

1 Introduction
Metal complexes are important and have found applications in different fields such as catalysis, biology, and material sciences [1–4]. These complexes may have different geometries; therefore, they are potentially biologically active [5]. Metal complexes such as Cu(II), Zn(II), Ni(II), and Co(II) have been studied extensively because of their attractive chemical and physical properties [6,7]. Copper, nickel, cobalt, and zinc are sometimes called biometals because they help catalyze biochemical processes in humans [8].
Lapshin and Alekseev [9] reported that biometals such as copper ions form stable complexes in the human body upon simultaneous administration of metal-containing vitamins and antibiotics such as ampicillin, amoxicillin, and cephalexin as most contain an amino group [10]. These antibiotics promote the removal of metals such as copper from the body, whereas the metal also catalyzes the hydrolysis of antibiotics, and this can reduce the effect of antimicrobial therapy [10]. Strachunskii and Kozlov confirmed that Zn(II) mediates vancomycin (a natural product glycopeptide antibiotic) polymerization, thereby enhancing its binding affinity toward d-Ala-d-Lac precursors [11].
To the best of our knowledge this is the first study to report that zinc(II) complex of 2,2′:6′,2″-terpyridine (Tpy) and pyridine-2,6-dicarboxylic acid (Pydc) has been prepared and its interaction with antibiotics has been studied. The objective of this work was to present the synthesis and theoretical study on electronic and molecular structure of the zinc(II) complex of 2,2′:6′,2″-terpyridine (Tpy) and pyridine-2,6-dicarboxylic acid (Pydc), [Zn(II)(Tpy)(Pydc)·4H2O] and to determine relationship between the molecular structure of the compound and theoretical interaction efficiency with amoxicillin. We studied the interaction of [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin theoretically to examine how the nature of a central metal ion influences the geometry and electronic characteristics (electrostatic potential and spin density distribution, highest occupied molecular orbital (HOMO), and lowest unoccupied molecular orbital (LUMO) structure), the absolute electronegativity (χ) values, and the fraction of electrons transferred (ΔN) between metal complexes and amoxicillin.
2 Experimental section
2.1 Materials and physical measurements
Chemicals and solvents were acquired from Sigma–Aldrich Company and used without further purification. Fourier transform infrared (FT-IR) and elemental analyses of [Zn(II)(Tpy)(Pydc)·4H2O] were determined using a PerkinElmer spectrophotometer and PerkinElmer CHN analyzer 2400 series II, respectively. Thermogravimetric analysis (TGA) was carried out using TA Instruments TA-Q500 under dry nitrogen gas at a flow rate of 60 cm3 min−1. Samples with a mass in the range of 1–5 mg were placed in an open platinum crucible and heated at a rate of 10 °C min−1.
2.1.1 Crystallographic data collection and structural analysis
Single crystals suitable for X-ray crystallography were obtained by slow evaporation of aqueous/alcoholic solution. Details of the crystal parameters, data collection, and refinements are listed in Table 1. Data were collected using a Bruker KAPPA Apex II Dual Source diffractometer with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation at 293 K. The structure was solved by direct methods in SHELXS and refined by full-matrix least-squares methods based on F2 using SHELXL [12]. Table 2 lists selected molecular parameters.
Crystal data and structural refinement details of [Zn(II)(Tpy)(Pydc)·4H2O].
| Crystallographic data | [Zn(II)(Tpy)(Pydc)·4H2O] |
| Empirical formula | C22H22N4O4Zn |
| Formula weight | 535.81 |
| Crystal system | Triclinic |
| Space group | P1¯ |
| a (Å) | 8.431 (3) |
| b (Å) | 12.696 (4) |
| c (Å) | 21.701 (7) |
| α | 85.465 (7)0 |
| β | 84.808 (6)0 |
| γ | 82.950 (7)0 |
| V (Å3) | 2290.6 (13) |
| Z | 4 |
| F (000) | 1104 |
| Dx (Mg m−3) | 1.554 |
| Diffraction radiation type | Mo Kα |
| λ (Å) | 0.71073 |
| μ (mm−1) | 1.13 |
| Crystal description | Needle |
| Temperature (K) | 173 |
| Crystal size (max × mid × min) | 0.14 × 0.09 × 0.07 |
| Measurement device | Bruker APEX II diffractometer |
| Absorption correction type | Multiscan |
| Radiation monochromator | Graphite |
| Radiation source | Fine focus sealed tube |
| No. of F values used [I > 2σ(I)] | 6125 |
| Index ranges ±h, ±k, ±l | ±10, ±16, ±27 |
| No. of unique reflections | 9878 |
| Rint | 0.096 |
| Weighting expression w | 1/[σ2(Fo2) + (0.0585P)2 + 1.1595P], where P = (Fo2 + 2Fc2)/3 |
| Tmin/Tmax (absorption correction) | 0.858/0.925 |
| R[F2 > 2σ(F2)] | 0.0554 |
| wR(F2) | 1.1527 |
| S (goodness of fit) | 1.034 |
| (Δ/σ)max (refine_ls_shift/su_max) | 0.000 |
| Final Δρmax/Δρmin (e Å−3) | 0.64/−0.47 |
Selected bond distances (Å) and angles (°) for [Zn(II)(Tpy)(Pydc)·4H2O] (A) and [Zn(II)(Tpy)(Pydc)·4H2O] (B).
| Bond length (Å)/angles | A | B |
| ZnN27 | 2.030 (3) | 2.020 (3) |
| ZnN12 | 2.072 (3) | 2.065 (3) |
| ZnO30 | 2.138 (3) | 2.180 (3) |
| ZnN1 | 2.173 (3) | 2.161 (3) |
| ZnN13 | 2.173 (3) | 2.161 (3) |
| ZnO19 | 2.268 (3) | 2.213 (3) |
| N27ZnN12 | 163.89 (13) | 166.32 (13) |
| N27ZnO30 | 77.03 (12) | 76.58 (12) |
| N12ZnO30 | 118.30 (12) | 116.90 (12) |
| N27ZnN13 | 99.90 (13) | 101.78 (13) |
| N12ZnN1 | 75.83 (13) | 75.92 (13) |
| O30ZnO19 | 151.74 (11) | 151.93 (11) |
2.2 Theory and computational detail
Density functional theory (DFT) methods were used in this study. All of the calculations were done by Gaussian 09 software [13], using the B3LYP functional [14,15] and a LanL2dz basis set. The B3LYP, a version of DFT method [16], uses Beche's three-parameter functional (B3) and includes a mixture of HF with DFT exchange terms associated with the gradient-corrected correlation functional of Lee, Yang, and Parr [14,15].
Molecular interactions between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin were calculated at 298 K. Ab initio calculations such as HOMO energy (EHOMO), LUMO energy (ELUMO), LUMO–HOMO energy gap (ΔE), hardness (ŋ), softness (σ), electronegativity (χ), and chemical potential (μ), I = −EHOMO, A = −ELUMO were calculated using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
Finally, LUMO–HOMO energy gap (ΔE) was calculated using the following formula: ΔE = ELUMO − EHOMO.
2.3 Synthesis of [Zn(II)(Tpy)(Pydc)·4H2O]
The solvothermal synthesis of [Zn(II)(Tpy)(Pydc)·4H2O] was performed by reacting a mixture of Zn(NO3)2·6H2O (1.89 g, 0.01 mol), 2,2′:6′,2″-terpyridine (2.3 g, 0.01 mol), and pyridine-2,6-dicarboxylic acid (1.67 g, 0.01 mol), which had been dissolved in 30 mL mixture of water, ethanol, and methanol (1:1:1), respectively. The dissolved mixture was transferred into a reaction vessel in an oven and set to 160 °C for 5 days (Scheme 1). The reaction was slow-cooled gradually and left in the oven at room temperature for another 24 h to obtain colorless needles of [Zn(II)(Tpy)(Pydc)·4H2O] (Scheme 1). Yield: 76%. CHN analysis; Anal Found (Calcd) % for C22H22N4O8Zn: C, 49.31 (49.24); H, 4.14 (4.08); N, 10.46 (10.44).
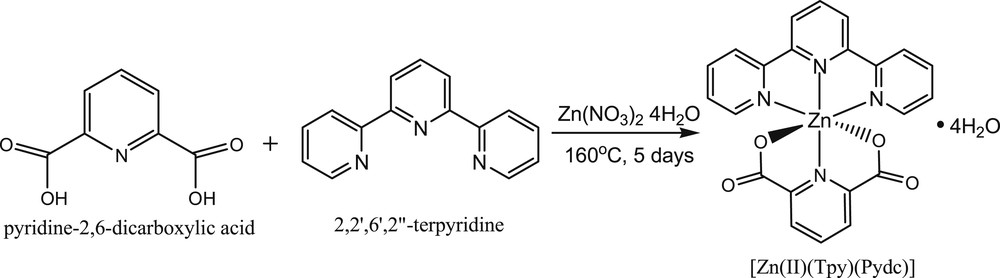
Synthesis of [Zn(II)(Tpy)(Pydc)·4H2O].
2.4 Interaction studies
For the experimental interaction studies, 0.05 g of [Zn(II)(Tpy)(Pydc)·4H2O] was brought in contact with a 2 mL solution containing 5.47 × 10−3 M of 3.99 mg of amoxicillin (Sigma–Aldrich) dissolved in 0.2 mL DMSO, followed by adding 1.8 mL H2O. The [Zn(II)(Tpy)(Pydc)·4H2O] was separated from the resultant mixture via centrifugation at 2000 rpm for 5 min, and the resultant solution was analyzed using a UV–vis spectrophotometer at 256 nm at regular time intervals (every 30 min).
3 Results and discussion
The crystal is clearly different in color as compared with any of the starting materials. Image of the colorless needle like crystals of [Zn(II)(Tpy)(Pydc)·4H2O] is shown in Fig. S1.
3.1 FT-IR spectra
FT-IR of 2,2′:6′,2″-terpyridine, pyridine-2,6-dicarboxylic acid, and [Zn(II)(Tpy)(Pydc)·4H2O] shows significant changes in absorption frequencies due to disappearance of some characteristic frequencies that occurred at coordination sites of 2,2′:6′,2″-terpyridine and pyridine-2,6-dicarboxylic acid upon complex formation (Fig. 1). Vibration frequencies at 1685, 3100, and 1585–1550 cm−1 were ascribed to v(CO), v(OH), and v(CN), respectively, for pyridine-2,6-dicarboxylic acid, whereas 2,2′:6′,2″-terpyridine displayed an interesting v(CN) frequency at around 1595–1550 cm−1.

FT-IR of spectrum of 2,2′:6′,2″-terpyridine, pyridine-2,6-dicarboxylic acid, and [Zn(II)(Tpy)(Pydc)·4H2O].
The characteristic absorption band OH of pyridine-2,6-dicarboxylic acid at 3100 cm−1 disappeared upon coordination with Zn(II) salt [17]. With a formation of noncoordinated water molecules observed at 3454 cm−1, H2O molecules arise from the zinc(II) salts and it was also observed in the crystal structure of [Zn(II)(Tpy)(Pydc)·4H2O] [18]. Other characteristic absorption bands such as v(CO) and v(CN) shifted to lower region on the complex, [Zn(II)(Tpy)(Pydc)·4H2O], spectrum due to coordination with zinc(II) ion [19,20]. New bands at 516–553 cm−1 in the crystal give inference about ѵ(M–O) and ѵ(M–N) bonding.
3.2 Crystal structure of [Zn(II)(Tpy)(Pydc)·4H2O]
In the complex [Zn(II)(Tpy)(Pydc)·4H2O], Zn(II) is coordinated to both 2,2′:6′,2″-terpyridine and pyridine-2,6-dicarboxylate (Fig. 2). The structure of the compound showed an octahedral geometry with N(1A)N(12A)N(13A)N(27A)O(19A)O(30A) and N(1B)N(12B)N(13B)N(27B)O(19B)O(30B) planes from the two molecules bisecting each other at an angle of 33.35°. In both crystal structural compounds, the Zn(II) atom is coordinated to four nitrogen atoms (ZnA–N27A, ZnA–N12A, ZnA–N1A, and ZnA–N13A) with a bond distance ranging from 1.335 to 2.173 Å, comparable with the range usually observed in Zn(II) compounds with similar coordination mode [16]. The two carboxylate oxygen coordinated to the zinc(II) ion via Zn1A–O30A and Zn1A–O19A with bond distances of 2.179 and 2.213 Å, respectively. Four water molecules were found for each zinc(II) compound and are held by intermolecular hydrogen bond of the type OHO. The bond angles N27A–Zn1A–N12A, N27A–Zn1A–O30A, N12A–Zn1A–O30A, N27A–Zn1A–N1A, N12A–Zn1A–N1A, and O30A–ZnN1A are ranged within 90°–180° and are in accordance with the results obtained for octahedral complexes [16]. The complete crystallographic data and the structure refinement parameters are summarized in Table 1. Selected bond distances and angles (Å) for [Zn(II)(Tpy)(Pydc)·4H2O] are given in Table 2.
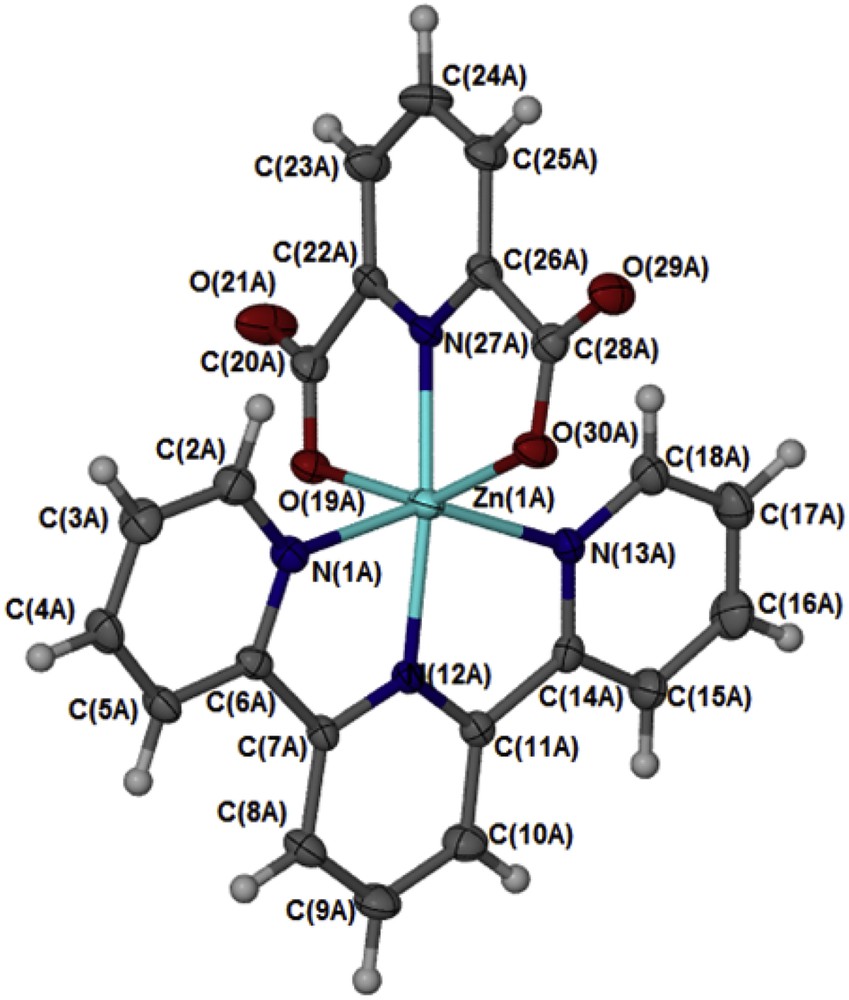
An Oak Ridge Thermal Ellipsoid Plot (ORTEP) view of [Zn(II)(Tpy)(Pydc)·4H2O] with ellipsoids drawn at 50% probability level. One of the two independent molecules is shown here; the other molecule has atomic labels ending B.
The structure of the compound [Zn(II)(Tpy)(Pydc)·4H2O] confirms that the compounds are held together by hydrogen bonding (Fig. 3) linking the crystallographically independent A and B molecules via water bridges, OO distances are between 2.73 and 2.83 Å. These layers pack with minimal interdigitation (Fig. 4). A space volume of 292.52 Å3 was observed.
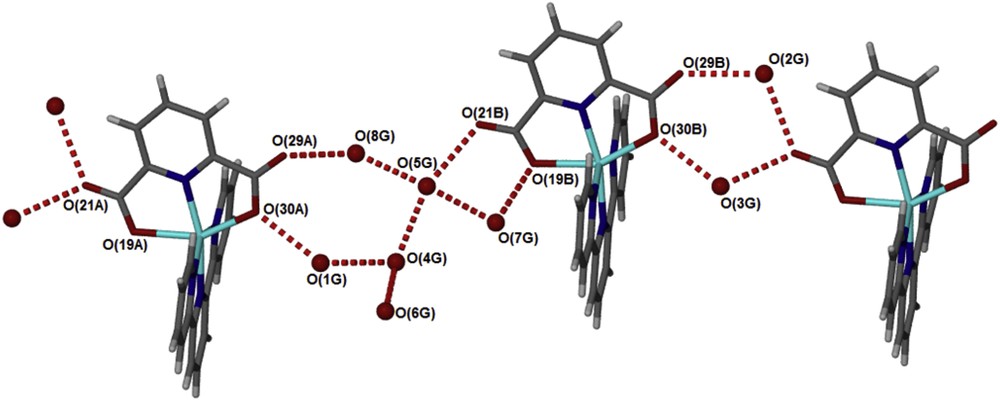
Hydrogen bonds connect the water guest molecules to alternating A and B molecules of [Zn(II)(Tpy)(Pydc)·4H2O].
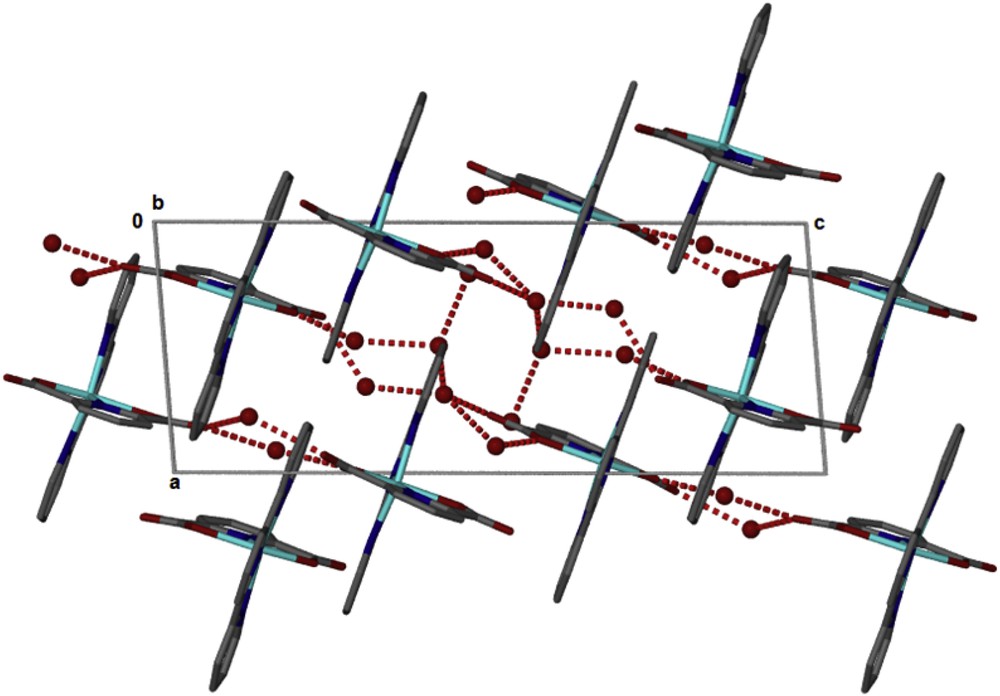
Packing in [Zn(II)(Tpy)(Pydc)·4H2O].
3.3 Thermogravimetric analysis
TGA of zinc(II) complex, [Zn(II)(Tpy)(Pydc)·4H2O], is presented in Fig. 5. A seven-step decomposition pattern was observed for the complex, [Zn(II)(Tpy)(Pydc)·4H2O]. An 8% weight loss was clearly observed around 81 °C, this was immediately followed by a gradual weight loss of 4% up to 288 °C. All water molecules (calculated for the unit) were lost in the temperature range between 81 and 288 °C (obsd 13.4%, calcd 12.0%). The decomposition of co-ordinated ligands 2,2′:6′,2″-terpyridine (Tpy) and pyridine-2,6-dicarboxylic acid (Pydc) was observed to begin at 290 °C with successive weight losses of 10%, 4%, 15%, and 10% at 312, 336, 357, and 448 °C, respectively. The final weight loss of 35% resulted in the formation of zinc oxide (∼15%) at 552 °C. TGA plots of ligands, 2,2′:6′,2″-terpyridine (Tpy) and pyridine-2,6-dicarboxylate (Pydc), are presented in Supplementary data (Figs. S2 and S3).
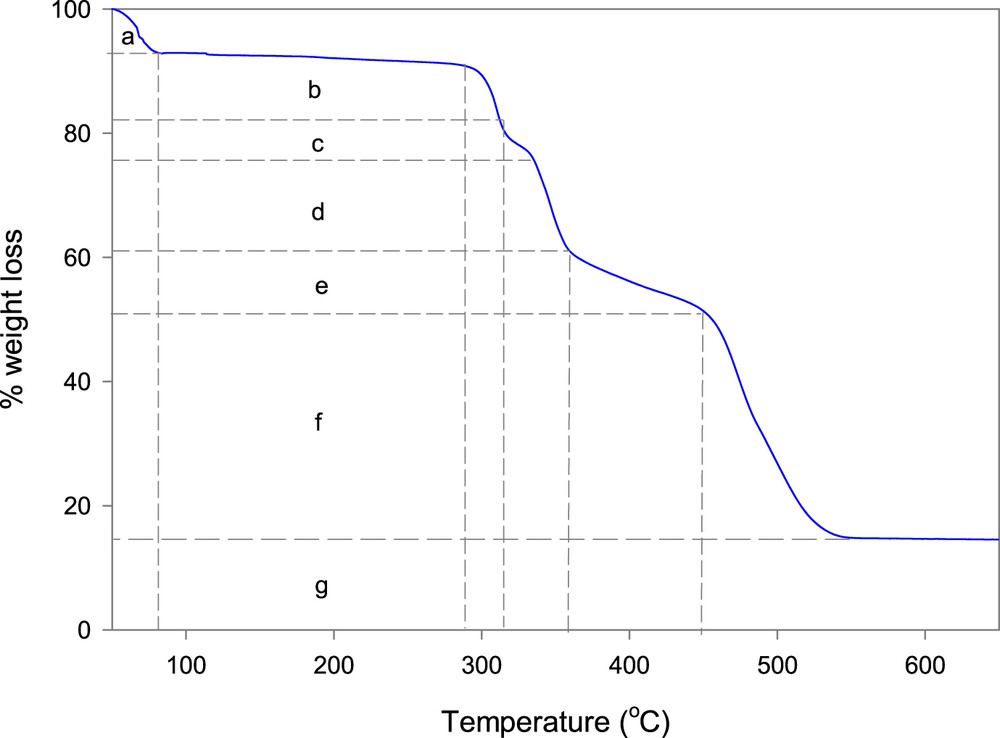
TGA plot of [Zn(II)(Tpy)(Pydc)·4H2O], where a = 8% wt. loss (Temp = 81 °C); b = 4% wt. loss (Temp = 288 °C); c = 10% wt. loss (Temp = 312 °C); d = 4% wt. loss (Temp = 336 °C); e = 15% wt. loss (Temp = 357 °C); f = 10% wt. loss (Temp = 448 °C) and g = 15% wt. loss (Temp = 552 °C).
3.4 Luminescence properties
Luminescence emission spectra of 2,2′:6′,2″-terpyridine, pyridine-2,6-dicarboxylic acid, and [Zn(II)(Tpy)(Pydc)·4H2O] were obtained in DMSO at room temperature as shown in Fig. 6. 2,2′:6′,2″-Terpyridine and pyridine-2,6-dicarboxylic acid exhibited blue fluorescent emission bands at 424 nm (λex = 330) and 489 nm (λex = 346), respectively. Emissions at these wavelengths are assigned to intraligand (π → π∗) transition [20,21]. A second weak emission band for pyridine-2,6-dicarboxylic acid at 372 nm was attributable to n → π∗ transition. Shifts in the emission wavelengths were observed for [Zn(II)(Tpy)(Pydc)·4H2O] at 412 and 434 nm (λex = 346) when compared with those of the free ligands, this was ascribed to coordination of the Zn(II) with nitrogen and oxygen donor atoms of 2,2′:6′,2″-terpyridine and pyridine-2,6-dicarboxylic acid (metal-to-ligand charge transfer). The UV spectra (λex = 346) in DMSO indicate that the solid-state octahedral geometry of [Zn(II)(Tpy)(Pydc)·4H2O] is preserved in solution. The luminescent emission of [Zn(II)(Tpy)(Pydc)·4H2O] indicates that it may be a potential compound for optical and blue-light emitting materials. These findings are in good agreement with our previous study on Zn(II)-related complexes [22].
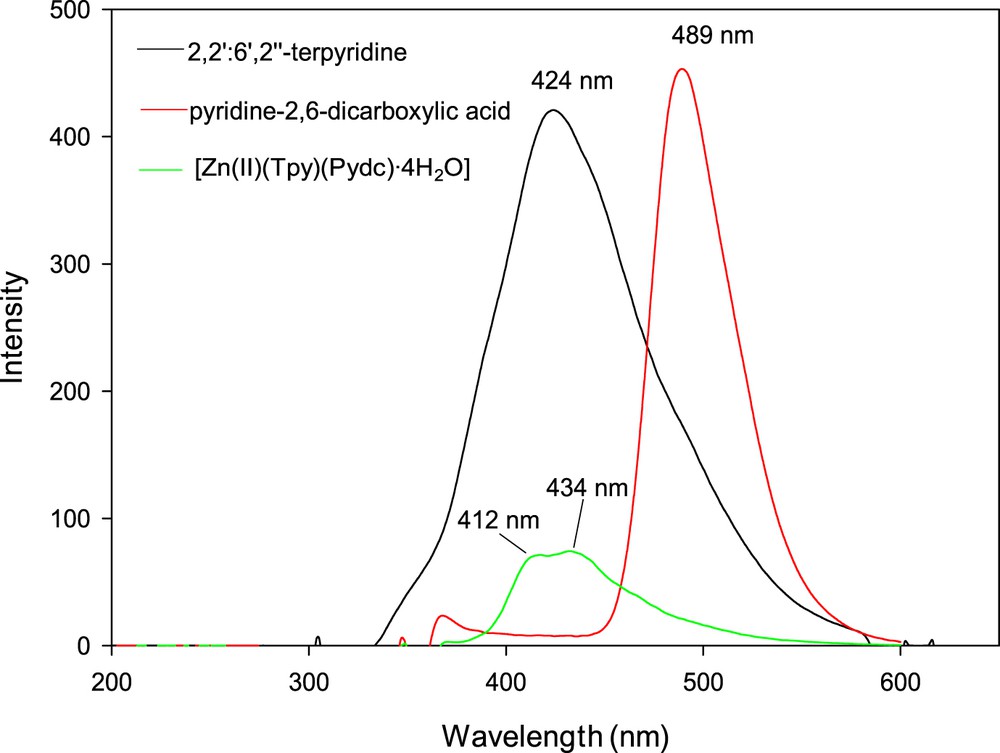
Emission spectra of 2,2′:6′,2″-terpyridine, pyridine-2,6-dicarboxylic acid, and [Zn(II)(Tpy)(Pydc)·4H2O].
3.5 Hirshfeld surface analyses
The molecular Hirshfeld surface [23] (dnorm, curvedness, and shape index for [Zn(II)(Tpy)(Pydc)·4H2O]) is demonstrated in Fig. 4 and mapped over dnorm ranges −0.3346 to 1.4318 Å, shape index ranges −0.9993 to 0.9977 Å, and curvedness ranges −4.5383 to 0.8944 Å. The dnorm mapping indicates the strong hydrogen bond interactions, such as OH⋯O hydrogen bonding between coordinated/lattice water and carboxylate oxygen, which gives the indication that the main interaction between complexes occurs around the region seen as a bright red area in the Hirshfeld surface area (Fig. 7). The two-dimensional fingerprint plots for [Zn(II)(Tpy)(Pydc)·4H2O] (Fig. 8) show that the intermolecular H⋯H, OH⋯O, and CH···π interactions are well dominated and are in complement to the Hirshfeld surface area.
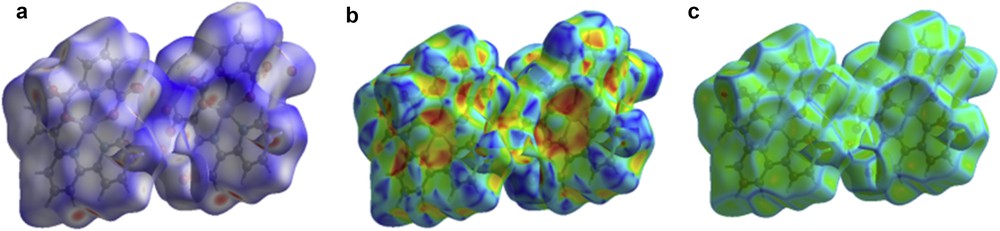
Molecular Hirshfeld surface area: (a) dnorm, (b) shape index, and (c) curvedness for [Zn(II)(Tpy)(Pydc)·4H2O].
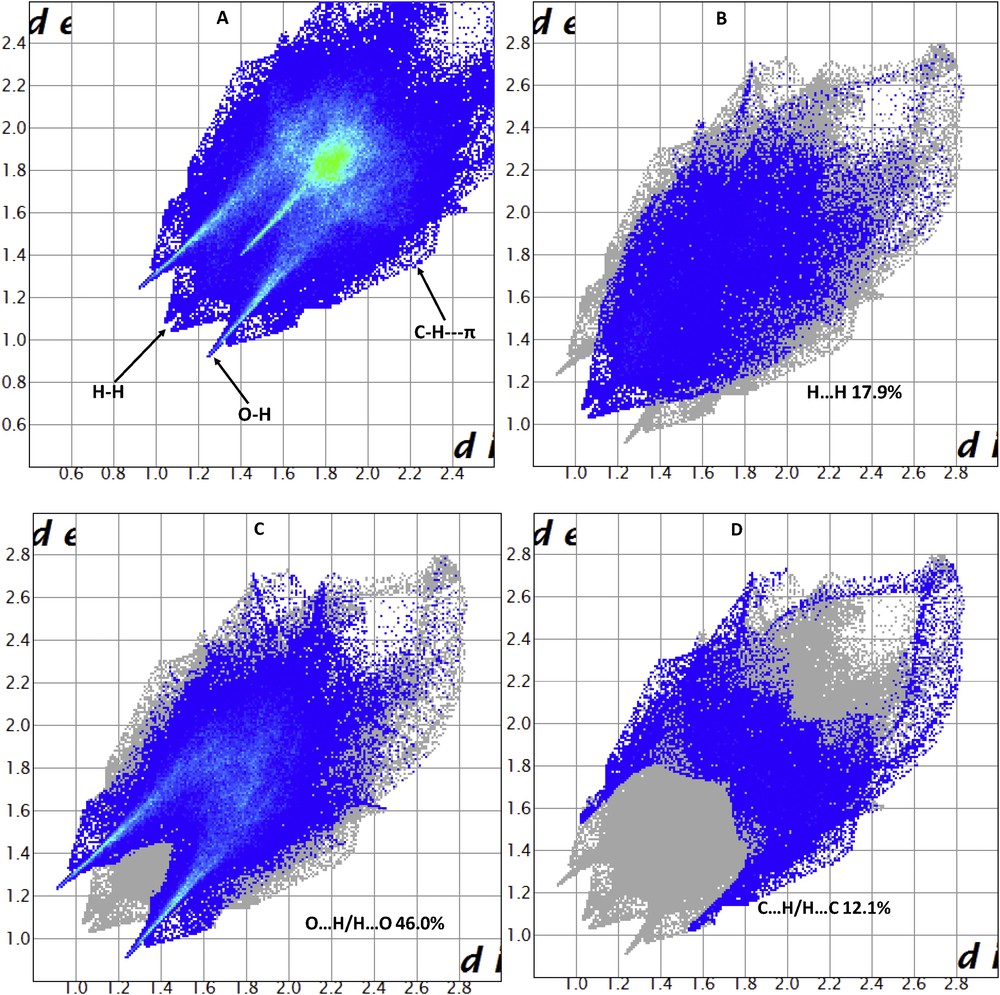
Fingerprint plot of [Zn(II)(Tpy)(Pydc)·4H2O]: (a) full and resolved into (b) H⋯H, (c) OH/HO, and (d) H⋯C contacts showing the percentages of contacts contributed to the total Hirshfeld surface area.
Shit et al. [24] reported that the shape index is the most sensitive to very subtle changes in surface shape, the red triangles on them (above the plane of the molecule) are represented by the concave regions indicating atoms of the π-stacked molecule above them, and the blue triangles are represented by the convex regions indicating the ring atoms of the molecule inside the surfaces [24]. The measure of the shape of the surface area of [Zn(II)(Tpy)(Pydc)·4H2O] is defined by the curvedness [24]. The flat areas of the surface correspond to low values of curvedness, whereas sharp curvature areas correspond to high values of curvedness and usually tend to divide the surface into patches, indicating interactions between neighboring molecules. The large flat region, which is demarcated by a light blue outline, refers to the π···π stacking interactions. Two sharp spikes pointing toward lower left of the plot are typical OH⋯O hydrogen bonds. This portion corresponds to OH/HO interactions comprising 46.0% of the total Hirshfeld surface area for [Zn(II)(Tpy)(Pydc)·4H2O]. CH···π interactions were observed in the fingerprint plots of HC/CH contacts comprising 12.1% of the total Hirshfeld surface area for [Zn(II)(Tpy)(Pydc)·4H2O]. They correspond to all CH⋯C interactions of which CH···π appears in the same fingerprint plot. The broad region bearing short and narrow spikes at the middle of the plot is reflected as H⋯H interaction in [Zn(II)(Tpy)(Pydc)·4H2O] comprising 17.9% of the total Hirshfeld surface area for [Zn(II)(Tpy)(Pydc)·4H2O]. Apart from these, the presence of C⋯O, O⋯O, C⋯C, N⋯H, and O⋯C less important interactions is observed and summarized in Table S1.
3.6 Interaction between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin
To demonstrate the potential medical application of [Zn(II)(Tpy)(Pydc)·4H2O], the interaction property between amoxicillin and [Zn(II)(Tpy)(Pydc)·4H2O] was examined. Amoxicillin was chosen as it is widely used for bacterial sinusitis. This technique was used to quantify the amount of amoxicillin adsorbed into the [Zn(II)(Tpy)(Pydc)·4H2O]. Interaction studies were ascertained using a UV–vis spectrophotometer at 256 nm (Fig. 9). Calibration curve obtained for amoxicillin solution at 256 nm is presented in Fig. S4. The complex, [Zn(II)(Tpy)(Pydc)·4H2O], remained undissolved in amoxicillin solution (in H2O) during the study.

UV spectra of amoxicillin dissolved in water showing absorbance decreases with time.
About 97.5% of amoxicillin interacted with [Zn(II)(Tpy)(Pydc)·4H2O] (Table 3). This was attributed to hydrogen bonding, CH–π interaction, and electrostatic interaction at a molecular level between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin.
Absorbance, concentration, percentage, and mass of amoxicillin before and after interaction with [Zn(II)(Tpy)(Pydc)·4H2O] at 30 ± 2 °C.
| Parameters | Before contact | After contact (filtration) |
| Absorbance of Amoxicillin | 0.236 | 0.006 |
| Concentration of amoxicillin | 5.47 × 10−3 M | 1.40 × 10−4 M |
| Percentage of amoxicillin | 100% | 2.50% |
To further illustrate this finding, theoretical studies on the electronic properties of [Zn(II)(Tpy)(Pydc)·4H2O] were carried out.
3.7 Theoretical studies
Molecular interactions between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin were modeled. HOMO and LUMO positions on amoxicillin are presented in Fig. 10. HOMO centers around the hydroxy phenolic ring of amoxicillin, whereas LUMO is located around heptane-24-carboxylic acid end of amoxicillin.
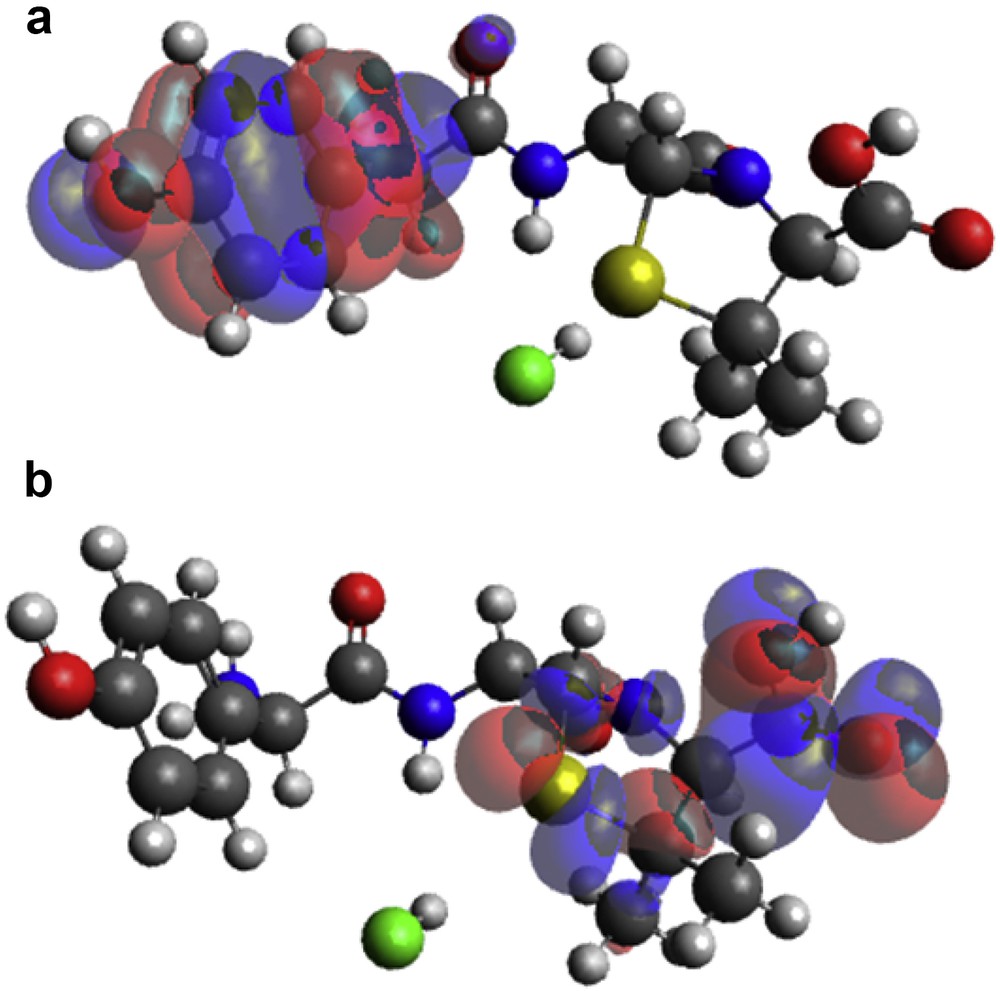
(a) HOMO and (b) LUMO locations on modeled amoxicillin.
Zinc(II) complexes of 2,2′:6′,2″-terpyridine and pyridine-2,6-dicarboxylic acid, [Zn(II)(Tpy)(Pydc)·4H2O], showed HOMO centers around the metal center and pyridine-2,6-dicarboxylate group, whereas LUMO is situated around 2,2′:6′,2″-terpyridine ring (Fig. 11).
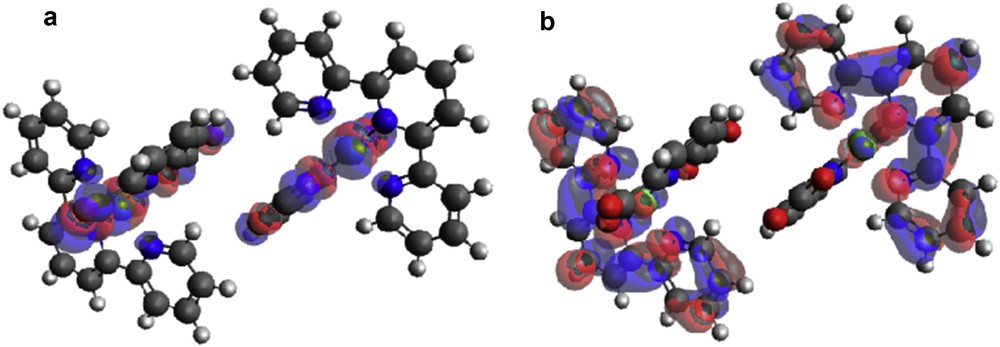
(a) HOMO and (b) LUMO locations on modeled [Zn(II)(Tpy)(Pydc)·4H2O].
An adduct was formed when [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin were brought in contact with each other. In this, the HOMO position originates from the hydroxy phenolic ring of amoxicillin and LUMO is situated around the 2,2′:6′,2″-terpyridine ring of [Zn(II)(Tpy)(Pydc)·4H2O] (Fig. 12a). This clearly indicated that the interaction between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin is through electron transfer from the HOMO positions to LUMO atoms (Fig. 12b). Electron transfer further confirms the possibility of forming π–π interaction between aromatic rings of mostly terpyridine rings of [Zn(II)(Tpy)(Pydc)·4H2O] and the hydroxy phenolic ring of amoxicillin.
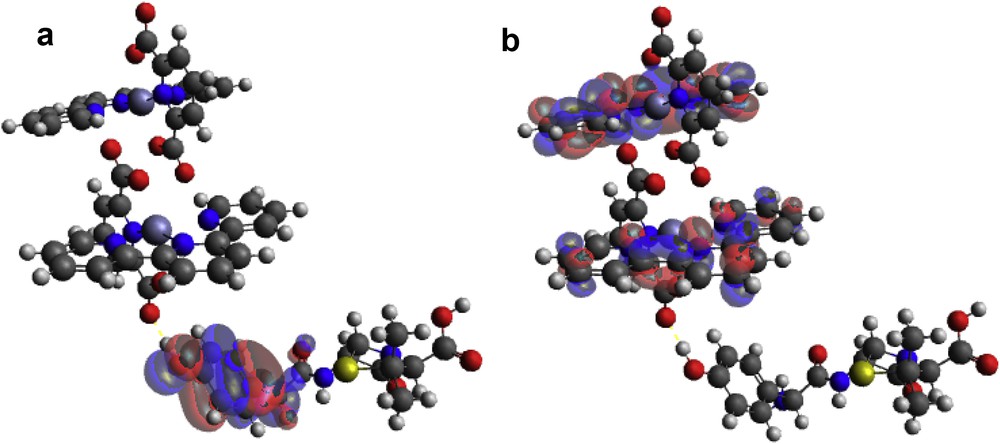
(a) HOMO and (b) LUMO locations on modeled [Zn(II)(Tpy)(Pydc)·4H2O]–amoxicillin adduct.
3.7.1 Energy gap (ΔE)
The HOMO–LUMO gap describes the stability and resistance of molecules, and it also predicts activity between species by providing the electrical transport properties as well as electron carrier and mobility in molecules [25]. HOMO and LUMO energies of amoxicillin, [Zn(II)(Tpy)(Pydc)·4H2O], and [Zn(II)(Tpy)(Pydc)·4H2O]–amoxicillin adduct are presented in Fig. 13. Low HOMO energy (better electron donor) describes the observed high ionization potential observed, whereas high LUMO energy (better electron acceptor) confirms high electron affinity. The energy gaps of amoxicillin, [Zn(II)(Tpy)(Pydc)·4H2O], and [Zn(II) (Tpy) (Pydc)·4H2O]–amoxicillin adduct were 4.945, 4.248 and 3.205 eV, respectively. The observed energy, 3.205 eV, is indicative of a favorable interaction between amoxicillin and [Zn(II)(Tpy)(Pydc)·4H2O].
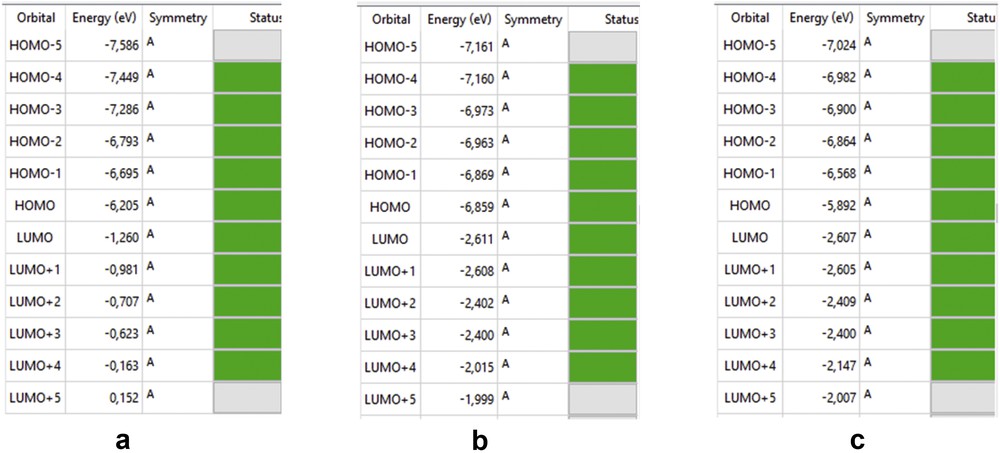
HOMO and LUMO energies of (a) amoxicillin, (b) [Zn(II)(Tpy)(Pydc)·4H2O], and (c) [Zn(II)(Tpy)(Pydc)·4H2O]–amoxicillin adduct.
3.7.2 Electrostatic charge transfer
The charge transfer was investigated by natural bond orbital analysis by looking at the partial charge difference in amoxicillin, [Zn(II) (Tpy) (Pydc)·4H2O], and [Zn(II) (Tpy) (Pydc)·4H2O]–amoxicillin adduct before and after the interaction. Some atomic partial charges on atoms of amoxicillin were observed to shift slightly after the formation of adduct, thus further indicating that charge transfer occurs from amoxicillin to [Zn(II)(Tpy)(Pydc)·4H2O].
Partial charges on sulfur (S), oxygen (from OH), oxygen (from COOH), and nitrogen (on amoxicillin) before interaction with [Zn(II)(Tpy)(Pydc)·4H2O] were 0.081, −0.757, −0.741, and (−0.530, −0.671, −0.944), respectively. After interaction with [Zn(II)(Ter)(Pydc)·4H2O] to form an adduct, new partial charges on sulfur (S), oxygen (from OH), oxygen (from COOH), and nitrogen (on amoxicillin) were 0.099, −0.795, −0.738, and (−0.527, −0.943, −0.673), respectively. This confirms the transfer of electron from amoxicillin to [Zn(II)(Tpy)(Pydc)·4H2O] for adduct formation to take place.
Oxygen atoms on [Zn(II)(Tpy)(Pydc)·4H2O] before and after the interaction with amoxicillin were (−0.815, −0.812, −0.753, −0.747) and (−0.816, −0.771, −0.757, −0.747), respectively. A slight change was also observed with nitrogen atoms with partial charges becoming less negatively charged upon forming adduct. The zinc(II) ion becomes more positively charged upon forming adduct (from 1.363 to 1.377).
Electronic identifiers calculated indicated that amoxicillin is slightly softer than [Zn(II)(Tpy)(Pydc)·4H2O] and vice versa with hardness, notwithstanding within close range. Complex, [Zn(II)(Tpy)(Pydc)·4H2O], is highly electronegative as compared with amoxicillin, thus attracting electron from amoxicillin for adduct formation to take place (Table 4).
Some electronic structure identifiers of the studied adducts.
| Parameters | Amoxicillin | [Zn(II)(Tpy)(Pydc)·4H2O] | Adduct |
| Hardness (ŋ) | 2.4725 | 2.124 | 1.6025 |
| Softness (σ) | 0.4044 | 0.4708 | 0.6240 |
| Electronegativity (χ) | 3.7325 | 4.7350 | 4.2495 |
| Chemical potential (μ) | −3.7325 | −4.7350 | −4.2495 |
3.7.3 Electronic properties (UV adsorption analysis)
The electronic properties of [Zn(II)(Tpy)(Pydc)·4H2O], amoxicillin, and adduct were calculated using the time-dependent density functional theory (TD-DFT) approach on the previously optimized ground-state compounds in the gas phase (Fig. S5). The excitation wavelengths (λ) of [Zn(II)(Tpy)(Pydc)·4H2O], amoxicillin, and adduct were 334, 268, and 551 nm, respectively. The observed shift in wavelength confirmed the formation of adducts.
4 Conclusions
We have reported the synthesis of a novel complex [Zn(II)(Tpy)(Pydc)·4H2O] via a solvothermal method. X-ray crystallography confirmed the formation of octahedral zinc(II) complex of 2,2′:6′,2″-terpyridine (Ter) and pyridine-2,6-dicarboxylic acid (Pydc), [Zn(II)(Tpy)(Pydc)·4H2O]. Luminescent emission of [Zn(II)(Tpy)(Pydc)·4H2O] indicated that the complex may have potential application as sensors for optical and blue–light emitting materials. Structural characterization of [Zn(II)(Tpy)(Pydc)·4H2O] revealed that the net supramolecular arrangements are dictated by H⋯H, O⋯H, and CH⋯π interactions. Experimental and theoretical studies clearly indicated that [Zn(II)(Tpy)(Pydc)·4H2O] dimer interacts with amoxicillin via electron transfer, π–π interaction, hydrogen bond interaction, and van der Waals forces. The observed interaction between [Zn(II)(Tpy)(Pydc)·4H2O] and amoxicillin shows possible development of the complex for medicinal applications.
Acknowledgments
M.D.O. acknowledges financial support from the Organization for Women in Science for the Developing World (OWSD). The authors thank the Centre for High Performance Computing (CHPC), Cape Town, South Africa for providing the platform in carrying out the molecular modeling studies on the Gaussian09 software.