

1 Introduction
Alzheimer's disease (AD) is the main cause of neurodegenerative disorders. Today, dementia is estimated to affect more than 46 million people worldwide, with the number doubling every 20 years, and the World Health Organization estimates that the number may reach 131 million by 2050 [1]. The profound irreversible memory loss and progressive cognitive dysfunction, together with impaired language skill and personality changes, make AD a terrible disease for patients and their families, and render it a potential social and economic crisis of 21st century [2].
Currently, the approved drugs (four acetylcholine inhibitors and memantine, a noncompetitive antagonist of N-methyl-d-aspartate receptor) are weakly effective and their efficiency/cost ratios are questionable. Therefore, the design of new efficient drugs to stop AD evolution at the early stages has become one of the most important challenges in medicinal chemistry.
A normal brain requires metal ions for a number of important cellular processes. As such, the brain contains relatively high concentrations of transition metals, especially Fe, Zn, and Cu, that take part in the neuronal activity within the synapses (Zn(II) in particular) and ensure the function of various metalloproteins and metalloenzymes. However, the accumulation of Cu, Fe, and Zn within senile plaques reaches 400, 950, and 1100 μM, respectively, three to five times the concentration observed in a normal brain [3]. The strong affinity of these metal ions for Aβ amyloids promotes Aβ aggregation [4], which is considered as a main factor for amyloid toxicity in AD or other pathological condition [5]. Moreover, the activation of copper–amyloid by an endogenous reductant is probably at the origin of the redox stress tightly correlated with AD pathogenesis [6]. Iron–amyloids are weaker promoters of an oxidative stress, mainly due to iron precipitation [7]. Consequently, we focused our attention on the design of specific copper chelators to restore metal homeostasis in AD brain by extraction of copper ions from pathological sinks (amyloids being the major one) and transfer them to copper-carrier proteins, to recycle them in their physiological role [8]. On the basis of such hypothesis, we designed a series of ligands that are expected to (1) extract Cu(II) from the Cu–Aβ complex, (2) release the copper ion under physiological conditions, and (3) inhibit the production of reactive oxygen species induced by Cu–Aβ in the presence of a physiological reductant. Therefore, they may be able to regulate copper homeostasis and reduce the oxidative stress in AD brain. These ligands must be selective for copper with respect to zinc chelation, to avoid the depletion of zinc proteins by an exogenous ligand as potential drug candidate. In fact, the lack of selectivity for copper and the coordination of zinc by clioquinol and PBT2 might be at the origin of their failure in clinical trials [9]. On the basis of our knowledge on tetradendate ligand PA1637, which suffer from a low solubility [10], we decided to design new tetradentate copper ligands based on a mono-8-aminoquinoline motif to reduce the size and the molecular weight of the ligands and consequently facilitate their passage through the blood–brain barrier.
Here, we report the syntheses and the characterization of prototype ligands of this new series, named TDMQ (tetradendate monoquinolines). For the sake of stability, these ligands were prepared as hydrochloric salts. In a preliminary study of complexation, TDMQ5 was found to efficiently chelate copper(II) as a 1/1 metal/ligand complex, whereas zinc(II) was not chelated in the same conditions.
2 Results and discussion
These TDMQ ligands consist of an 8-aminoquinoline nucleus and a bidentate amine side-chain attached at C2 of the quinoline moiety, both fragments taking part in copper chelation to provide the square planar tetradentate coordination sphere, which is preferred for efficient chelation and selectivity for copper(II).
2.1 Syntheses
Structural modulations of the 8-aminoquinoline series were carried out by preparing compounds having (1) different substituents on the quinoline nucleus, a chlorine atom at C5 and C7 positions for TDMQ5 and TDMQ20, or CF3 group at C6 for TDMQ16 and TDMQ22, and (2) a side chain of variable length: one methylene group in the proximal part of the side chain for TDMQ5 and TDMQ16, or two methylenes for TDMQ20 and TDMQ22. The structures of these ligands are summarized in Scheme 1.
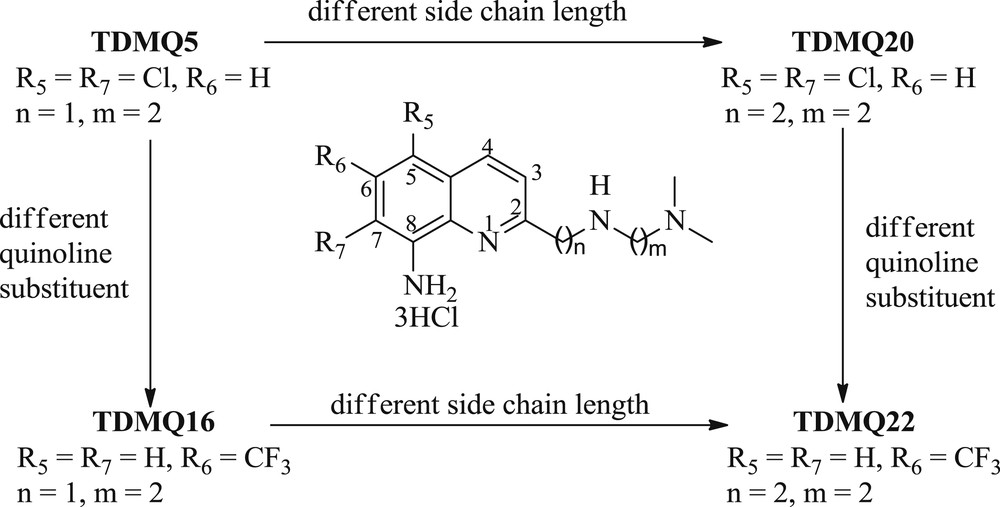
Structure of TDMQ ligands having different side chain lengths and different quinoline substituents.
2.1.1 Synthesis of TDMQ5 and TDMQ16 (short side chain, n = 1)
TDMQ5 was prepared starting from 3,5-dichloroaniline as presented in Scheme 2 (A = 3,5-dichloro-2-nitroaniline). The quinoline ring was synthesized in the presence of acetaldehyde in hydrochloric acid to yield 5,7-dichloro-2-methylquinoline (B). Nitration of B in a mixture of HNO3/H2SO4 produced 8-nitro-5,7-dichloro-2-methylquinoline (D). Reduction of the nitro group was achieved by iron/acetic acid in ethanol. After protection of the amino group, the 2-methyl substituent was oxidized by selenium dioxide, and the side chain was introduced by reductive amination of the aldehyde function by N,N-dimethyl-1,2-ethanediamine in the presence of triacetoxyborohydride. The resulting ligand was then protonated in a diethyl ether solution of hydrochloric acid.
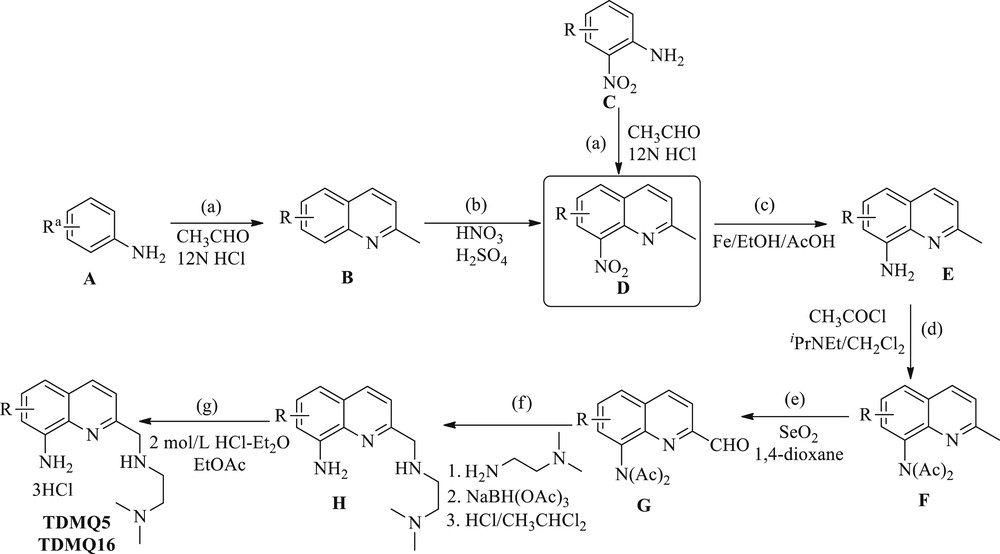
Synthesis of TDMQ5 Rand TDMQ16R having a short proximal side chain (n = 1). aR stands for 5,7-dichloro- for TDMQ5 and 6-trifluoromethyl- for TDMQ16. (a) CH3CHO/12NHCl, 4 h; (b) HNO3/H2SO4, rt, 2 h; (c) Fe, AcOH, EtOH, 80 °C, 1 h; (d) CH3COCl, iPrNEt, CH2Cl2, rt, 12 h; (e) SeO2, 1,4-dioxane, 80 °C, 4 h; (f) NH2(CH2)2N(CH3)2, CH3CHCl2, Ar, 1 h, then NaBH(OAc)3, 12 h, then HCl, CH2Cl2; and (g) 2 mol/L HCl-Et2O, EtOAc.
TDMQ16 was synthesized starting from 2-nitro-4-(trifluoromethyl)aniline (C, Scheme 2), and the quinoline ring was synthesized as described above for TDMQ5, then providing 2-methyl-8-nitro-6-(trifluoromethyl)quinoline (D). The following steps of the synthesis of TDMQ16 were the same as for TDMQ5 (reduction of 8-nitro, protection of amino group, oxidation of 2-methyl, introduction of the side chain, and protonation). The overall yields were 7% and 13% for TDMQ5 and TDMQ16, respectively, from commercially available starting materials.
2.1.2 Synthesis of TDMQ20 and TDMQ22 (long side chain, n = 2)
TDMQ20 was synthesized starting from 5,7-dichloro-2-methyl-8-nitroquinoline (D, Scheme 3). Subsequent vinylation of D was carried out in a mixture of ferric chloride and potassium persulfate in N,N-dimethylacetamide (DMA) to yield 5,7-dichloro-8-nitro-2-vinylquinoline (K). Introduction of N,N-dimethyl-1,2-ethylenediamine as the side chain afforded compound L. Reduction of the nitro group of L by iron/acetic acid in ethanol provided the 8-aminoquinoline (M). This ligand was then protonated in a diethyl ether solution of hydrochloric acid to yield TDMQ20.
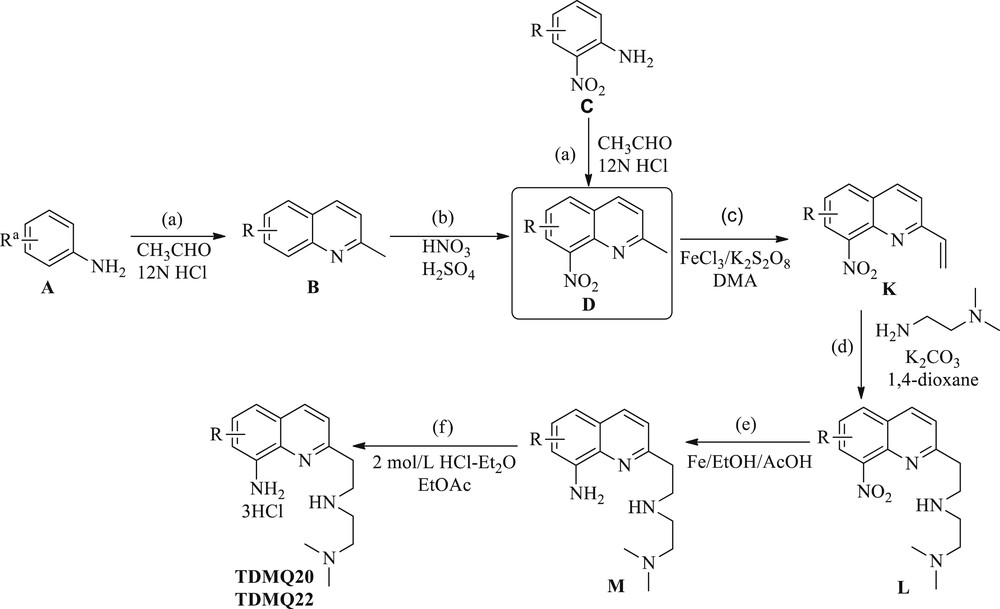
Synthesis of TDMQ20 and TDMQ22, having a long proximal side chain (n = 2). aR stands for 5,7-dichloro- for TDMQ20 and 6-trifluoromethyl- for TDMQ22. (a) CH3CHO/12NHCl, 4 h; (b) HNO3/H2SO4, rt, 2 h; (c) FeCl3, K2S2O8, DMA, 110 °C, 15 min for K-TDMQ26, 3 h for K-TDMQ20; (d) NH2(CH2)2N(CH3)2, K2CO3, 1,4-dioxane, 1 h; (e) Fe, AcOH, EtOH, 80 °C, 1 h; and (f) 2 mol/L HCl-Et2O, EtOAc.
TDMQ22 was synthesized starting from 2-nitro-4-(trifluoromethyl)aniline (C, Scheme 3). The quinoline ring was synthesized as described above for TDMQ5 and provided D. The following steps of the synthesis of TDMQ22 were the same as for TDMQ20: vinylation of the 2-methyl group, introduction of the side chain, reduction of the nitro group at C8, and protonation.
The overall yields were 25% and 22% for TDMQ20 and TDMQ22, respectively, from commercially available starting materials.
2.2 NMR characterizations
Each TDMQ ligand was fully characterized by correlation spectroscopy (COSY), heteronuclear multiple-quantum correlation (HMQC), and heteronuclear multiple-bond correlation (HMBC) 2D NMR experiments, thus allowing to confirm the coupling between the quinoline nucleus and the side chain on one side and the protonation sites on the other side. As example, a selection of NMR data for TDMQ20 is presented in Scheme 4. The quin–CH2 methylene group was detected at 3.44 ppm in proton and 33.72 ppm in carbon-13, and was coupled with a CH2 detected at 3.65/45.35 ppm. The C3 of the quinoline ring was detected at 123.85 ppm, whereas the H3 was detected at 7.65 ppm. The CH2 carbon at 33.72 ppm was coupled with the H3 (7.65 ppm), and conversely, the C3 (123.85 ppm) was coupled with the CH2 (3.44 ppm), as a result of 3J correlation in HMBC analyses. This result confirms the linking between the quinoline and the diamine side chain.
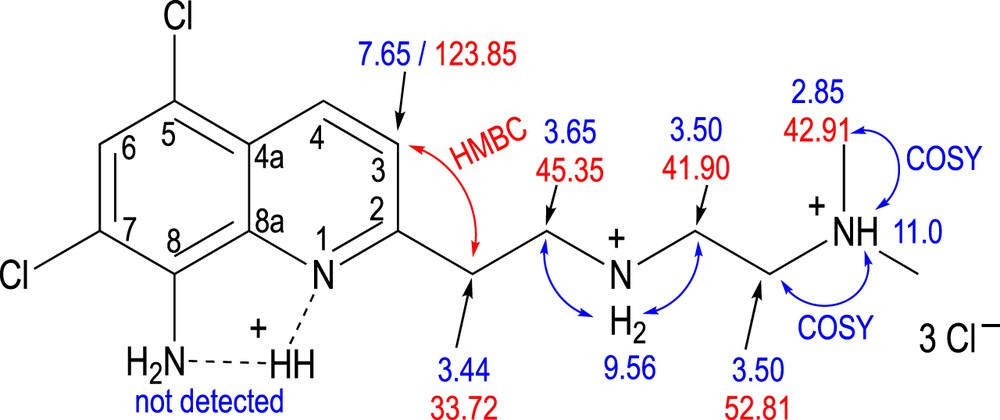
Selected NMR data of TDMQ20 in DMSO-d6. Chemical shifts are given with respect to tetramethylsilane. 1H chemical shifts are in blue, and 13C chemical shifts are in red. COSY correlations are indicated as blue double arrows; HMBC correlations are indicated as a red double arrow.
The protons of the side chain exhibited chemical shifts that were consistent with protonation of both proximal and distal amine functions. For instance, the distal [NH+(CH3)2] protons were detected at 2.85 ppm, with a low field shift of ca. 0.6 ppm with respect to a neutral N(CH3)2 terminus (δ = 2.22 ppm in M-TDMQ20). Similarly, the methylene protons of the quinCH2CH2NH+ fragment were detected at 3.44 and 3.65 ppm, strongly deshielded with respect to the corresponding methylene in the neutral ligand M-TDMQ20 (2.77 and 2.44 ppm). Furthermore, mobile protons were detected as broad singlets at 11.0 and 9.56 ppm, accounting for one and two protons, respectively. The signal at 11.0 ppm exhibited COSY correlations with the protons detected at 2.85 ppm [6H, (CH3)2], and with those detected at 3.50 ppm [4H, CH2-CH2-NH+(CH3)2]. It was then assigned to the terminal ternary ammonium NH+(CH3)2. The signal at 9.56 ppm exhibited COSY correlations with the protons detected at 3.65 and 3.50 ppm, assigned to the methylene located at α-position of the secondary amine CH2NH2+CH2. It was then assigned to the secondary ammonium.
For both TDMQ16 and TDMQ22 series, the trifluoromethyl substituent and C6 gave rise to expected JC-F coupling constants of 270–272 Hz for 1J, 30–32 Hz for 2JC6-F, and ca. 3 Hz for 3JC5-F. For all protonated TDMQs ligands, the NH protons of the quinoline moiety were not detectable in the 1H spectrum, whereas they were identified in the spectra of the precursor-free base ligands (for instance, at 5.40 ppm in the spectrum of the precursor-free base M-TDMQ20). This is likely due to protonation of the 8-aminoquinoline function and subsequent exchange of the proton between the two nitrogen sites. This is consistent with the fact that the 8-aminoquinoline cycle exhibits a single pKa value at 3.1–3.3 [10b].
3 Chelation of copper(II)
The ability of TDMQ5 to chelate metals Cu(II) and Zn(II) was monitored by using UV–visible titration of the aminoquinoline ring (230–400 nm). Spectral modifications upon addition of aliquots of concentrated aqueous solutions of Cu2+ or Zn2+ salts were recorded and considered significant for the metal complexation by the TDMQ ligand.
Upon addition of Cu2+ into a solution of TDMQ5 (45 μM in Tris·HCl buffer, pH 7.4), spectral changes were immediately observed from 0.2 to 1 mol equivalent of Cu2+, as the result of a fast and complete complexation (no free Cu2+ in solution). No further changes were observed when an excess of Cu2+ was added (up to 2–4 mol equivalents with respect to the ligand). Observation of isosbestic points is in agreement with the formation of a single Cu(II) complex with a stoichiometry metal/ligand = 1/1. The titration of TDMQ5 by Cu2+ is presented in Scheme 5. The absorbance at 261 and 344 nm of the free ligand disappeared in favor of a band at 244 nm assigned to the Cu(II)–TDMQ5 complex. Isosbestic point detected at 335 nm was well defined. Isosbestic points at 253 and 281 nm were well defined in the range 0–1 mol equivalent of Cu, but poorly defined in the presence of an excess of metal salt, due to the own absorbance of free Cu2+ below 300 nm. The plot of absorbance at 350 nm versus the number of Cu2+mol equivalent (Scheme 5c) confirms that the stoichiometry of the complex is metal/ligand = 1/1, as previously described for bis(8-aminoquinolines) [10b–d].
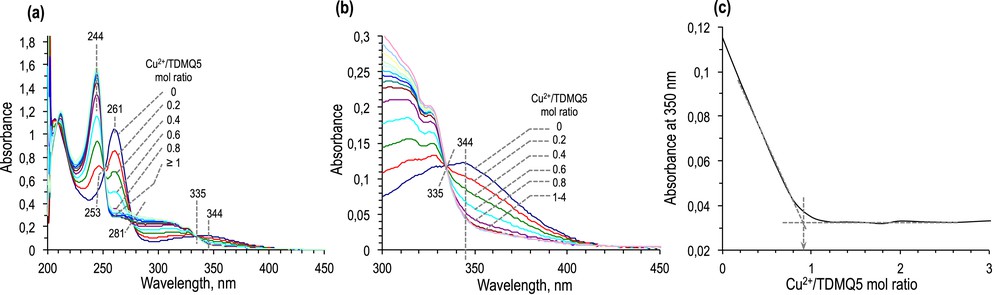
(a) Titration of TDMQ5 (45 μM) by CuCl2 (0–4 mol equivalent) in Tris saline buffer, pH 7.4, monitored by UV–visible spectrometry. (b) Magnification of the region 200–450 nm. (c) Plotting of the absorbance at 350 nm with respect to the CuCl2/TDMQ5 mol ratio.
When a similar titration was carried out by addition of ZnCl2, no significant spectral change occurred upon addition of 1–3 mol equivalent of metal salt, indicating that the affinity of TDMQ5 for Zn(II) was far below its affinity for Cu(II). This result confirms the selectivity for Cu of ligands having a potential N4 chelating structure [10b], whereas ligands in which oxygen atoms are involved in the coordination structure (like 8-hydroxyquinolines) are not metal selective [9,11].
4 Conclusions
In this study, the synthesis of a series of new tetradentate ligands based on a mono-8-aminoquinoline scaffold, which are potentially selective chelators of Cu(II), is reported. They have been designed as putative drugs to regulate copper homeostasis in the brain of patients with AD, without disturbing zinc. Interestingly, the preliminary study reported here indicate that TDMQ5 is able to efficiently chelate copper(II) as a 1/1 metal/ligand complex, whereas zinc(II) does not do so in the same conditions.
Because the substituents of the quinoline nucleus and the length of the side chain are expected to modulate the metal affinity and selectivity of TDMQ ligands, complete complexation studies of these ligands, including structure of the complexes, measure of affinity constants for Cu(II) and Zn(II), and ability of these ligands to transfer copper from the pathological Cu–amyloids to the physiological Cu–glutathion complex, are currently under investigation.
5 Experimental section
5.1 General methods
All reagents were used as received from commercial sources or prepared according to the literature. Analytical thin-layer chromatography (TLC) was performed with 0.20 mm silica gel 60F plates and visualized by ultraviolet light or by treatment with a spray of the Pancaldi reagent ((NH4)6MoO4, Ce(SO4)2, H2SO4, H2O). Column chromatographic purification of products was carried out on silica gel (300–400 mesh). 1H NMR spectra were measured in CDCl3 or DMSO-d6 (with TMS as standard). The TDMQ ligands and their synthesis intermediates have been prepared according to the general protocols depicted in Schemes 2 and 3. Each intermediate compound was numbered as “X-TDMQn”, where X stands for the letter of the given intermediate in Schemes 2 and 3, followed by the target TDMQ, with n = 5, 16, 20, and 22.
5.2 Synthesis of TDMQ5
The synthesis of TDMQ5 (Scheme 1, R5 = Cl, R6 = H, R7 = Cl) is reported in the following sections.
5.2.1 5,7-Dichloro-2-methylquinoline (B-TDMQ5)
To a solution of 3,5-dichloroaniline (3.24 g, 20 mmol) in HCl (12 M, 15 mL) at 0 °C acetaldehyde was added dropwise, under stirring. The reaction medium was kept at 0 °C for 15 min and the temperature was gradually increased to 80 °C. The mixture was stirred at 80 °C for 4 h. The resulting mixture was poured into ice-cold water and neutralized with aqueous ammonium. After extraction with CH2Cl2, the organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude mixture was purified by silica gel flash column chromatography (ethyl acetate/petroleum ether, 1/20, v/v). After evaporation of the eluent, B-TDMQ5 was obtained as a yellow solid (2.88 g, 68%). 1H NMR (400 MHz, CDCl3): δ = 8.40 (d, J = 8.0 Hz, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.55 (d, J =2.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 2.76 (s, 3H).
5.2.2 5,7-Dichloro-2-methyl-8-nitroquinoline (D-TDMQ5)
To a stirred solution of B-TDMQ5 (2.12 g, 10 mmol) in neat sulfuric acid (10 mL) was added fuming nitric acid (2.0 mL) dropwise over a 1 h period at an ambient temperature. The resulting mixture was stirred for an additional hour and was then poured onto ice. The mixture was allowed to warm to ambient temperature, neutralized with aqueous ammonium hydroxide. After extraction with CH2Cl2, the organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude mixture was purified by silica gel flash column chromatography (ethyl acetate/petroleum ether, 1/10, v/v). Evaporation of the solvent afforded D-TDMQ5 as a yellow solid (2.34 g, 91%). 1H NMR (400 MHz, CDCl3): δ = 8.43 (d, J = 8.0 Hz, 1H), 7.64 (s, 1H), 7.50 (d, J = 8.0 Hz, 1H), 2.76 (s, 3H).
5.2.3 5,7-Dichloro-2-methylquinolin-8-amine (E-TDMQ5)
Iron (838 mg, 15 mmol) and acetic acid (3 mL) were added to a solution of 5,7-dichloro-2-methyl-8-nitroquinoline (D-TDMQ5, 1.28 g, 5 mmol) in ethanol (9 mL). The mixture was stirred under reflux for 4 h. The reaction mixture was poured into an aqueous saturated NaHCO3 solution (200 mL) and extracted with CH2Cl2 (3 × 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (ethyl acetate/petroleum ether, 1:10, v/v). Evaporation of the solvent afforded E-TDMQ5 as a yellow solid (1.02 g, 90%). 1H NMR (400 MHz, CDCl3): δ = 8.31 (d, J = 8.0 Hz, 1H), 7.42 (s, 1H), 7.35 (d, J = 8.0 Hz, 1H), 5.36 (br s, 2H), 2.76 (s, 3H).
5.2.4 N-Acetyl-N-(5,7-dichloro-2-methylquinolin-8-yl)acetamide (F-TDMQ5)
To a solution of compound E-TDMQ5 (900 mg, 3.96 mmol) in CH2Cl2 (25 mL) was added acetyl chloride (1.2 mL) and N,N-diisopropylethylamine (5.9 mL) at 0 °C. The reaction mixture was kept at 0 °C for 15 min and stirred overnight at room temperature. After the removal of solvents under reduced pressure, the crude product was purified by silica gel flash column chromatography (ethyl acetate/petroleum ether, 1:20, v/v). After evaporation of the eluent, F-TDMQ5 was obtained as a yellow solid (924 mg, 75%). 1H NMR (400 MHz, CDCl3): δ = 8.41 (d, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.42 (d, J = 8.0 Hz, 1H), 2.71 (s, 3H), 2.29 (s, 6H).
5.2.5 N-Acetyl-N-(5,7-dichloro-2-formylquinolin-8-yl)acetamide (G-TDMQ5)
To a solution of compound F-TDMQ5 (850 mg, 2.73 mmol) in 1,4-dioxane (10 mL) was added selenium dioxide (908 mg, 8.19 mmol). The reaction mixture was heated, and the temperature was increased to 80 °C and stirred for 12 h. The reaction mixture was filtered through a Celite pad and the product was eluted with dichloromethane. The combined filtrates were evaporated under reduced pressure to dryness. The residue was purified by silica gel flash column chromatography (ethyl acetate/petroleum ether, 1:20, v/v). After evaporation of the eluent, G-TDMQ5 was obtained as a yellow solid (621 mg, 70%). 1H NMR (400 MHz, CDCl3): δ = 10.13 (s, 1H), 8.76 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.92 (s, 1H), 2.32 (s, 6H).
5.2.6 N1-((8-Amino-5,7-dichloroquinolin-2-yl)methyl)-N2,N2-dimethylethane-1,2-diamine(H-TDMQ5)
To a solution of G-TDMQ5 (550 mg, 1.69 mmol) in 1,2-dichloroethane (30 mL) was added N,N-dimethyl-1,2-ethanediamine (596 mg, 738 μL, 6.76 mmol) under argon. The reaction mixture was stirred at room temperature for 1 h, and sodium triacetoxyborohydride (1.43 g, 6.76 mmol) was added. The resulting mixture was stirred for additional 12 h and diluted with 100 mL CH2Cl2, followed by the addition of saturated aqueous sodium bicarbonate solution (40 mL). The organic phase was separated. The aqueous phase was added 2 mL ammonium hydroxide, extracted with CH2Cl2 (3 × 30 mL). The organic phase was combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. To the crude product in CH2Cl2 (5 mL) was added 6NHCl (2 mL), which was stirred at 50 °C for 4 h. Water (50 mL) was added, followed by the addition of extra aqueous 25% ammonium hydroxide. The mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash chromatography (ethyl acetate/isopropanol/ammonium hydroxide (25%), 5:1:0.2, v/v/v). Evaporation of the solvent afforded H-TDMQ5 as a yellow solid (683 mg, 71%). 1H NMR (400 MHz, CDCl3): δ = 8.35 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 8.8 Hz, 1H), 7.43 (s, 1H), 5.38 (br s, 2H), 4.10 (s, 2H), 2.75 (t, J = 6.0 Hz, 2H), 2.55 (br s, 1H), 2.48 (t, J = 6.0 Hz, 2H), 2.22 (s, 6H).
5.2.7 TDMQ5
To a solution of H-TDMQ5 in ethyl acetate was added excess hydrogen chloride solution (2.0 N in diethyl ether). The precipitate was filtered, washed with diethyl ether, and dried under reduced pressure to give TDMQ5 as a yellow solid (98%).
Data for TDMQ5: decomposes at ∼269.5 °C; 1H NMR (500 MHz, DMSO-d6): δ = 11.14 [br s, 1H, N+H(CH3)2)], 10.07 [br s, 2H, CH2-NH2+CH2), 8.48 (d, J = 8.5 Hz, 1H, H4), 7.70 (d, J = 8.5 Hz, 1H, H3), 7.68 (s, 1H, H6), 4.68 (tr, 2H, J = 5 Hz, QuinCH2NH2+), 3.60 (m, 2H, NH2+CH2CH2), 3.59 (m, 2H, NH2+CH2CH2), 2.87 [d, J = 4 Hz, 6H, NH+(CH3)2]; 13C NMR (125.7 MHz, DMSO-d6): δ = 151.40 (C2), 141.93 (C8), 136.94 (C8a), 134.25 (C4), 128.31 (C6), 124.46 (C4a), 121.93 (C3), 114.57 (C5), 112.20 (C7), 52.95 [CH2-NH+(CH3)2], 50.79 (QuinCH2NH2+), 42.80 [NH+(CH3)2], 41.95 [NH2+CH2CH2NH+(CH3)2]; IR (KBr): 3426, 3253, 2947, 2715, 1630, 1596, 1465, 1373, 1322, 995, 947, 861, 712, 539 cm−1; HRMS (EI) calcd for C14H18Cl2N4: 312.0909 ([M]+); found: 312.0900; Elemental Anal. Calcd (%) for C14H18Cl2N4·3HCl·0.6H2O·0.02NH4Cl (apparent MW = 434.48): C, 38.70; H, 5.17; N, 12.96. Found: C, 38.75; H, 5.18; N, 13.07.
5.3 Synthesis of TDMQ16
The synthesis of TDMQ16 (Scheme 1, R5 = H, R6 = CF3, R7 = H) is reported in the following sections.
5.3.1 2-Methyl-8-nitro-6-(trifluoromethyl)quinoline (D-TDMQ16)
The procedure was similar to that described for the preparation of B-TDMQ5, but using 2-amino-5-trifluoromethyl nitrobenzene (C, Scheme 1). D-TDMQ16 was obtained as a light yellow solid (59%). 1H NMR (400 MHz, CDCl3): δ = 8.28 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 8.12 (s, 1H), 7.54 (d, J = 8.4 Hz, 1H), 2.81 (s, 3H).
5.3.2 2-Methyl-6-(trifluoromethyl)quinolin-8-amine (E-TDMQ16)
The procedure was similar to that described for the preparation of E-TDMQ5. E-TDMQ16 was obtained as a light yellow solid (74%). 1H NMR (400 MHz, CDCl3): δ = 8.02 (d, J = 8.4 Hz, 1H), 7.39 (s, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.01 (s, 1H), 5.15 (br s, 2H), 2.74 (s, 3H).
5.3.3 N-(2-Methyl-6-(trifluoromethyl)quinolin-8-yl)acetamide (F-TDMQ16)
The procedure was similar to that described for the preparation of F-TDMQ5. F-TDMQ16 was obtained as a light yellow solid (78%). 1H NMR (400 MHz, CDCl3): δ = 9.85 (s, 1H), 8.97 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.76 (s, 1H), 7.42 (d, J = 8.4 Hz, 1H), 2.79 (s, 3H), 2.38 (s, 3H).
5.3.4 N-(2-Formyl-6-(trifluoromethyl)quinolin-8-yl)acetamide (G-TDMQ16)
The procedure was similar to that described for the preparation of F-TDMQ5. G-TDMQ16 was obtained as a light yellow solid (50%). 1H NMR (400 MHz, CDCl3): δ = 10.28 (s, 1H), 9.77 (s, 1H), 9.12 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.90 (s, 1H), 2.43 (s, 3H).
5.3.5 N1-((8-Amino-6-(trifluoromethyl)quinolin-2-yl)methyl)-N2,N2-dimethylethane-1,2-diamine (H-TDMQ16)
The procedure was similar to that described for the preparation of H-TDMQ5. H-TDMQ16 was obtained as a light yellow solid (85%). 1H NMR (400 MHz, CDCl3) δ = 8.07 (d, J = 8.4 Hz, 1H), 7.48 (d, J = 8.4 Hz, 1H), 7.39 (s, 1H), 7.01 (s, 1H), 5.23 (br s, 2H), 4.11 (s, 2H), 2.77 (t, J = 6.0 Hz, 2H), 2.55 (br s, 1H), 2.48 (t, J = 6.0 Hz, 2H), 2.23 (s, 6H).
5.3.6 TDMQ16
The protonation procedure was similar to that described for the preparation of TDMQ5. TDMQ16 was obtained as a yellow solid (88%).
Data for TDMQ16: decomposes at ∼209 °C; 1H NMR (400 MHz, DMSO-d6): δ = 11.27 [br s, 1H, N+H(CH3)2)], 10.19 [br s, 2H, CH2-NH2+CH2), 8.47 (d, J = 8.4 Hz, 1H, H4), 7.65 (d, J = 8.4 Hz, 1H, H3), 7.59 (br s, 1H, H5), 7.15 (d, 4JHH = 1.7 Hz, 1H, H7), 4.67 (t, J = 5 Hz, 2H, QuinCH2NH2+), 3.67 [m, 2H, CH2-NH+(CH3)2], 3.61 (m, 2H, NH2+CH2CH2), 2.87 [d, 6H, NH+(CH3)2]; 13C NMR (125.7 MHz, DMSO-d6): δ = 152.13 (C2), 145.43 (C8), 137.76 (C8a), 138.79 (C4), 128.34 (2JCF = 30 Hz, C6), 124.93 (q, 1JCF = 272 Hz, F3C-C6), 121.85 (C3), 127.53 (C4a), 112.02 (3JCF = 3 Hz, C5), 105.30 (C7), 52.96 [CH2-NH+(CH3)2], 51.10 (QuinCH2NH2+), 42.78 [N+H(CH3)2], 42.05 [NH2+CH2CH2NH+(CH3)2]; IR (KBr): 3374, 3302, 3169, 2976, 2794, 2718, 1630, 1604, 1508, 1439, 1365, 1292, 1265, 1231, 1159, 1141, 1110, 1041, 1020, 925, 865, 849, 835, 790, 712 cm−1; HRMS (EI) calcd for C15H19F3N4: 312.1562 ([M]+); found: 312.1559; Elemental Anal. Calcd (%) for C15H19F3N4·2.9HCl·0.4H2O·0.08CH3COOC2H5 (apparent MW = 432.32): C, 42.56; H, 5.44; N, 13.18. Found: C, 42.53; H, 5.42; N, 13.18.
5.4 Synthesis of TDMQ20
The synthesis of TDMQ20 (Scheme 2, R5 = Cl, R6 = H, R7 = Cl) is reported below.
5.4.1 5,7-Dichloro-8-nitro-2-vinylquinoline (K-TDMQ20)
To a solution of D-TDMQ5 (Scheme 2, R5 = Cl, R6 = H, R7 = Cl, 1.0 g, 3.9 mmol) in dimethylacetamide (DMA, 5 mL) was added FeCl3 (19.0 mg, 0.117 mmol) and K2S2O8 (1.05 g, 7.8 mmol). The mixture was stirred at 110 °C for 1.5 h and quenched with water. After extraction with CH2Cl2, the organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude mixture was purified by silica gel flash column chromatography (EtOAc/petroleum ether, 1:10, v/v). After evaporation of the eluent, compound K-TDMQ20 was obtained as a brown solid (430.3 mg, 41%). 1H NMR (400 MHz, CDCl3): δ = 8.52 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.8 Hz, 1H), 7.65 (s, 1H), 6.97 (dd, J = 17.6, 10.8 Hz, 1H), 6.46 (d, J = 17.6 Hz, 1H), 5.79 (d, J = 10.8 Hz, 1H).
5.4.2 N1-(2-(5,7-Dichloro-8-nitroquinolin-2-yl)ethyl)-N2,N2-dimethylethane-1,2-diamine (L-TDMQ20)
To a mixture of K-TDMQ20 (780 mg, 2.9 mmol) and K2CO3 (481 g, 3.48 mmol) in 1,4-dioxane (10 mL) was added N,N-dimethyl-1,2-ethanediamine (511.2 mg, 5.8 mmol). The reaction mixture was stirred at room temperature for 1 h and quenched with water. After extraction with CH2Cl2, the organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude mixture was purified by silica gel flash column chromatography (ethyl acetate/isopropanol/25% ammonium hydroxide, 8/2/0.5, v/v/v). After evaporation of the eluent, L-TDMQ20 was obtained as yellow solid (777 mg, 75%). 1H NMR (400 MHz, CDCl3): δ = 8.46 (d, J = 8.8 Hz, 1H), 7.66 (s, 1H), 7.54 (d, J = 8.8 Hz, 1H), 3.22 (t, J = 6.0 Hz, 2H), 3.12 (t, J = 6.0 Hz, 2H), 2.74 (t, J = 6.4 Hz, 2H), 2.43 (t, J = 6.4 Hz, 2H), 2.23 (s, 6H).
5.4.3 N1-(2-(8-Amino-5,7-dichloroquinolin-2-yl)ethyl)-N2,N2-dimethylethane-1,2-diamine (M-TDMQ20)
The procedure was similar to that described for the preparation of E-TDMQ5. M-TDMQ20 was obtained as a light yellow solid (90%). 1H NMR (400 MHz, CDCl3): δ = 8.33 (d, J = 8.8 Hz, 1H), 7.43 (s, 1H), 7.37 (d, J = 8.8 Hz, 1H), 5.40 (br s, 2H), 3.19–3.12 (m, 4H), 2.77 (t, J = 6.0 Hz, 2H), 2.44 (t, J = 6.0 Hz, 2H), 2.22 (s, 6H), 1.98 (br s).
5.4.4 TDMQ20
The procedure was similar to that described for the preparation of TDMQ5. TDMQ20 was obtained as a yellow solid (90%).
Data for TDMQ20: mp 189–191 °C; 1H NMR (500 MHz, DMSO-d6): δ = 11.0 [br s, 1H, N+H(CH3)2)], 9.56 [br s, 2H, CH2-NH2+CH2), 8.39 (d, J = 8.4 Hz, 1H, H4), 7.65 (d, J = 8.4 Hz, 1H, H3), 7.64 (s, 1H, H6), 3.65 (dt, J = 6.8 Hz, 2H, QuinCH2CH2), 3.50 [m, 4H, NH2+CH2CH2NH+(CH3)2], 3.44 (t, J = 6.8 Hz, 2H, QuinCH2), 2.85 [s, 6H, NH+(CH3)2); 13C NMR (125.7 MHz, DMSO-d6): δ = 156.71 (C2), 141.40 (C8), 137.52 (C8a), 133.65 (C4), 127.48 (C6), 123.85 (C3), 123.90 (C4a), 115.25 (C5), 112.11 (C7), 52.81 [CH2-NH+(CH3)2], 45.35 (QuinCH2CH2), 42.91 [NH+(CH3)2], 41.90 [NH2+CH2CH2NH+(CH3)2], 33.72 (QuinCH2); IR (KBr): 3441, 3354, 3194, 3015, 1676, 1632, 1593, 1545, 1468, 1403, 1376, 1310, 763 cm−1; HRMS (ESI) calcd for C15H21Cl2N4: 327.1143 ([M+H]+); found: 327.1148; Elemental Anal. Calcd for C15H20Cl2N4·3HCl·0.2H2O (apparent MW = 440.24): C, 40.93; H, 5.36; N, 12.73. Found C, 40.71; H, 5.24; N, 12.78.
5.5 Synthesis of TDMQ22
The synthesis of TDMQ22 (Scheme 2, R5 = H, R6 = CF3, R7 = H) is reported below.
5.5.1 8-Nitro-6-(trifluoromethyl)-2-vinylquinoline (K-TDMQ22)
The procedure was similar to that described for the preparation of K-TDMQ20, but starting from D-TDMQ16. K-TDMQ22 was obtained as a brown solid (45%). 1H NMR (400 MHz, CDCl3): δ = 8.30–8.28 (m, 2H), 8.15 (s, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.02 (dd, J = 17.6, 10.8 Hz, 1H), 6.52 (d, J = 17.6 Hz, 1H), 5.82 (d, J = 10.8 Hz, 1H).
5.5.2 N1,N1-Dimethyl-N2-(2-(8-nitro-6-(trifluoromethyl)quinolin-2-yl)ethyl)ethane-1,2-diamine (L-TDMQ22)
The procedure was similar to that described for the preparation of L-TDMQ20. L-TDMQ22 was obtained as a yellow solid (80%). 1H NMR (400 MHz, CDCl3): δ = 8.29 (s, 1H), 8.24 (d, J = 8.8 Hz, 1H), 8.16 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 3.26 (t, J = 6.4 Hz, 2H), 3.16 (t, J = 6.4 Hz, 2H), 2.77 (t, J = 6.4 Hz, 2H), 2.44 (t, J = 6.4 Hz, 2H), 2.22 (s, 6H).
5.5.3 N1-(2-(8-Amino-6-(trifluoromethyl)quinolin-2-yl)ethyl)-N2,N2-dimethylethane-1,2-diamine (M-TDMQ22)
The procedure was similar to that described for the preparation of M-TDMQ20. M-TDMQ22 was obtained as a yellow solid (54%). 1H NMR (400 MHz, CDCl3): δ = 8.04 (d, J = 8.4 Hz, 1H), 7.38 (s, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.01 (s, 1H), 5.24 (br s, 2H), 3.21–3.11 (m, 4H), 2.78 (t, J = 6.0 Hz, 2H), 2.45 (t, J = 6.0 Hz, 2H), 2.21 (s, 6H), 1.97 (br s).
5.5.4 TDMQ22
Protonation of M-TDMQ22 was achieved in the same way as reported above for TDMQ5. TDMQ22 was obtained as a yellow solid (90%).
Data for TDMQ22: mp 180–182 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.41 [br s, 2H, CH2-NH2+CH2), 8.36 (d, J = 8.4 Hz, 1H, H4), 7.58 (d, J = 8.4 Hz, 1H, H3), 7.49 (s, 1H, H5), 7.06 (d, 4JHH = 1.9 Hz, 1H, H7), 3.66 (dt, J = 6.8 Hz, 2H, QuinCH2CH2), 3.50 [m, 4H, NH2+CH2CH2NH+(CH3)2], 3.42 (t, J = 6.8 Hz, 2H, QuinCH2), 2.86 [s, 6H, NH+(CH3)2); 13C NMR (125.7 MHz, DMSO-d6): δ = 157.11 (C2), 146.32 (C8), 137.97 (C8a), 138.18 (C4), 127.60 (2JCF = 31 Hz, C6), 124.90 (1JCF = 270 Hz, F3C-C6), 123.71 (C3), 126.91 (C4a), 111.19 (C5), 103.88 (C7), 52.88 [CH2-NH+(CH3)2], 45.39 (QuinCH2CH2), 42.92 [N+H(CH3)2], 42.07 [NH2+CH2CH2NH+(CH3)2], 33.88 (QuinCH2); IR (KBr): 3423, 3140, 2962, 2762, 2560, 1647, 1604, 1535, 1479, 1427, 1359, 1287, 1260, 1215, 1165, 1123, 1087, 986, 903, 834, 790, 715, 504 cm−1; HRMS (ESI) calcd for C16H22F3N4: 327.1797 ([M+H]+); found: 327.1792; Elemental Anal. Calcd for C16H21F3N4·3HCl·0.9H2O·0.1NH4Cl (apparent MW = 457.32): C, 42.02; H, 5.78; N, 12.56. Found C, 41.95; H, 5.52; N, 12.49.
5.6 Metal complexation studies
The UV–visible cuvette contained a solution of 45 μM of TDMQ ligand in 20 mM Tris·HCl pH 7.4, 150 mM NaCl. Aliquots of aqueous solutions of CuCl2 or ZnCl2 (9 mM) were added (1–4 mol equivalent) under magnetic stirring at 25 °C. UV–visible spectrum was recorded after each addition of metal salt. Total variation in volume in the cuvette was below 2% upon addition of 4 mol equivalent of metal. CuCl2 and ZnCl2 were selected as metal salts because chloride is, by far, the more concentrated anion in biological medium ([Cl–] = 100 mM in human serum and 125 mM in cerebrospinal fluid).
Acknowledgments
This work was supported by the CNRS (France), the Guangdong Province (grant 2050205), and the Guangdong University of Technology (grant 220418037) (PR China).