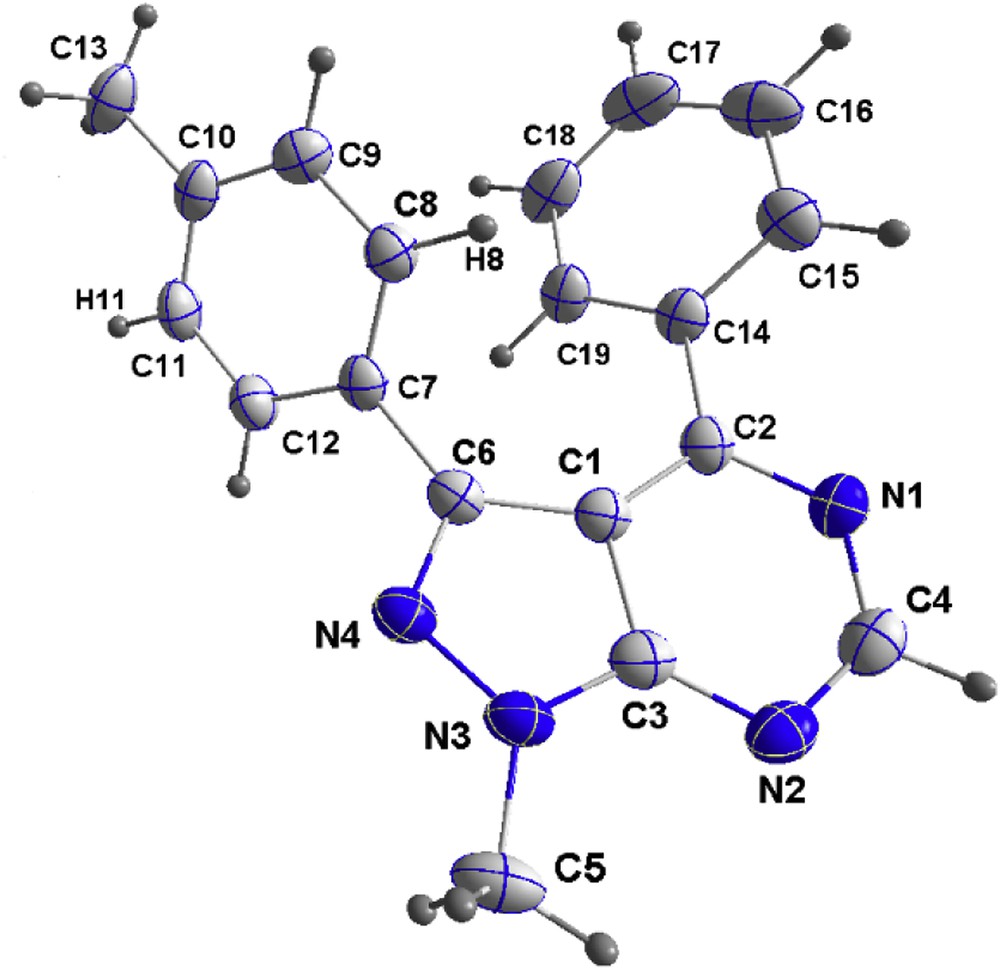
X-ray structure of compound 7.
Afterward, we prepared the compound 4-methoxy-1-methyl-1H-pyrazolo[3,4-d]pyrimidine [15] 14 by reacting 4-chloro-1-methyl-1H-pyrazolo[3,4-d]pyrimidine 3 with sodium methoxide and methyl alcohol in THF, then we tested the optimal conditions found (Table 1, entry 8) on these compounds functionalized at position 4 (3 and 14). No C-3 arylation was detected, and only degradation was observed (Scheme 2).
C-3 arylation of 3 and 14.
On the basis of the newly reported studies on the C-3 arylation reaction [11b,11c], a probable mechanism for the C-3 arylation on pyrazolopyrimidine is put forward (Scheme 3). Pd(OAc)2 and ligand form a Pd(II) intermediate (complex 1, Scheme 3) followed by a Pd(II)–pyrazolopyrimidine complex (complex 2, Scheme 3). Then an oxidative addition of aryl halide (X = I or Br) results in a Pd(IV) complex (complex 3, Scheme 3). After reductive elimination, the required compound 7 is achieved, and complex 4 is formed. In the end, complex 4 is transformed into complex 1.

Possible mechanism for C-3 arylation.
3 Conclusion
It has been shown for the first time that Pd-catalyzed regioselective direct C-3 arylation of substituted 1H-pyrazolo[3,4-d]pyrimidine is possible by using 1,10-phenanthroline as a ligand. Satisfactory to excellent yields were achieved for a wide range of aryl coupling partners with both electron-rich and electron-poor substituents, providing access to a library of various 1H-pyrazolo[3,4-d]pyrimidine compounds.
4 Experimental section
4.1 Materials and instrumentation
All reagents were purchased from commercial suppliers and were used without further purification. Microwave-assisted reactions were carried out in a Biotage Initiator microwave synthesis instrument and temperatures were measured by an IR sensor. The reactions were monitored by thin-layer chromatography analysis using silica gel (60 F254) plates. Compounds were visualized by UV irradiation. Flash column chromatography was performed on silica gel 60 (230–400 mesh, 0.040–0.063 mm). Melting points (mp [°C]) were taken on samples in open capillary tubes and are uncorrected. 1H and 13C NMR spectra were recorded on a spectrometer at 250 MHz (13C, 62.9 MHz) or 400 MHz (13C, 100 MHz). Chemical shifts are given in parts per million from tetramethylsilane as an internal standard. The following abbreviations are used for the proton spectra multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; qt, quintuplet; and m, multiplet. Coupling constants (J) are reported in Hertz (Hz). High-resolution mass spectra (HRMS) were performed on a Maxis Bruker 4G by the “Federation de Recherche” ICOA/CBM (FR2708) platform.
4.2 General procedure for direct arylation
A mixture 1-methyl-4-phenyl-1H-pyrazolo[3,4-d]pyrimidine 4 (0.1 g, 0.47 mmol), aryl iodide 5b (2 equiv), Cs2CO3 (1.42 mmol, 3 equiv), K3PO4 (1.18 mmol, 2.5 equiv), 1,10-phenanthroline (0.19 mmol, 0.4 equiv), and Pd(OAc)2 (0.094 mmol, 0.2 equiv) in DMA (3 mL) was prepared under an atmosphere of argon. The resulting mixture was flushed with argon and heated to 165 °C for 48 h. After completion of the reaction, the mixture was then allowed to cool to room temperature, and the solvent was removed under reduced pressure, water (15 mL) was added, and the resulting aqueous phase was extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by column chromatography on silica gel (EtOAc/petroleum ether).
1-Methyl-4-phenyl-3-(p-tolyl)-1H-pyrazolo[3,4-d]pyrimidine (7): Yellow solid; Yield (0.114 g, 0.38 mmol), 80%; mp 128–130 °C. 1H NMR (400 MHz, methanol-d4) δ: 8.91 (s, 1H), 7.41–7.31 (m, 3H), 7.19 (dd, J = 6.8, 1.4 Hz, 2H), 6.95 (dd, J = 10.3, 3.0 Hz, 4H), 4.10 (s, 3H), 2.27 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ: 163.25, 154.1, 153.9, 145.5, 138.3, 136.2, 130.0, 129.7 (2C), 129.1, 129.0 (2C), 128.1 (2C), 127.7 (2C), 109.1, 32.8, 19.9. HRMS (ESI): calcd for C19H17N4 [M+H]+ 301.1448; found 301.1452.
3-(4-Methoxyphenyl)-1-methyl-4-phenyl-1H-pyrazolo[3,4-d]pyrimidine (8): Yellow solid; Yield (0.107 g, 0.34 mmol), 71%; mp 139–141 °C. 1H NMR (400 MHz, CDCl3-d) δ: 9.09 (s, 1H), 7.49 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.31–7.19 (m, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 4.23 (s, 3H), 3.79 (s, 3H). 13C NMR (101 MHz, CDCl3-d) δ: 163.3, 160.0, 154.9, 154.6, 145.3, 136.8, 130.8 (2C), 130.3, 130.2 (2C), 128.2 (2C), 125.0, 113,6 (2C), 109.6, 55.5, 34.1. HRMS (ESI): calcd for C19H17N4O [M+H]+ 317.1397; found 317.1396.
1-Methyl-4-phenyl-3-[4-trifluoromethyl)phenyl]-1H-pyrazolo[3,4-d]pyrimidine (9): White solid; Yield (0.109 g, 0.31 mmol), 65%; mp 145–147. 1H NMR (400 MHz, methanol-d4) δ: 9.04 (s, 1H), 7.43 (m, 5H), 7.36 (d, J = 8.0 Hz, 2H), 7.25 (dd, J = 7.5, 3.0 Hz, 2H), 4.22 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ: 164.6, 155.8, 155.5, 145.2, 137.6, 137.4, 131.65 (2C), 131.3 (q, 2JCq,F = 32.3 Hz), 131.1 (2C), 131.0 (2C), 129.2 (2C), 125.8 (q, 3JCH,F = 4 Hz), 125.5 (q,1JCq,F = 273 Hz), 110.8, 34.4. HRMS (ESI): calcd for C19H14F3N4 [M+H]+ 355.1165; found 355.1167.
1-Methyl-4-phenyl-3-(m-tolyl)-1H-pyrazolo[3,4-d]pyrimidine (10): Yellow oil; Yield (0.099 g, 0.33 mmol), 69%. 1H NMR (400 MHz, methanol-d4) δ: 8.95 (s, 1H), 7.43–7.32 (m, 3H), 7.21 (dd, J = 11.1, 4.7 Hz, 2H), 7.08–7.04 (m, 3H), 6.76 (s, 1H), 4.14 (s, 3H), 2.04 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ: 163.2, 154.1, 153.9, 145.5, 137.3, 136.2, 131.6, 130.3, 130.0, 129.7 (2C), 128.8, 127.6 (2C), 127.5, 125.9, 109.2, 32.8, 19.85. HRMS (ESI): calcd for C19H17N4 [M+H]+ 301.1447; found 301.1450.
1-Methyl-4-phenyl-3-(o-tolyl)-1H-pyrazolo[3,4-d]pyrimidine (11): Yellow solid; Yield (0.090 g, 0.30 mmol), 63%; mp 132–134 °C. 1H NMR (400 MHz, CDCl3-d) δ: 9.11 (s, 1H), 7.42 (t, J = 4.9 Hz, 2H), 7.24 (dd, J = 8.6, 4.0 Hz, 2H), 7.17–7.07 (m, 4H), 7.03 (d, J = 9.3 Hz, 1H), 4.24 (s, 3H), 1.84 (s, 3H). 13C NMR (101 MHz, CDCl3-d) δ: 163.5, 155.45, 154.5, 145.3, 137.8, 136.5, 133.1, 130.8, 130.65, 130.5, 123.0 (2C), 129.3128.2 (2C), 126.1, 111.3, 34.5, 20.1. HRMS (ESI): calcd for C19H17N4 [M+H]+ 301.1448; found 301.1447.
4-(1-Methyl-4-phenyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzonitrile (12): White solid; Yield (0.090 g, 0.29 mmol), 61%; mp 135–137 °C. 1H NMR (400 MHz, methanol-d4) δ: 9.08 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.49–7.44 (m, 3H), 7.39 (d, J = 8.0 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 4.25 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ: 162.5, 156.3, 154.9, 149.1, 140.2, 138.0, 134.7 (2C), 134.5 (2C), 133.1, 130.8 (2C), 130.5 (2C), 119.5, 113.1, 112.9, 34.5. HRMS (ESI): calcd for C19H14N5 [M+H]+ 312.1244; found 312.1243.
Methyl 4-(1-methyl-4-phenyl-1H-pyrazolo[3,4-d]pyrimidin-3-yl)benzoate (13): White solid; Yield (0.097 g, 0.28 mmol), 59%; mp 158–160 °C. 1H NMR (400 MHz, methanol-d4) δ: 9.07 (s, 1H), 7.83–7.80 (m, 2H), 7.48–7.43 (m, 4H), 7.35–7.30 (m, 3H), 4.24 (s, 3H), 3.91 (s, 3H). 13C NMR (101 MHz, methanol-d4) δ: 167.5, 164.9, 155.75, 155.5, 147.1, 139.9, 137.8, 131.6, 131.3 (2C), 130.7, 130.6 (2C), 129.75 (2C), 129.3 (2C), 110.7, 52.7, 34.4. HRMS (ESI): calcd for C20H17N4O2 [M+H]+ 345.1345; found 345.1346.