

 CC-BY 4.0
CC-BY 4.0
The story of the beginning of the extension of the periodic table beyond uranium is closely related to nuclear physics and radiochemistry practiced in some laboratories in Europe and the United States from 1935 to 1945. In 1944, Seaborg advanced the concept of actinides. An article that encompasses this topic has been published in Comptes Rendus de Physique [1]. This article in Comptes Rendus de Chimie more explicitly handles the chemistry that led to the discoveries of the first four transuranic elements (neptunium, plutonium, americium, and curium) and their place in the periodic table within the actinide series, analogous to the lanthanide series.
Evolution of the principle of classification of chemical elements
The periodic table of chemical elements was built between 1869 and 2016. In 1869, Mendeleev proposed a system for classifying the sixty or so elements that were well-known at that time based on their atomic mass relative to that of hydrogen. This system consisted of arranging the elements in series (horizontal) and groups (vertical) so as to reveal the chemical behaviour analogies of the elements classified into groups. At that time, the properties of their oxides were well known [2]. The resulting table, even if preceded by other corresponding tables with other ways of classifying the elements, was retained as the first "periodic table" because of its predictive power. Indeed, it had empty boxes for unknown elements that were discovered subsequently and whose properties have been found to conform to Mendeleev's predictions. In 2016, the IUPAC gave names to the last elements of which some ephemeral isotopes had been discovered between 1968 and 2010. All this is well known and was widely commented upon during the International Year of the Periodic Table in 2019 [3].
Today, the periodic table has 118 boxes divided into seven periods and 18 columns, each containing the isotopes of the 118 elements. The elements in each column are homologues. Each element is formed of atoms. Each atom is characterised by two numbers attached to its nucleus: Z, the number of protons; and A, the sum of the numbers of protons and neutrons, N, (A = Z + N). All isotopes of an element have the same Z value, and values of N vary. The value of Z is the atomic number of the element. The number of isotopes of an element is determined by the values of N. The number of electrons in an atom is equal to Z. At each value of Z, for each element, there is a corresponding electronic structure defined in terms of filling atomic orbitals of types s, p, d and f. Since May 2019, the atomic mass of an AZX isotope is the mass of N of them, N being Avogadro's number, a universal constant (N = 6.02 × 1023). The atomic mass of an element is the weighted mass of the isotopes that constitute it.
The nuclei are stable for fixed values of Z and N. If N has only one value, the element is monoisotopic. If the values of Z and N are different of these fixed values or if N is much greater than or much less than Z or if Z is greater than 83, the nuclei disintegrate. The corresponding atoms are radioactive, and they then disappear according to their radioactive half-life, T1/2, emitting various radiations. Elements whose isotopes are all radioactive are called radioelements (Z > 83). Beyond Z = 98, radioelements exist only in immeasurable quantities. The heaviest are only produced on the scale of a few atoms, which disintegrate as soon as they are created.
All these notions are taught. They are recalled here because in the following text, the relationship between a stable or radioactive isotope and a chemical element is constantly referred to.
Before 1913, a chemical element was understood as an elementary entity that was preserved in chemical reactions. Its atomic mass was measured by determining the proportions in which it was bound to hydrogen and oxygen, using the mass of hydrogen as a unit of measurement. In 1930, the modern notion of an element was developed, and the principle of the construction of the periodic table could start based on the nuclear and electronic structures of the elements. For a long time, chemists have only experimented on matter using macroscopic quantities. With the synthesis of radioactive isotopes (also called radionuclides) with very short half-lives, radiochemists have conducted experiments on immeasurable quantities of matter whose behaviour poses a scale problem of several powers of ten to deduce the behaviour of the element that it represents [4].
Construction of the periodic table
Assigning elements to the boxes in the periodic table was performed during four phases (Figure 1) corresponding to the discoveries of the following: (1) natural elements (Antiquity-1925); (2) natural radioelements in filiation with 238U, 235U, and 232Th (1896–1939); (3) artificial radioelements products with accelerated light ions (1937–1961); and finally (4) artificial radioelements produced with accelerated heavy ions (1968–2010). The last three were initiated by major discoveries: natural radioactivity (1896) for phase 2; artificial radioactivity (1934) and acceleration of ions by cyclotrons starting from 1930 for phases 3 and 4. Phase 4 ended in 2010, as since that date and for a long time, the search for the element Z = 119 has been in vain. The discovery of natural radioactivity made it possible to have the first ions accelerated to sufficient energies to discover the nuclei of the atoms and transform them via nuclear reactions. The construction of the periodic table is related to the evolution of the notion of a chemical element.
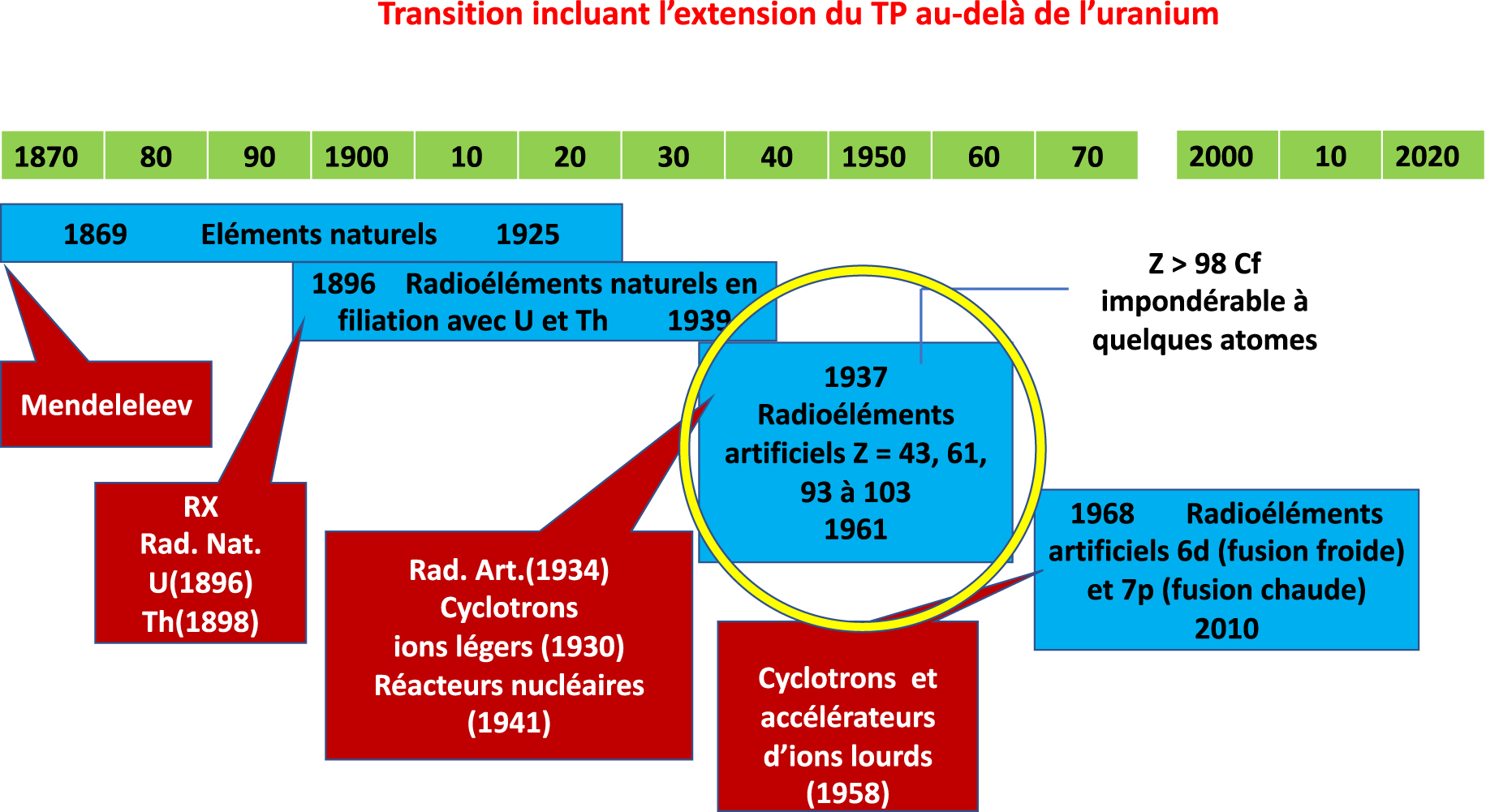
Start and end dates of the transitions in the construction of the periodic table. The initiator of phase 1, for the discovery of natural elements, is D. Mendeleev. The discoveries of X-rays (W. Roëntgen) and radioactivity (H. Becquerel) initiated phase 2, corresponding to the discovery of natural radioelements. The discovery of artificial radioactivity (I. and F. Joliot) initiated phase 3, during which the radioactive isotopes of the artificial radioelements were produced using cyclotrons and nuclear reactors (subject of this article). During phase 4, the isotopes of the elements 6d and 7p were produced using heavy ion accelerators. Masquer
Start and end dates of the transitions in the construction of the periodic table. The initiator of phase 1, for the discovery of natural elements, is D. Mendeleev. The discoveries of X-rays (W. Roëntgen) and radioactivity (H. Becquerel) initiated phase ... Lire la suite
Beyond Uranium
The first transuranium radioelement, neptunium, was discovered in 1940 by Edwin McMillan at Berkeley after observations on the radioactivity produced by irradiation of uranium with neutrons, irradiations that had begun in 1934, just after the discovery of artificial radioactivity by Irene and Frédéric Joliot [5]. They had shown that the alpha rays of polonium transformed monoisotopic 27Al into 30P according to the following nuclear reaction: 27 Al(α, n) 30P, and that 30P disintegrated by beta plus emission (β +) with a half-life of 2.5 minutes giving table 30Si. They also irradiated other light elements with alpha particles and showed that it was possible to produce radioactive isotopes decaying by beta minus (β−) emission, a type of radioactivity well known for natural radionuclides. They also had Po/Be neutron sources, like radiochemistry laboratories. Since the Joliots' discovery, Enrico Fermi, the first to do so, irradiated uranium in Rome with neutrons for the purpose of synthesising isotopes of transuranian elements. He believed that certain forms of radioactivity isolated using chemistry from a multitude of others were the signatures of two radioactive isotopes of elements with Z > 92. His results have been criticised and his experiments were repeated at the Institut du Radium in Paris and at the Kaiser Wilhem Gesellshaft Institute in Berlin. In fact, the experiments of Irene Joliot-Curie (Paris) and Otto Hahn (Berlin) led Otto Hahn to discover the fission of uranium [6], a major discovery in the history of humanity. By studying the products of fission, Joliot made an essential observation, repeated by McMillan.
Before discussing McMillan's experiences, here is a brief overview of what was known about nuclear physics towards 1935 when it came to synthesising nuclei beyond Z = 92 and what means were available to experimenters for identifying radioactive isotopes and assigning them to an element.
Nuclear synthesis
The nuclear process: AZX (n, γ) A+1ZX (β−) A+1Z+1X, is the capture of a neutron by a heavy isotope with emission of a gamma photon followed by a β − decay of the new isotope formed allows to gain a unit in the Z scale, that is to say to go from an isotope of one element to an isotope of the next element in the periodic table. It is the addition of the neutron to AZX which makes A+1Z+1X a β− emitter in general. It is also possible to gain two units in Z via irradiating of AZX with alpha particles: AZX (He2+, n) A+3Z+2X. Since 1935, these routes were the easiest to follow as there were sources of alpha particles and neutrons available. The first sources were based on 212Po (α, 138 d) or 226Ra (α, 1600 a) salts or 222Rn (α, 3.8 d) gas and their daughters. They could have an alpha activity of 1 Ci. Others, mixtures of the salts of 226Ra and beryllium, yielded about 107 neutrons/s/Ci/4π via the process 10Be(α, n) 13C. The fast neutrons emitted by this process could be slowed down in paraffin.
In 1939, the two cyclotrons at Berkeley in the USA produced light ions: p (1H +), d (2H +) and α (4He2 +), but also fast neutrons, by bombarding a sheet of Be with deuteron, which gave rise to the reaction 10Be (d, n) 11B. One could hope to gain one or two units in Z by reactions such as: AZX (p, n) AZ+1X, AZX (d, n) A+1Z+1X, AZX (d,2n) AZ+1X or AZX (He2+,2n) A+2Z+2X. The hope of being able to synthesise and detect dN2 radionuclides by irradiating the N1 isotopes (stable or radioactive) of a target, during dt, with a flux Φ of particles, is proportional to the cross section of the reaction σ = dN2/ΦN1 dt, and to the yield R of detection of the activity of the radionuclides formed, AN2 = R[0.7/T1/2N2], with R depending on the instrumentation. Joliot had proved the formation of 30P on 2.5 × 104 atoms (100 Bq), which corresponds more or less to the production of a few atoms per mCi of the 210Po in the source.
At the end of 1939, it was known that the irradiation of uranium, 238U and 235U (0.7%), with neutrons emitted by the available sources, gave a dreadful mixture of radioactivities. However, the effective production cross sections of the radionuclides responsible for these radioactivities were very low, to the point that it was necessary to irradiate targets for a long time with sources based on natural radionuclides. The production of artificial radionuclides was dramatically increased with accelerated particle beams or cyclotron neutrons because the fluxes were out of all proportion to those of the sources. At Berkeley, the chemists had access to two cyclotrons of 37 and 60 inches, built by Ernest Lawrence and Stanley Livingston. The neutron flux produced by 16 MeV and 10−3 microampere deuteron beams bombarding a sheet of Be was equivalent to that of a 0.1 Ci alpha-neutron source. Intensities of several hundred microamperes were common.
Means of detecting radioactivity
By 1939, the means of detection and measurement of the intensity of alpha, beta, or gamma radiation were the ionisation chamber and the Geiger Muller (GM) counter. The radiation energies were measured by absorption in metal screens. The proportional amplifier ionisation chamber, used for measuring the energy of alpha radiation, was in its infancy. The Wilson (cloud) chamber gave a photographic image of the tracks of ionised particles left from radiation in a water saturated atmosphere. GM yields were variable.
Chemical identification
Since Marie Curie, the only method of identifying the chemical nature of a radionuclide in an immeasurable quantity was co-precipitation [7], which consists in carrying down a trace element (radionuclide) present in a solution with an element in a measurable amount (with similar chemical properties), then dissolving the obtained precipitate and restarting the co-precipitations in order to enrich the successive precipitates in trace elements (Figure 2). The essential results of the laws of co-precipitation were as follows: if by carrying down one element by another we can enrich the precipitates, we are in the presence of two elements; if there is no enrichment, it means that there are two isotopes. In the application of this method, the idea was that the homologous elements of the periodic table have similar properties. It is easy to implement co-precipiation if the trace element is represented by a radionuclide that is easily detectable. Co-precipitation made it possible to identify all the daughters of 238U, 235U, and 232Th (second transition series of the periodic table).
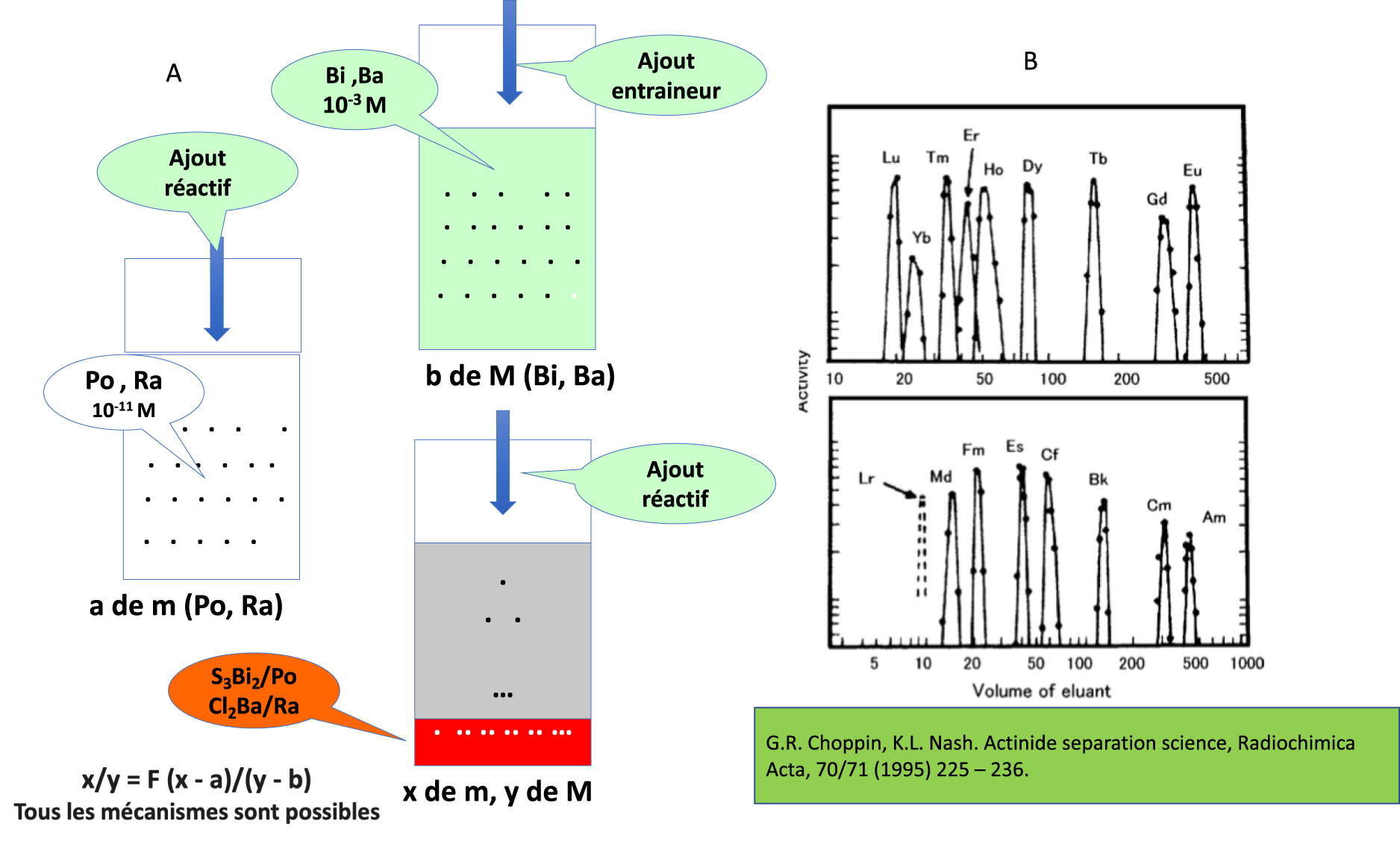
Illustrations of the identification of radionuclides produced in immeasurable quantity. (A) Co-precipitation. The addition of a reagent (H2S or HCl) to a 10−11 M solution of 210Po or 228Ra does not produce precipitates. After the addition of a Bi or Ba salt at 10−3 M, the addition of reagent (H2S or HCl) precipitates Si3Bi2 and Po or BaCl2 and Ra. The quantities x and y of the microcomponent and macrocomponent in the precipitates are linked to the separation factor F. These examples correspond to the discoveries of Po and Ra by M. Curie. B) Chromatography. Drip elution of the radionuclides as M3+ ions, Z = 63 to 71, and Z = 96 to 102 from a Dowex-50 cation exchange resin by ammonium alpha-hydroxy-isobutyrate. Masquer
Illustrations of the identification of radionuclides produced in immeasurable quantity. (A) Co-precipitation. The addition of a reagent (H2S or HCl) to a 10−11 M solution of 210Po or 228Ra does not produce precipitates. After the addition of a Bi or Ba salt ... Lire la suite
Status of the irradiation of uranium by neutrons in 1939
Experiments on the irradiation of uranium by slow or fast neutrons conducted before 1939 had led to many observations; for example, slow neutrons were more efficient than fast neutrons to induce fission and that fission was 15 times more likely than neutron capture. Two observations were particularly significant in the context of this article. The weak activities of radionuclides formed by irradiation of uranium with sources, measured using GM, were only 4 to 5 times higher than those of daughters of 238U: 234Th (β, 24.1 d) and 234mPa (β, 1.2 min) both beta emitters. Irene Joliot-Curie showed in 1938 [8] that if Th and Pa were separated from uranium, before irradiation, the identification of the radionuclides formed in uranium irradiation was facilitated. Also, after 1938, radiochemically pure uranium (i.e., free of its daughters) was irradiated, but for only twenty days. To make uranium radiochemically pure, Th and Pa were carried down by Fe(OH)3 in a uranium carbonate solution, and the uranium was precipitated as U2O7(NH4)2. Hahn followed this protocol to demonstrate U fission. He identified the fission product 140Ba (β, 12, 8 d) on 3.6 106 atoms co-precipitated with BaCl2. Joliot showed [9, 10] in 1939 that the fission of 238,235U by neutrons gave two to three free fast neutrons, and that the fission products escaped from a fine layer of U2O7(NH4)2 irradiated with neutrons, by carrying and sharing the 200 MeV of the fission energy in the form of kinetic energy.
Discovery of neptunium
Before 1939, all experiments on irradiation of uranium were conducted in Europe and looked upon by researchers at Berkeley. Confirming Joliot's experiments, McMillan noticed in the spring of 1940, that a radioactivity 𝛽 of 2.3 d, easily measurable with a GM, remained in the thin layer of U2O7(NH4)2 irradiated with neutrons [11]. Obviously, it was not due to a fission product, but it was compatible with the β emission of an AZX radionuclide in which the recoil energy of the daughter AZ +1X is almost zero (100 eV), for example, that of the 23992U beta emitter, formed by the radiative capture of a neutron by 23892U, and giving 23993X, itself a beta emitter. According to this hypothesis, 23993X should have been a radioactive isotope of an unknown element homologue of rhenium. McMillan and Philip Abelson [12] irradiated U2O7(NH4)2 with Berkeley's 60-inch cyclotron for an hour with neutrons produced by a beam of deutons of 100 microamperes. Under these conditions, about 107 atoms of new isotopes were formed.
Based on radioactivity measurements, they show that the activity of 2.3 d of X actually comes from the 𝛽 decay of the activity of 23 min, with lower energy than that of the 23 min. The 23 min activity was attributed to 239U, an activity already measured by Fermi as early as 1935. The formation process of 23993X according to the sequence: 23892U (n, γ) 23992U (β, 23 min, Emax = 1 MeV) giving 23993X (β, 2.3 d, Emax = 0.5 MeV) was confirmed. Until these experiments, the half-life of 239U was not clear; Fermi and Hahn had proposed different values.
To verify these findings and identify the chemical properties of 23993X = X (to simplify), they began to experiment with co-precipitation in the solution of dissolution of the target in HCl by taking Re as the carrier. To accomplish this, ReO4NH4 was added to the solution. The experiments were negative; X was neither carried by ReS2 (treatment by H2S) nor by metallic Re (reduction by Zn), nor even by volatilisation with ReO3 (oxidation by heating with H2SO4). Would X be atypical for not being classified as a homologue of Re? Would transuranic elements in general also be atypical, with properties closer to those of U than to those of the elements classified in columns 8 (Z = 94), 9 (Z = 95), and 10 (Z = 96) of the periodic table?
McMillan and Abelson thus compared the summary properties of X and U during a particular redox cycle in order to explore the degrees of oxidation or the valences of X. This was possible because the solution for dissolving the target contained 238U in measurable quantities as well as 239U and X in tracer quantities. They conducted co-precipitations knowing that acetic acid precipitates U(VI) in the form of double sodium acetate, that the Ce (III,IV) or Th(IV) fluorides respectively entrain the trivalent elements M(III) and M(IV), that BrO3K (
The addition of acetic acid to the dissolution solution of the target precipitates the double acetate of 238U(VI) and sodium, which carries down 239U; X remains in solution. The addition of BrO3K changes nothing in the behaviour of uranium isotopes with respect to acetic acid, but 238,239U(VI) acetate carries down X: this indicates that X is oxidised from X(?) to X(VI). Treatment of the solution with SO2 does not change the behaviour of uranium either, but X remains in solution; this does not indicate anything about the possible reduction of X.
For co-precipitation in the form of fluorides, the reaction medium is prepared by adding soluble salts of cerium and/or thorium to the dissolution solution. The addition of HF does not lead to any carrying of X by CeF3 or ThF4: X is neither trivalent nor tetravalent, but as it can be oxidised to X(VI), this shows that in HCl, it is in the form of X(V). After oxidation with bromate, the same applies: X is oxidised to X(VI). After reduction by bubbling SO2 into the medium, X is carried by ThF4: X is reduced to X(IV).
These simple experiments led to the establishment of 23993X (V), an isotope of a new element of which the stable oxidation degree is five and which can be oxidised and reduced. X was named neptunium by McMillan and Abelson.
They showed that we can separate Np and U from a uranium target irradiated with neutrons via a redox cycle: BrO3K + SO2, which brings Np to an oxidation degree 4, Np(IV). Np(IV) can then be co-precipitated with ThF4, giving Np, separated from the fission products.
Discovery of plutonium
Following the discovery of Np, McMillan and Abelson hypothesised a "Series of Uranides," and Seaborg [13] entered the race to the elements with Z > 93. The team suspected that 239Np (β, 2.3 d) could produce, via decay, an isotope of Z = 94, an α emitter. Indeed, a very low α activity with a very long period was detected with a proportional counter. However, to have enough of it to measure its activity, it was necessary to increase the production of 239Np. This was hopeless given the small cross section of the reaction of radiative capture of 238U and the neutron fluxes available in Berkeley's cyclotrons. To jump two units in Z from 238U, we must look for another way. For example, the irradiation of uranium with charged particles, a reaction (d,2n) followed by β emission, seemed to the team like a possible way to go from Z = 92 to Z = 94.
In August 1940, Glen Seaborg, Edwin McMillan, Joseph Kennedy, and Arthur Walh launched 5.5g irradiation of U3O8 (purified) with a mixed beam of neutrons and deuterons at the Berkeley's cyclotron: 1 hour and 200 microamperes. The idea was to produce at least 239Np (known) as a reference, formed by the neutrons, and other isotopes of Z = 94 formed by the deuterons and beta emissions.
The uranium target was treated with the known redox cycle BrO3K/SO2 in order to isolate Np in ThF4. After this treatment, Np is relatively pure, as explained above. After this treatment, Np is relatively pure.
In December 1940, they demonstrated on the basis of radioactivity measurements that ThF4 co-precipitates 239Np (β, 2.3 d, Emax = 1 MeV) well, but also co-precipates another radionuclide giving a beta activity X (β, 2,1 d, Emax = 1 MeV), which decays giving an α activity that increases with time: Y(α). Both radioactivities have approximately the same period and energy. They then compared the redox behaviours of the radionuclides associated with X (β, 2.1 d) and Y(α) with the known behaviour of 239Np (β, 2.3 d) as a reference.
The ThF4 precipitate was dissolved in a concentrated HCl medium. After oxidation of the solution by bromate, Np(β, 2.3 d) and X (β, 2.1 d) were not carried by ThF4, but Y(α) was; this indicated that X (β, 2.1 d) was probably an isotope of Np and that it was indeed oxidised to Np(VI) as Np(β, 2.3 d) while Y(α) was not. However, if the solution was treated with sodium persulphate in the presence of silver, Y(α) was not carried by ThF4 (and neither were the Np isotopes): Y(α) was oxidised to oxidation degree 6 but with more difficulty than Np. In a reducing medium, imposed by SO2, all the radionuclides were co-precipitated by ThF4: Y(α) was in oxidation degree 4 like the isotopes of Np. From these experiments, the Seaborg team deduced that the stable oxidation degree of Y(α) is 4. In 1940, the S2O8Na2/Ag reagent (
In February 1941, the conclusions of Seaborg's team were as follows: according to a physical point of view, X(β, 2.1 d) is the 238Np radionuclide formed by the process 238U(d,2n) 238Np; Y(α) is the daughter of 238Np; 23894Y(α) is an isotope of the second transuranic element. 238Np is a new isotope of Np. The formation of 239Np comes from the neutron pathway and the nuclear reaction 238U(d, n) 239Np. Chemically, 23894Y(𝛼) is tetravalent, while Np is pentavalent and it is much more difficult to oxidise than Np. The element corresponding to 23894Y(α, 80 a) would be named Pu in 1942, and the publication in the open literature on this subject dates to 1946 [14]. As early as 1941, research on element 94 was more or less secret. From the beginning of 1942, it was covered by military secrecy as part of the "Manhattan Project". From January 1941, Seaborg informed Washington of progress on element 94 by notes. In 1946, when the research was unveiled, more was known about the properties of Pu than the publication indicated.
Very quickly, Seaborg et al showed that we can separate Pu and Np from U and fission products via a two-step redox cycle: (1) dissolving the irradiated uranium in an acid medium, treatment with S2O8Na2 and then with SO2 followed by the co-precipitation of Np(IV) and Pu(IV) with ThF4; (2) taking up ThF4 in an acid medium and then oxidising Np(IV) to Np(V) with BrO3K and co-precipitation of Pu(IV) with ThF4, Np(V) remaining in solution.
The discovery of the chemical properties of 238Pu that were similar but different from those of Np and U supported the idea of the existence of a series of uranides, including U and the transuranic elements, but all experiments in this direction had involved immeasurable quantities. To obtain measurable quantities of Pu, Seaborg took the neutron path that led to 239Pu via the intermediary 239Np. In March 1941, 1.2 kg of UO2(NO3)2, enclosed in a paraffin block, was irradiated for two days with slow neutrons at the Berkeley cyclotron to obtain 239Np. Nitrate was dissolved in HNO3, uranium was extracted by a diethyl ether solvent, and the aqueous solution was oxidised by BrO3K. 239Pu, which remained in oxidation degree 4, was co-precipitated with LaF3. Enriching it with Pu was necessary. After 6 successive cycles of co-precipation-dissolution-co-precipation, the precipitate was almost purely 239PuF4, weighing 0.5 μg. Seaborg showed that 239Pu fissioned 50 times better than 235U. This was a discovery that suggests the possibility of making a nuclear device by a route other than obtaining 235U via the isotopic enrichment of 238,235UF6. 235U was discovered in 1935 and made visible in 1940. In September 1942, research into the chemical properties of Pu continued with the "Plutonium Project" in Chicago, an integral part of the Manhattan Project.
It begins with the irradiation of 10 kg of U3O8 in the Berkeley and St. Louis cyclotrons. The uranium oxide batches were treated in Chicago. Pu separation protocols suitable for large-scale manufacturing were studied. The first visible Pu compound obtained in 1942 was a sample of 2.8 μg of PuO2. It was obtained from 10 microliters of a concentrated solution of 239Pu. The total amount of 239Pu produced by cyclotron was only 2 mg.
In order to make a bomb, which requires a few kilograms of this isotope, the industrial production of 239Pu was achieved through treating the fuel of a plutonogenic nuclear reactor. At the end of 1944, in Hanford, three plutonium reactors and a processing plant produced 50 to 100 g/d of "military Pu," 97.3% pure 239Pu. The first Pu atomic bomb was tested in Alamogordo in July 1945. Today, 1 tonne of spent fuel from electronuclear reactors contains about 10 kg of "civil Pu" of variable isotopic composition, 238 to 243Pu, depending on the combustion rate [15].
Discoveries of Curium and Americium
In 1944, McMillan and Seaborg believed that the elements Z = 95 and Z = 96 would have the same properties as Np and Pu. They irradiated a 239Pu target at the Berkeley cyclotron with deuterons in the hope of producing an isotope of Z = 96, and by neutrons in the hope of producing an isotope of Z = 95. This certainly produced new forms of radioactivity, but they were insensitive to redox cycles. Seaborg thus hypothesised the existence of a series of elements similar to the lanthanides: the actinides. Thus the elements Z = 95 and Z = 96 are homologues of Eu and Gd and are trivalent [16]. This brilliant intuition would be confirmed.
In 1944, Seaborg, Ralph James, Leon Morgan, and Albert Ghiorso irradiated 239Pu with He2 + to obtain isotopes of the element Z = 96 because neutrons induced fission of 239Pu and complicated measurements. They identified the radionuclide 24296X (α, 163 d) by co-precipitation with LaF3. It is formed by the reaction: 239Pu (He2+,n) 242Cm. X was named curium (Cm). Cm is indeed trivalent. They also showed that 242Cm produces 238Pu by α emission, which was already known.In these experiments, it was not possible to isolate an isotope of Z = 95.
Meanwhile, an intense source of neutrons became available in Chicago: the heart of the historic Fermi nuclear reactor of 1942 that had been rebuilt.
In 1945, Ghiorso, James, Morgan, and Seaborg [17] irradiated 239Pu in this reactor. In a reactor, the nuclear processes are linked in succession, and everything becomes more complicated when it comes to sorting out the radionuclides. They identified 24195X (α, 443 a) by co-precipitation with LaF3. It was formed by the following processes: 239Pu(n, γ) 240Pu (n, γ) 241Pu(β, 14 a) 241Am. X was named americium. Am is indeed trivalent. They showed that 242Cm, which had just been discovered, was also produced in a reactor according to: 241Am(n, γ) 242Am(β, 14 a) 242Cm.
In 1945, Seaborg published a periodic table that included the beginning of the actinide series [16].
Discovery of the transcurium elements
Going into the details would require lengthy explanations. In 1945, the idea was that we could gradually synthesise isotopes of the elements of Z > 96 by appropriate nuclear reactions, all the while knowing that their production would decrease drastically with the increase in Z value. It was necessary to have the following: the right isotope to irradiate; intense beams of particles; appropriate and very sensitive detectors; faster methods for separation and identification than co-precipitation; and rapid nuclear techniques for chemistry at the atom scale. During the 10 years between 1949 and 1961, these conditions were gradually met. The elements Bk up to Lr were discovered (Table 1). It is worth noting here a major advance in the identification of the elements Am to Lr which also confirmed the actinide hypothesis: the order of elution of these elements to the degree of oxidation 3, attached to a column of ion exchange resin, by a highly selective complexing agent which was beginning to be used at the time, ammonium alpha-hydroxy-isobutyrate. Elution is carried out in reverse order from Z, as for the lanthanides from Eu to Lu. It is faster than co-precipitation.
Discovery conditions of the transcurium elements [13]
| Symbol and article references of the discovery | Discovery date | Discovery process | Discovery conditions | Discovery article |
|---|---|---|---|---|
| Bk [22] | 1949 | 241Am(He,2n) 243Bk(α, ) 241Am(He,n) 244Bk(CE, 4.5 h) EHe = 35 MeV, 10 μA/cm2, Berkeley | 7 mg of 241Am, several thousand Bk atoms | [22] |
| Cf [23] | 1950 | 242Cm(He, n) 245Cf(α, 44 min) EHe = 35 MeV, 10 μA/cm2, Berkeley | μg of 242Cm, 5000 Cf atoms | [23] |
| Es Fm [24] | 1955 | 238U(15n, 7e-) 253Es(α, 20 d) 238U(18n, 8e-) 255Fm(α, 22 h) Argonne National Laboratory, Chicago, Los Alamos Scientific Laboratory, Albuquerque | kg of 238U, hundreds of atoms of Es and Fm. Mike explosion, U absorbs many times 1024 neutrons in a few (a few moles of neutrons). Also produced by neutron irradiation of grams of Pu in the high-flux MTR reactor in Idaho. | [24] |
| Md [25] | 1955 | 253Es(He, n) 256Md(EC, 1.3 d) 256Md gives 256Fm(SF, 2.6 h) EHe = 41 MeV, 100 μA/cm2, Berkeley | 109 atoms of 253Es(α, 20 d) produced in Idaho. 17 atoms of 256Md produced “one atom at a time”. Parasitic reaction (α,n) on 244Cm giving 246Cf | [25] |
| No [26] | 1958 | 246Cm(12C,4n) 254No(55 s) and 244Cm(12C,4n) 252No(2.3 s). 254No produces 250Fm(α, 30 min) 252No produces 248Fm(α, 36 s) HILAC Berkeley | The Berkeley group unambiguously identifies 254No based on the existence of 250Fm (11 atoms detected at the correct elution position) and the decay characteristics of 252No. Other groups using 244Cm and 13C - Stockholm - or 239Pu and 16O - Dubna - did not identify the reaction. In 1964, the Dubna group identified 256No(α, 3 s) as a parent of 252Fm. The existence of No has been confirmed. | [26] |
| Lr [27] | 1961 | 249to252Cf(10,11B, xn) 257Lr(α,4.3 s) HILAC, Berkeley | 3 μg of Cf, a few atoms of 257Lr detected among other heavy radionuclides. Existence of 256Lr(α, 45 s) confirmed in 1965 in Dubna following the reaction 243Am(18O, 5n) | [27] |
Mendelevium
This is the last element of which isotopes that can be produced with light ions because beyond it, there is no more beta emission that can cause an increase in unit in Z. Electronic capture prevails when N ≫ Z, and it leads to a Z-1 radionuclide. . It is also the first element for which atoms are produced "one atom at a time" and for which statistical chemistry can apply (repetition of experiments). In 1955, the production output of the isotope 256Md (EC, 1.5 h) from 106 atoms of 253Es (α, 20 d) was two atoms for every three hours of irradiation. It was detected by the spontaneous fission of its daughter 256Fm (2, 3 h). In 1975, micrograms of 253Es were available and 106 atoms per hour were produced, outshining by far the chemistry performed on immeasurable quantities. Chemical properties at these concentration scales pose many instrumental, detection, and fundamental chemistry problems. Sharing experiences on Md(III) and Md(II) have made it possible to launch the equipment to identify and perform chemistry on elements with Z > 103.
Actinides
Numerous data now confirm the validity of the actinides series being analogous to the lanthanides series. This is accepted by the scientific community. The physicochemical properties of all actinides are well known [18]. However, there is no strict correspondence between lanthanides and actinides. For example, Th, Pa, and U are in fact homologues of Hf, Ta, and W; the electronic structures of actinides are based on the filling of relativistic subshells 5f5/2 and 5f7/2, whose energy and extension differ from the 5f subshells. The energies of these subshells are very close to those of the subshells 6d, whereas the subshells 4f and 5d are separated in energy. The first 5f electrons appear in the atom of Pa, while for the lanthanides they first appear in Ce. This results in a multiplicity of degrees of oxidation (Figure 3).
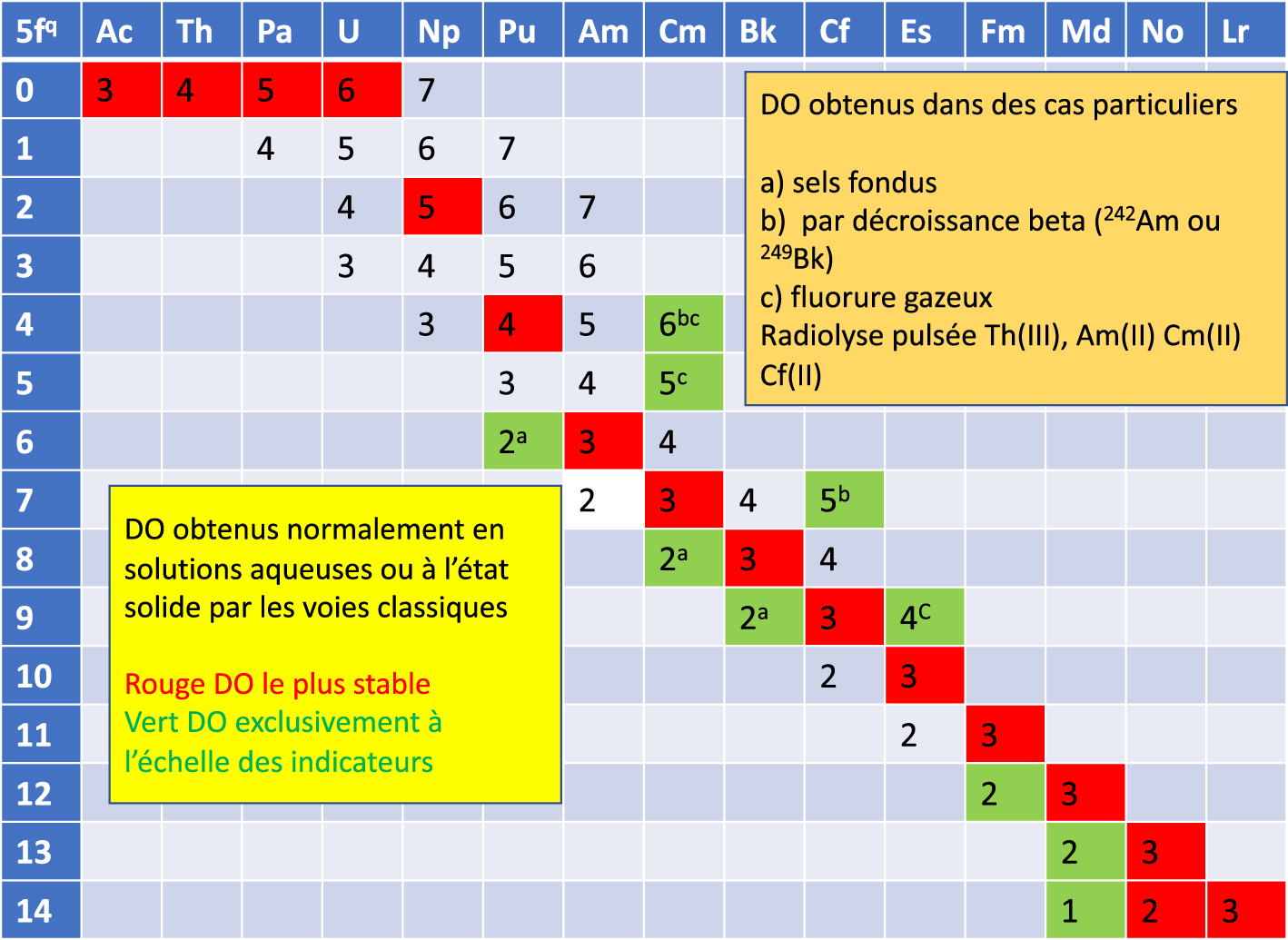
Degree of oxidation (DO) of actinides or elements 5f under various conditions.
Pu is in the best-known element in the media [19]. For physicochemists, this is a fascinating element. It has seven allotropic forms in the metallic state, and it forms alloys with many other metals. The delta phase, stabilised with Ga, Pu-Ga, is stable from ambient temperature to 550 °C. Its density increases with temperature and its compressibility is remarkable. These properties are used to trigger an explosive chain reaction in bombs with 239Pu via compression. Plutonium produces a wide variety of solid compounds and many ionic species in solution, with the possibility of creating up to twelve bonds. In an acidic medium, four degrees of oxidation (3, 4, 5, and 6) can coexist in equilibrium. 238Pu (α, 80a) is used as an energy source in space probes.
Conclusion
The discovery of the transuranic elements arranged in the actinide series was a short story (1943 to 1960), but in which radiochemistry played a major role, as well as heavy experimental means for irradiation such as cyclotrons and nuclear reactors. The period around 1939–1945 was exceptionally fertile in discoveries, but only revealed after the war. It was preceded by the period 1934–1939, no less rich in discoveries such as that of uranium fission. Several great scientists involved in "nuclear science" during these periods were awarded a Nobel Prize: I. and F. Joliot. (1935), E. Fermi (1938), E.O. Lawrence (1939), O. Hahn (1944), E. McMillan, and G.T. Seaborg (1951).
Historically, the period of the Second World War was the pivotal period during which nuclear science was passed from Europe to the United States, with the beginning of the Second World War. Technologically, this is the period of transition from radioactive sources to particle accelerators. At the scientific level (physics/chemistry), it is the period when we moved from natural elements to artificial elements, and when we opened many fields of research and applications. At the societal level, during this period, a pivotal element in the implementation of nuclear energy was produced: plutonium.
The discovery of actinides paved the way for the synthesis of trans-lawrencium elements, Z = 104 to 118 [20, 21]. The fourth construction phase of the periodic table began in 1968 and corresponded to the discoveries of elements 6d and 7p, the ephemeral elements.