

During embryogenesis, cells undergo transcriptional (a biological process of gene expression that transcribes the DNA sequence into the corresponding RNA biopolymer) alterations enabling them to adopt physical properties required for migration (a central mechanism in the development of multicellular organism and defines a physical process by which cells can migrate in tissues to disseminate and give rise to more specialized tissues). This process, known as epithelial-to-mesenchymal transition, is followed by the reverse mesenchymal-to-epithelial transition allowing these cells to give rise to specialized tissues in metazoan development. These cells are characterized by a metastable mesenchymal (mesenchymal stem cells define a state of multipotent undifferentiated cells that can give rise to more specialized cell types) state associated with high epigenetic plasticity (epigenetics defines heritable changes in gene expression that do not involve changes in the primary DNA sequence. Plasticity broadly defines the capacity of undifferentiated cells to give rise to multiple cell lineages exhibiting distinct functions at the organismal level) that regulates the appropriate transcriptional program [1]. It is now well established that in carcinoma (a type of cancer that develops from epithelial cells. Epithelial tissues represent one of the four types of animal tissues along with connective, muscle, and nervous tissues), cancer cells can take advantage of similar processes to detach from primary tumors, disseminate, and form metastases (a pathogenic spread of cancer cells from primary to a secondary site within the body of the host) at distant sites (Fig. 1a) [2]. A subset of cancer cells, so-called cancer stem cells (CSCs), contribute to the clonogenic heterogeneity (the presence of various cell types within a tissue exhibiting different physiological features) of tumors and are refractory to conventional chemotherapeutic agents, for which the efficacy is usually restricted to fast replicating (the chemical step through which genomic DNA is synthesized to give rise to a second identical copy before cell division) and dividing normal cancer cells. In contrast, CSCs exhibit a pronounced mesenchymal phenotype and are quiescent by nature making it difficult to completely eradicate neoplastic (neoplasm is an abnormal growth of tissues that leads to tumors) tissues using these drugs (Fig. 1a) [3]. Emerging strategies have been designed on the ground that combinatorial therapies involving debulking agents could reduce the size of the primary tumor, whereas anti-CSC agents could prevent self-renewal (the capacity of the cell to divide into two daughter cells while keeping an undifferentiated cell state) of tumor cells, migration, and metastasis formation (Fig. 1b) [3]. However, the repertoire of small molecules that selectively target mesenchymal cancer cells is currently limited, and druggable pathways specific to CSCs have not yet been clearly defined. The discovery of such compounds and establishment of pathways at work in CSCs have, at least partly, been hampered by the lack of stable CSC models that could allow for high throughput screening (a technological process through which a large number of small molecules can be evaluated in a biological assay representative of a pathology) and identification of drugs effective against these cells.
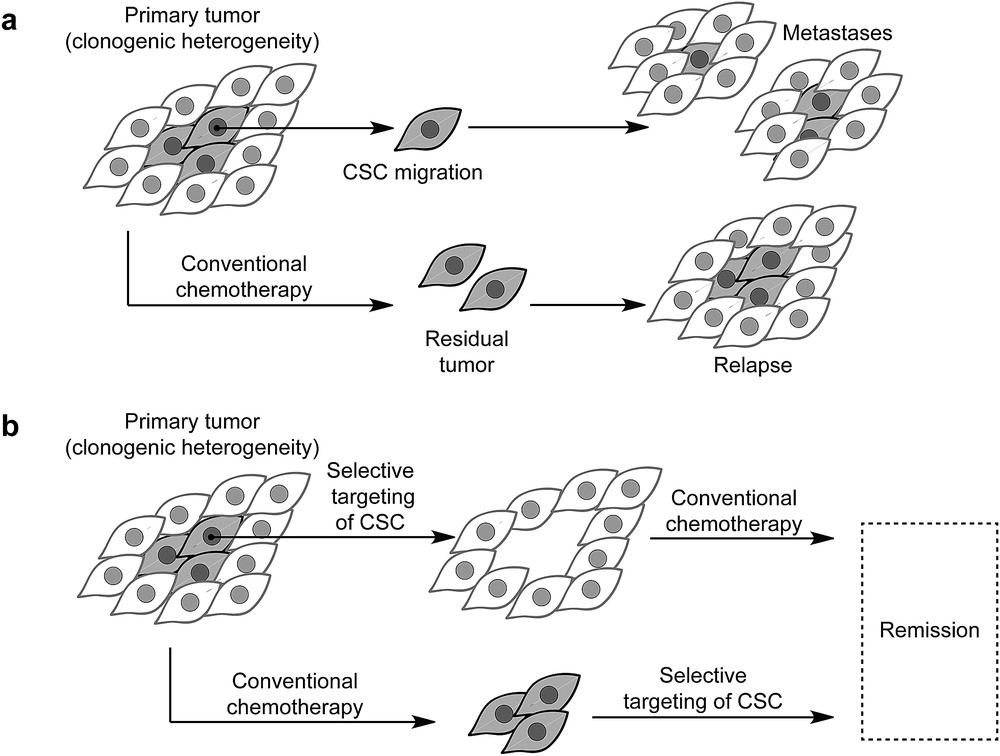
a) The paradigm of CSCs: they represent a subset of tumor cells. These cells are refractory to conventional treatment and can cause relapse. These cells can detach from primary tumors and form metastasis at distant sites. (b) Next-generation drugs can be used to eradicate CSCs and can be used along with conventional therapy to reduce the risk of relapse and enable remission.
To this end, artificial induction of a mesenchymal state was achieved by transforming (genetic transformation consists of genetic alterations of the cell genome to modify the physiological properties of this cell and evaluate the effect of alteration of specific genes) human mammary epithelial cells leading to HMLER CD44high/CD24low (cell surface markers that may characterize to some extent the level of cell differentiation) cells, a model that recapitulates the principal characteristics of CSCs including the formation of three-dimensional spheres (a three-dimensional growth of tissues) in nonadherent (adherent conditions defines a technical aspect of tissue culture whereby cells have the ability to attach to the substrate of the culture dish and grow in two dimensions as opposed to growing in suspension within the cell culture media) cell culture conditions, as well as self-renewal and tumor-seeding (the capacity of a single cell to give rise to a solid tumor on its own) capacity in animal models [4]. This and other models have successfully been used to screen for and to identify agents that could selectively kill CSCs in vitro and patient-derived tissues. In particular, Gupta et al. [5] have discovered that the natural product salinomycin could selectively kill CSCs in vitro and in vivo (Fig. 2a). This polyether ionophore was proposed to exert its activity by carrying alkali metals inside cells by passive diffusion through the lipidic plasma membrane (a spheroid lipid bilayer that separates the inner part of a cell from the outside), thereby leading to a cellular ionic imbalance causing mitochondrial (mitochondria are double-membrane bound cellular components where respiration and energy production is taking place) membrane depolarization and cell death [5,6]. Furthermore, salinomycin has been shown to exert a pleiotropic activity through inhibition of Wnt signaling (a group of signal transduction pathways made of proteins that pass signals into a cell through cell surface receptors), inhibition of autophagic flux (autophagy is a biological process whereby a cell can degrade its own cellular components to replenish the pull of building blocks essential for life), and induction of endoplasmic reticulum (a cellular compartment where translation, the conversion of RNA into proteins, is taking place) stress among many other dysfunctions [7–9]. The absence of a unifying mechanism prompted us to study the biology of salinomycin. Indeed, although the “ionophore theory” appeared to be sound and appealing, a clear rationale illuminating the selective activity of salinomycin against CSCs versus normal cancer cells had not been documented.
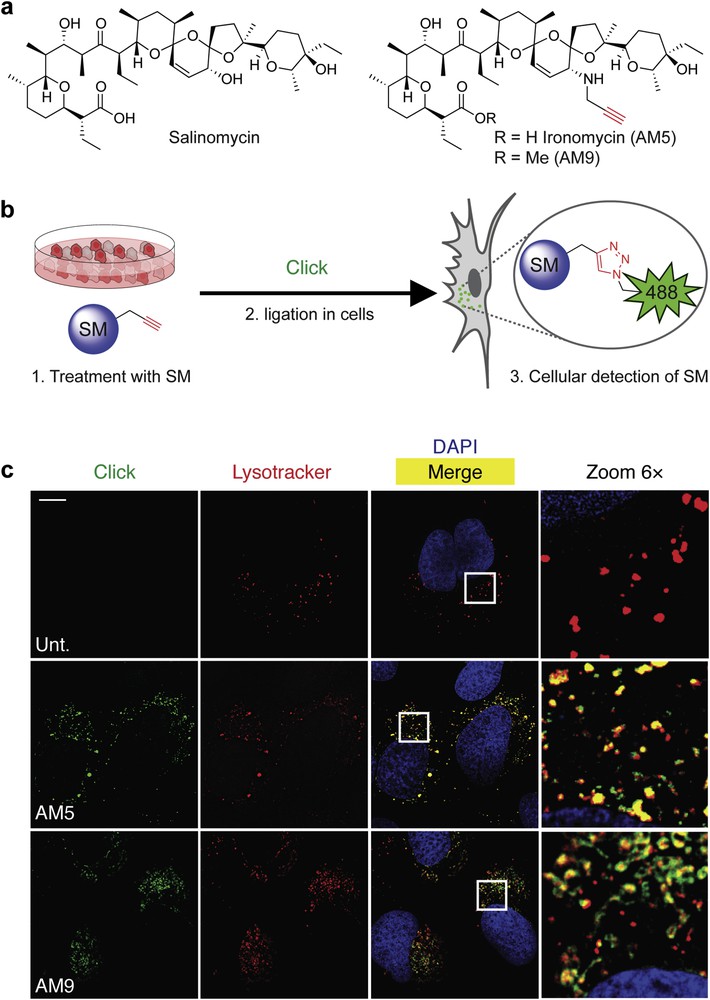
a) Molecular structures of salinomycin (Sal), biologically active derivative ironomycin (AM5), and negative control AM9. (b) Schematic illustration of labeling small molecules (SM) in cells using click chemistry (Click). (c) Fluorescence microscopy images showing the subcellular localization of labeled Sal derivatives in U2OS osteosarcoma cancer cells treated as indicated. Lysotracker (red) stains the lysosomes, 4′,6-diamidino-2-phenylindole (DAPI, blue) stains nuclear DNA, and Sal derivatives were labeled by means of click chemistry (green). Scale bar, 10 μm.
Delineating mechanisms of action of small molecules in disease-relevant models are limited by our ability to visually identify primary sites of phenotypic (observable changes or traits including morphology, biochemical, and physiological morphology of a cell) induction in cells [10]. To overcome this recurrent limitation, our laboratory has established a robust methodology based on the chemical labeling of small molecules in cells using in situ click chemistry. This strategy consists of introducing a minimal chemical modification (e.g., alkyne, strained alkene, and azide) onto the drug of interest by means of molecular editing with the view to produce a clickable surrogate drug that phenocopies (the induction of a comparable phenotype) the drug it was designed after. For instance, we generated the alkyne-containing derivatives AM5 and its methylated counterpart AM9 (Fig. 2a) [11]. These two derivatives were conveniently obtained in a two/three-step procedure involving a chemoselective allylic oxidation of salinomycin using manganese dioxide followed by a stereoselective reductive amination in the presence of propargyl amine and sodium cyanoborohydride, whereas methylation of the carboxylate was performed using methyl iodide [11]. It was found that AM5 was about 10-fold more potent than the natural product itself against two models of CSCs in vitro and in vivo against patient-derived xenografts of tumors that were resistant to conventional treatments, including the reference drug docetaxel [11]. In comparison, the methylated derivative AM9 was not active, which indicated that the free carboxylate was required to mediate the activity of salinomycin derivatives against CSCs. Strikingly, we found that at effective doses of AM5, no sodium transport could be detected using the hybrid crown ether–cryptand aromatic dye SBFI [12], challenging the original hypothesis proposing that salinomycin kills CSCs by carrying alkali metals in cells.
Following this, we labeled active and inactive alkyne-containing salinomycin derivatives AM5 and AM9 in cells using click chemistry. This technology, virtually applicable to any type of structures, has previously been used by us to identify the subcellular localization of active compounds operating through a wide range of mechanisms including small molecules that covalently react with or reversibly bind to nuclear DNA or protein targets (Fig. 2b) [13–17]. Interestingly, we discovered that salinomycin derivatives preferentially accumulated in the lysosomal compartments (acidic cellular components of cells filled with proteases that can degrade other cellular components including biomolecules such as proteins for recycling) regardless of their biological activity (Fig. 2c) [11]. Remarkably, we observed that although labeling of the inactive AM9 was effective and reproducible, active AM5 was rather sluggish. Given that labeling was performed by means of click chemistry relying on the reduction of inert copper(II) into an active copper(I) catalyst, it occurred to us that the mechanism of action of AM5 and salinomycin may involve redox processes, whereby the active analog AM5 induced a phenotype that could interfere with the redox properties of the reagent required for click labeling whereas inactive AM9 induced no detectable phenotype but could easily be labeled. This led us to consider an alternative mechanism where the functional target of salinomycin and free carboxylate-containing AM5 interacted with iron in lysosomes. In line with this, salinomycin and AM5, but not AM9, induced a phenotype consistent with the induction of cytosolic (the liquid part of the cell) iron depletion that was characterized by increased levels of the iron responsive element-binding protein 2 (IRP2), transferrin receptor (TfR), and reduction of the iron storage protein complex ferritin (FTH) [18]. Furthermore, we found that salinomycin and AM5 blocked the translocation of iron(II) from the lumen of lysosomes to the cytosol as visualized by the reduction of rhodamine N-oxide RhoNox-1, an iron(II) selective probe [19]. In addition, NMR spectroscopy confirmed a loose interaction of salinomycin and AM5 with iron(II) as defined by line broadening and shifting of specific 1H NMR signals of these compounds upon titration with iron(II) that could be reversed through the introduction of the tight iron binder bipyridine. Altogether, these data indicated that salinomycin derivatives exhibited lysosomotropic (the ability of a substance to accumulate in the lysosomal compartment at the expense of other cellular organelles) properties and sequestered iron in the lysosomal compartment, leading us to rename our active analog ironomycin (AM5) [11].
Further study showed that accumulation of iron in the lysosomal compartment led to the production of reactive oxygen species (ROS) over time through Fenton-type chemistry [20], which was followed by peroxidation and lysis of the lipid membrane of lysosomes and eventually led to lysosomal membrane permeabilization [21]. Then, we found that the sudden release of lysosomal ROS in the cytosol led to lipid peroxidation throughout the cell and finally to ferroptosis (a recently characterized cell death mechanism i.e., reliant on iron and mediated by ROS) (Fig. 3) [22].
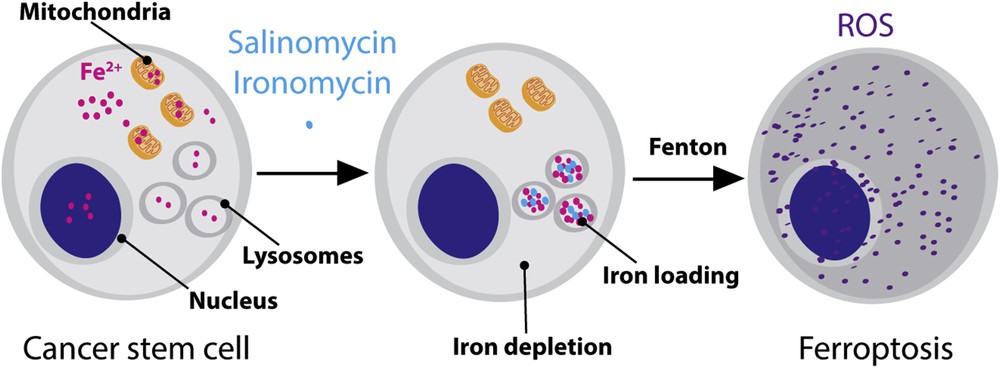
Schematic illustration of reprogramming the reactivity of iron in CSCs. In normal conditions, iron ensures diverse cellular functions including in the nucleus and mitochondria. Drugs such as salinomycin and ironomycin can induce a redistribution of iron with higher loading in lysosomes. Lysosomal iron promotes the production of ROS and ferroptosis.
Importantly, this investigation revealed that HMLER CSCs upregulated iron homeostasis and contained a higher level of total iron compared to their normal HMLER non-stem counterpart, which provided a rationale for the heightened sensitivity of CSCs to salinomycin derivatives [11]. Furthermore, this study raised a more pressing question concerning putative roles of iron in cancer [23] and more specifically in the maintenance of CSCs. Hence, we propose a new model whereby iron is required to regulate cellular plasticity through the oxidative demethylation of epigenetic methyl marks (methylation of DNA and protein residues of chromatin that regulate organization of the genome, DNA repair, and gene expression) that regulate transcriptional changes observed during the epithelial-to-mesenchymal transition program. Indeed, it has been found that specific residues such as H3K4me3, H3K9me3, and H3K27me3 (H3KX defines lysine residues of histone, one of the protein that forms chromatin around which genomic DNA is wrapped) are altered during tumor progression [24], and that iron-dependent jumonji demethylases (jumonji enzymes are specific proteins containing an iron catalyst that can promote the oxidative demethylation of methyl marks) are upregulated in cancer with poor prognosis [25], supporting the contention that iron is required in aggressive cancers to mediate changes at the epigenetic and chromatin (chromatin is a supramolecular complex of proteins and genomic DNA for which the primary function is to repress gene expression) levels. Reshuffling the cellular distribution of iron (e.g., cytosolic depletion and lysosomal loading) and reprogramming the chemical reactivity of this metal can be achieved by means of small molecule intervention. Switching from a prevalent role as a cofactor in Fe-S clusters of proteins involved in the mitochondrial electron transport chain or as a catalyst in the regulation of epigenetic marks through oxidative demethylation, to the production of deleterious ROS in lysosomes through Fenton-type chemistry can be exploited to elicit cell death preferentially in CSCs where the natural abundance of this metal is higher. These findings and work hypotheses will pave the way toward the development of a new generation of drugs to exploit cellular plasticity for therapeutic benefits.
Acknowledgments
R.R. thanks the European Research Council (grant no. 647973), Émergence Ville de Paris, and the “Ligue nationale contre le cancer” for funding. R.R. thanks the CNRS for support and P. Codogno for fruitful discussions.