



 CC-BY 4.0
CC-BY 4.0
1. Introduction
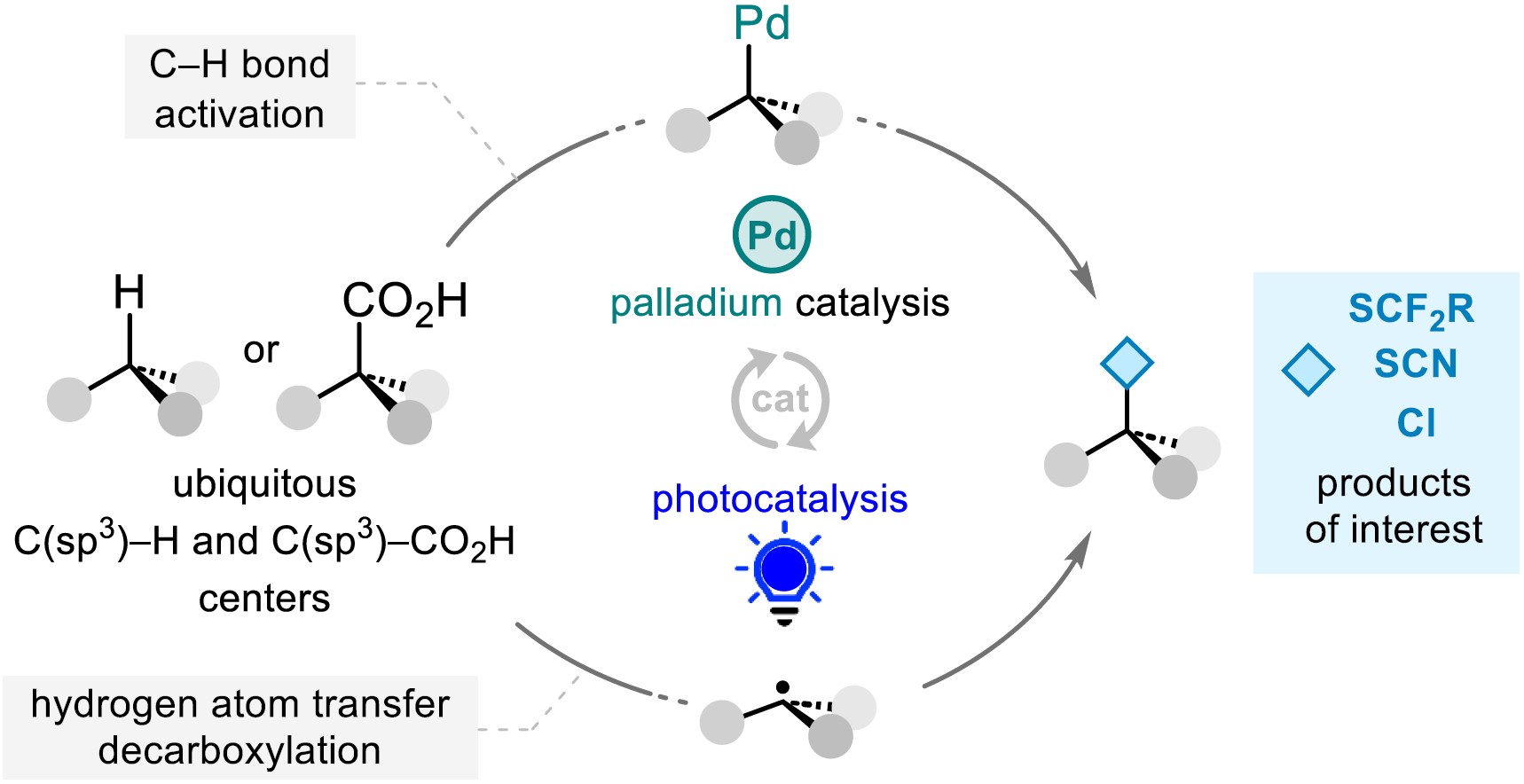
The quest for molecular complexity has garnered the interest of the scientific community over the years. To meet this synthetic challenge, modern tools have been designed, providing straightforward and efficient synthetic routes. Although major advances have been made for the functionalization of C(sp2) centers, the direct and selective functionalization of C(sp3) centers remains challenging and has driven the curiosity of scientists to push forward in this direction with innovative strategies [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22]. Given the challenges that the direct functionalization of C(sp3) centers represents and considering the omnipresence of sulfur- and fluorine-containing molecules in pharmaceuticals and agrochemicals, among others [23], this account summarizes our recent contributions to selectively and efficiently forge C–S bonds thanks to strategies based on transition metal catalysis and photochemistry, with a special focus on C(sp3)–S bond formation.
2. Transition-metal-catalyzed C(sp3)–H bond functionalization
2.1. Pd-catalyzed trifluoromethylthiolation of C(sp3) centers
Benefiting from the recent breakthroughs made in homogeneous catalysis and especially how transition-metal-catalyzed C–H bond functionalization has revolutionized the way molecules can be made [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22], we first focused on the direct construction of C–S bonds by transition metal catalysis via C–H bond activation on aliphatic molecules. This task is challenging considering the inert character of non-acidic C(sp3)–H bonds. Merging our interest in homogeneous catalysis and organofluorine chemistry, special attention has been paid to the SCF3 moiety [24, 25, 26, 27], an emerging fluorinated group possessing high electron-withdrawing character and good ability to enhance molecule lipophilicity (Hansch hydrophobic parameter: π = 1.44 vs π = 0.88 for CF3) [28, 29]. Hence, Pd-catalyzed C(sp3)–H bond trifluoromethylthiolation of alkyl amides at the β-position, under mild conditions, was developed using electrophilic sources (Scheme 1) [30]. Optimization of the reaction conditions revealed that the presence of a bidentate directing group (amide derived from 8-aminoquinoline) and pivalic acid as an additive was crucial to ensure the selectivity and efficiency of the process (note that a flow process has then been developed, further improving the efficiency of the reaction [31]). Various aliphatic amides having primary β-C(sp3)–H bonds were functionalized. From α,α-dialkyl substituted amides, the reaction was then applied to α,α-dimethylated hydrocinnamic acid derivatives. Even amides prepared from phenylacetic acid derivatives or bearing α-C–H bonds were smoothly converted into the corresponding fluorinated product, allowing the synthesis of the SCF3-containing analogues of bioactive molecules such as ibuprofen and naproxen. Note that a total control of the selectivity was observed: monofunctionalization at the β-position only, even in the presence of competitive sites (e.g., benzylic positions). For the Munavalli reagent I, playing both the roles of oxidant and SCF3 source, a plausible mechanism is depicted in Scheme 1. After the coordination of the palladium catalyst with the bidentate directing group (intermediate int1), the metallacycle int2 was formed (C–H bond activation event). The latter would undergo oxidative addition in the C–SCF3 bond leading to the putative Pd(IV) species int3, which after reductive elimination would yield the desired product and release the catalyst. Interestingly, the directing group can be easily cleaved under basic reaction conditions and the SCF3 moiety is converted into the corresponding SOCF3 group when treated with m-chloroperbenzoic acid (m-CPBA).
Trifluoromethylthiolation of C(sp3) centers under palladium catalysis.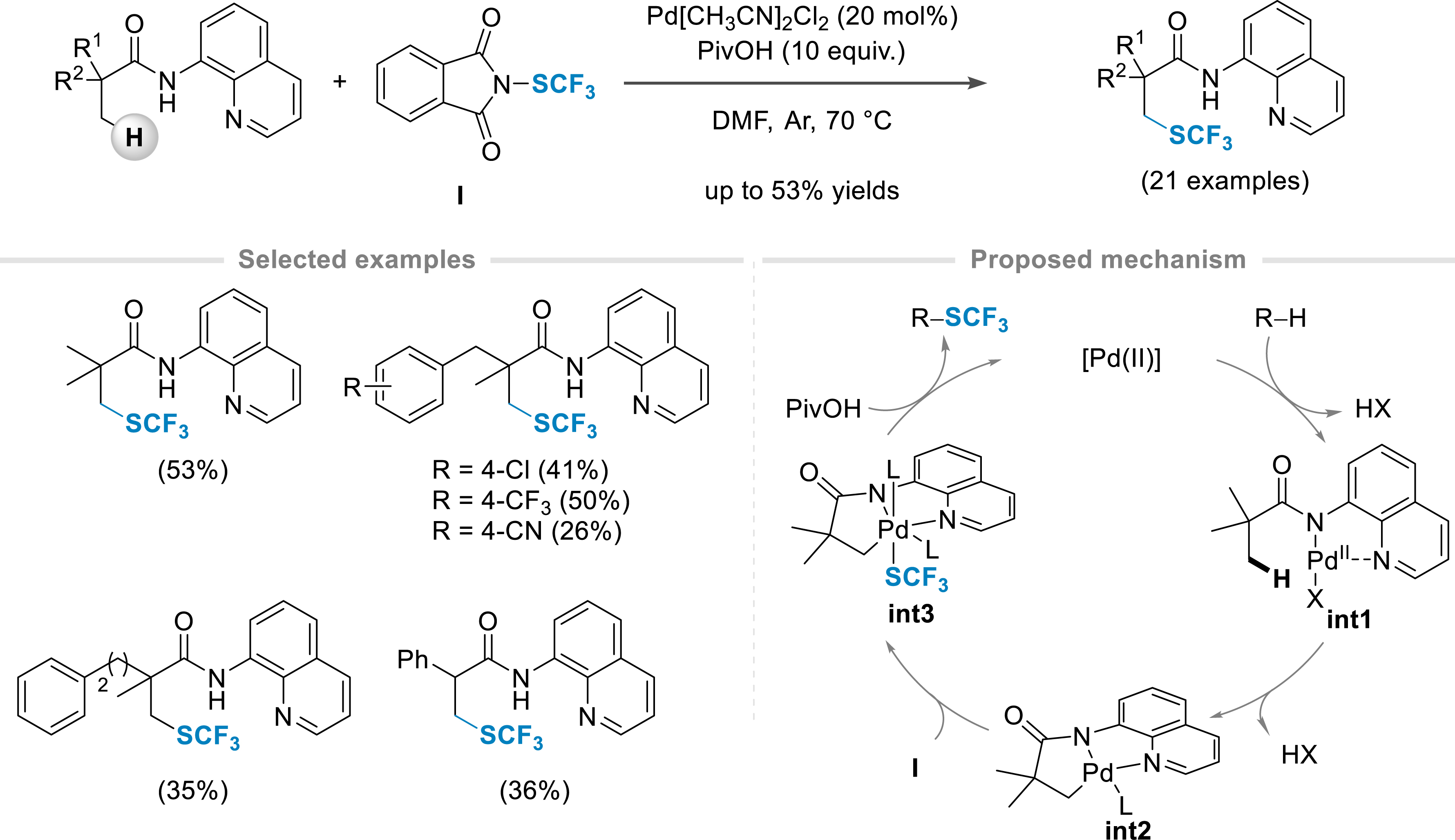
2.2. Directed chlorination of amides under palladium catalysis [32]
As the versatility of the above-depicted approach to functionalize C(sp3) centers was intriguing, the chlorination reaction was investigated. The Pd-catalyzed directed halogenation of non-activated C(sp3)–H is challenging as the reductive elimination is known to be difficult and the newly formed C–X bond might be reactive in the presence of the catalyst. Thanks to a Pd(II)/Pd(IV) process, known to facilitate the reductive elimination step, the Pd-catalyzed chlorination of aliphatic amides was developed using the readily available N-chlorosuccinimide (NCS) as the electrophilic Cl source (Scheme 2). The transformation proceeded smoothly at room temperature and favored the formation of mono- and di-chlorinated compounds, depending on the quantity of NCS. It is worth mentioning that when a mixture was obtained, the two compounds were easily separated by column chromatography. Thanks to this synthetic handle, the synthesis of β-lactams was realized by the treatment of chlorinated amides under basic conditions. This approach provided an access to molecules of interest for pharmaceutical and agrochemical companies. Moreover, under oxidative conditions in the presence of ceric ammonium nitrate, the amide derived from 8-aminoquinoline was smoothly converted into the corresponding primary amide.
Directed chlorination of aliphatic amides at room temperature. Overall yields are given, and all mono- and di-chlorinated compounds are isolated separately. a Using 10 equiv of NCS.
3. Photoinduced and photocatalyzed C(sp3)–H functionalization
3.1. Functionalization of C(sp3) centers by photocatalysis [33]
Pursuing our efforts to access trifluoromethylthiolated molecules in a sustainable manner, the photocatalytic α-trifluoromethylthiolation of N-acyl amines was studied in the presence of the Munavalli reagent I (Scheme 3). Despite major advances made for the C–H functionalization at the α-position of an amide by photocatalysis [34, 35, 36, 37, 38, 39, 40, 41, 42], there was no report dealing with the formation of a C–S bond. We hypothesized that the elusive acyclic fluorinated thioaminals would be synthesized by the catalytic generation of a carbon-centered radical at the α-position of a nitrogen under visible light irradiation in the presence of a suitable photocatalyst, followed by a reaction with a suitable SCF3 source. Using the low catalytic loading of an organophotocatalyst, the functionalization of aromatic and aliphatic amides was achieved in high yields (up to 83% yield, 20 examples). The transformation was tolerant to a large panel of diversely substituted benzamides as well as various functional groups (halogens, ester, cyano). Interestingly, the reaction was completely selective towards the formation of the desired trifluoromethylthiolated products even in the presence of a benzyl or a methyl group at the α-position of the amide carbonyl.
Photochemical trifluoromethylthiolation of N-acyl amines using the Munavalli reagent.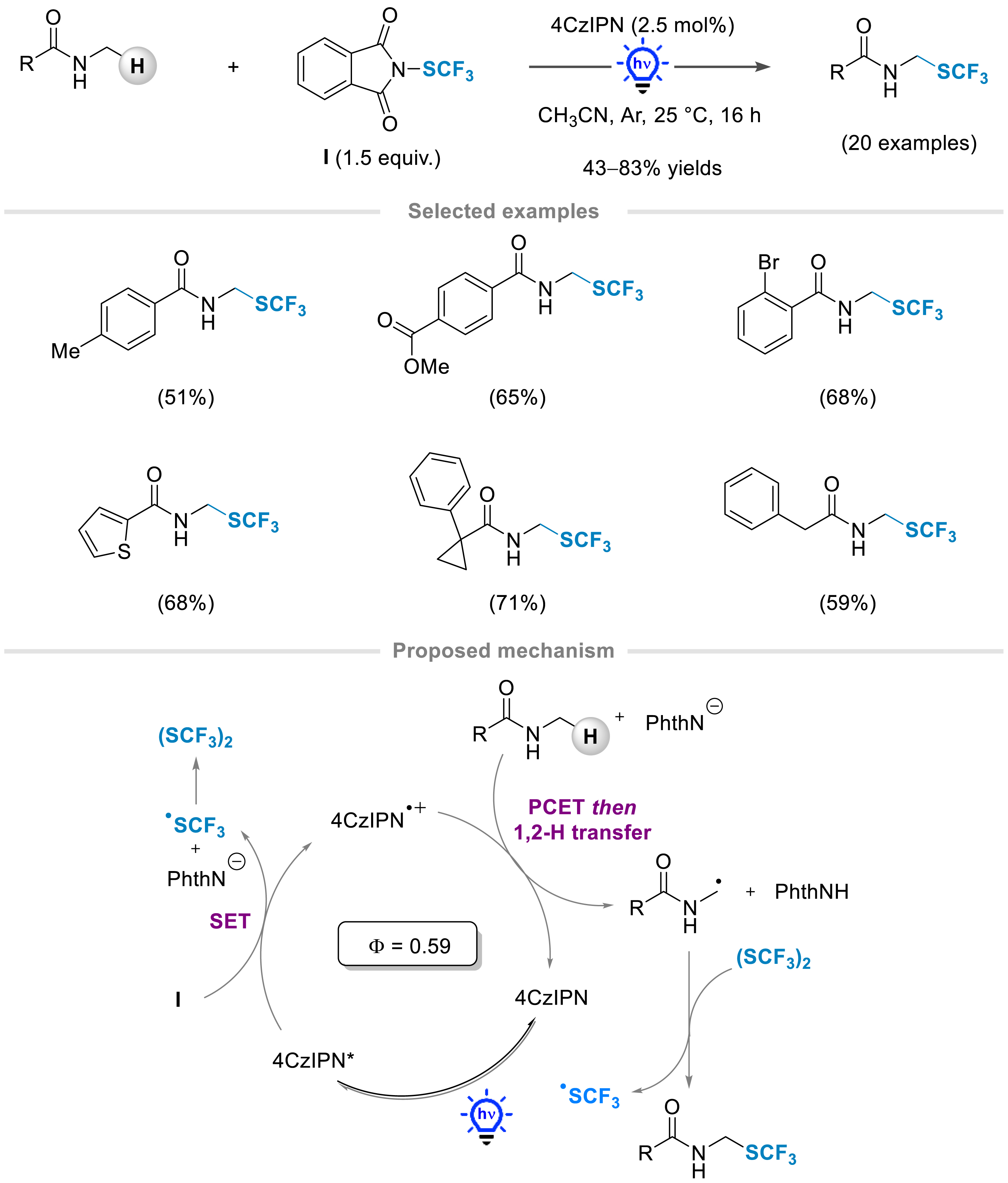
Detailed mechanistic studies were conducted. They revealed that the active SCF3 species was the in situ generated SCF3 dimer and supported a proton-coupled electron transfer (PCET). Indeed, the following plausible mechanism for this trifluoromethylthiolation reaction was suggested. The irradiation induces the excited state of the tetrakis(9H-carbazol-9-yl) isophthalonitrile (4CzIPN) photocatalyst, which reduces the electrophilic SCF3 source via a single electron transfer, leading to the generation of a SCF3 radical that can recombine to form the (SCF3)2 dimer along with the oxidized photocatalyst (4-CzIPN∙+). In the presence of the released phthalimide anion, a PCET followed by 1,2-hydrogen atom transfer (HAT) occurred, allowing the formation of a primary radical at the α-position of the nitrogen. The latter would react with the (SCF3)2 dimer to furnish the desired product along with the ∙SCF3 radical.
Over the last few years, our group has also been interested in developing new synthetic methods to incorporate a thiocyanate moiety on various substrates [43, 44, 45, 46, 47]. Thiocyanates are not only highly biorelevant compounds (for general reviews on thiocyanates, see [48, 49, 50, 51, 52, 53, 54, 55]), as this functional group is present in several bioactive molecules [23], but are also valuable synthetic linchpins that can be used to access various sulfur-containing products of biological interest [56, 57, 58]. In this context, in collaboration with the Lebel group, we have designed a divergent synthesis of alkyl thiocyanates and isothiocyanates from readily available carboxylic acids via a photocatalyzed decarboxylative transformation [47]. The formation of thiocyanates or their isothiocyanate isomers can be tuned by the choice of additives. In the presence of 2,4,6-triphenylpyrylium tetrafluoroborate as the organophotocatalyst, under blue light irradiation, N-thiocyanatosaccharin II as the thiocyanating agent, and a catalytic amount of sodium carbonate, a large panel of aliphatic carboxylic acids were smoothly converted into the corresponding alkyl thiocyanates; most yields were above 60% (Scheme 4). The reaction conditions were shown to be compatible with a wide range of functional groups, with ketones, halides, heterocycles, and azides remaining inert. Besides, highly biorelevant thiocyanates could be synthesized through this method as well, starting from bioactive carboxylic acids, such as the synthesis of an analogue of ibuprofen.
Divergent synthesis of alkyl thiocyanates and isothiocyanates from readily available carboxylic acids by photocatalysis.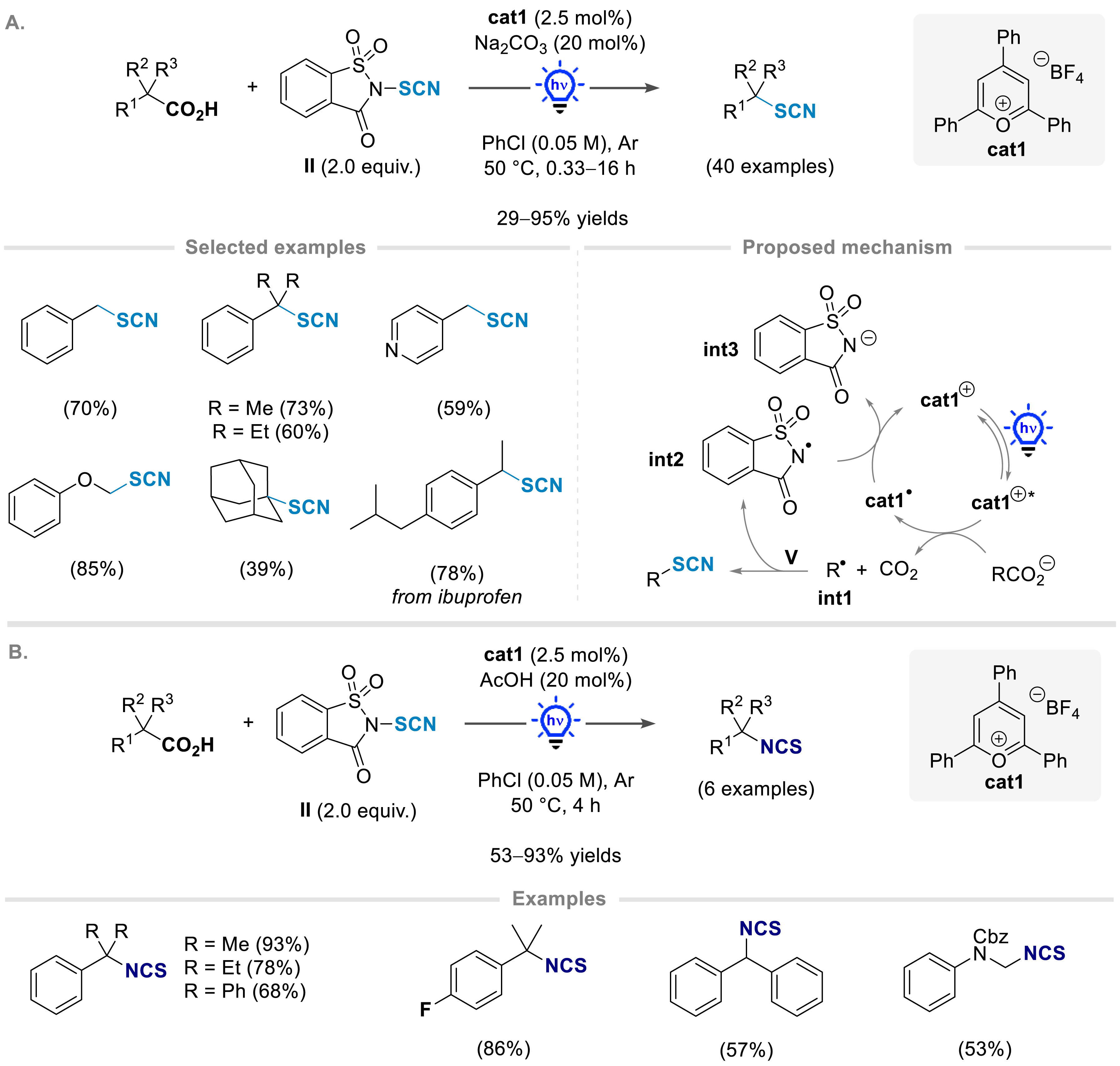
The mechanism of this transformation was proposed to involve a single-electron oxidation of the carboxylate formed by deprotonation of the corresponding carboxylic acid, leading to the radical intermediate int1 upon decarboxylation. Reaction with the thiocyanate source II then allowed the formation of the final thiocyanate product and the intermediate int2, which was subsequently reduced to int3, regenerating cat1. A radical chain propagation process involving HAT with int2 was also shown to be possible under the reaction conditions.
In the presence of a catalytic amount of acetic acid instead of sodium carbonate and under otherwise identical reaction conditions, the isothiocyanate products were obtained in high yields (53–91% yields). Their formation likely arose from the isomerization of the thiocyanates under these reaction conditions.
3.2. A light-driven process for the distal functionalization of C(sp3) centers
Our group has also been involved in the development of new synthetic methods for the straightforward introduction of various fluorinated functional groups and, in particular, emerging fluorinated moieties. Although considerable progress has been recently made for the functionalization of various substrates with biorelevant SCF2R functional groups [59, 60, 61, 62, 63], significant challenges still remained for such functionalization of non-activated C(sp3)–H positions on aliphatic chains. In this context, a radical reaction represents an attractive tool for the regioselective functionalization of C(sp3)–H centers with fluorinated moieties [59].
Inspired by the use of hypervalent iodide reagents as radical promoters from alcohols [64], we have developed a new synthetic approach for the regioselective δ-functionalization of free alcohols with various SCF2R moieties under mild reaction conditions, enabled by 1,5-HAT [65, 66]. In the presence of phenyliodine diacetate (PIDA) and the reagents III–VI under blue light irradiation, free alcohols (primary, secondary, and tertiary) were converted into the corresponding products bearing the fluorinated moiety at the δ-position from the alcohols (Scheme 5). This approach was successfully applied for the introduction of SCF3 [32], SCF2PO(OEt)2 [66], SCF2CO2Et [66], and SCF2H [66] moieties in the presence of the corresponding sulfonyl-SCF2R reagents. The reaction conditions were shown to be compatible with various functional groups, such as azides, protected amines, and esters. The mechanism of the reaction was proposed to involve the formation of the hypervalent iodide intermediate int1 from PIDA and the alcohol substrate, followed by its homolysis under blue light irradiation producing the alkoxy radical int2. The latter may subsequently undergo 1,5-HAT to furnish the C-centered radical intermediate int3, which can react with the sulfonyl–SCF2R reagent (III–VI) to form the functionalized alcohol. For purification purposes, the free alcohol products were in situ acetylated to obtain the final products.
1,5-HAT enabled the construction of a carbon–SCF2R bond on aliphatic alcohols at a remote position.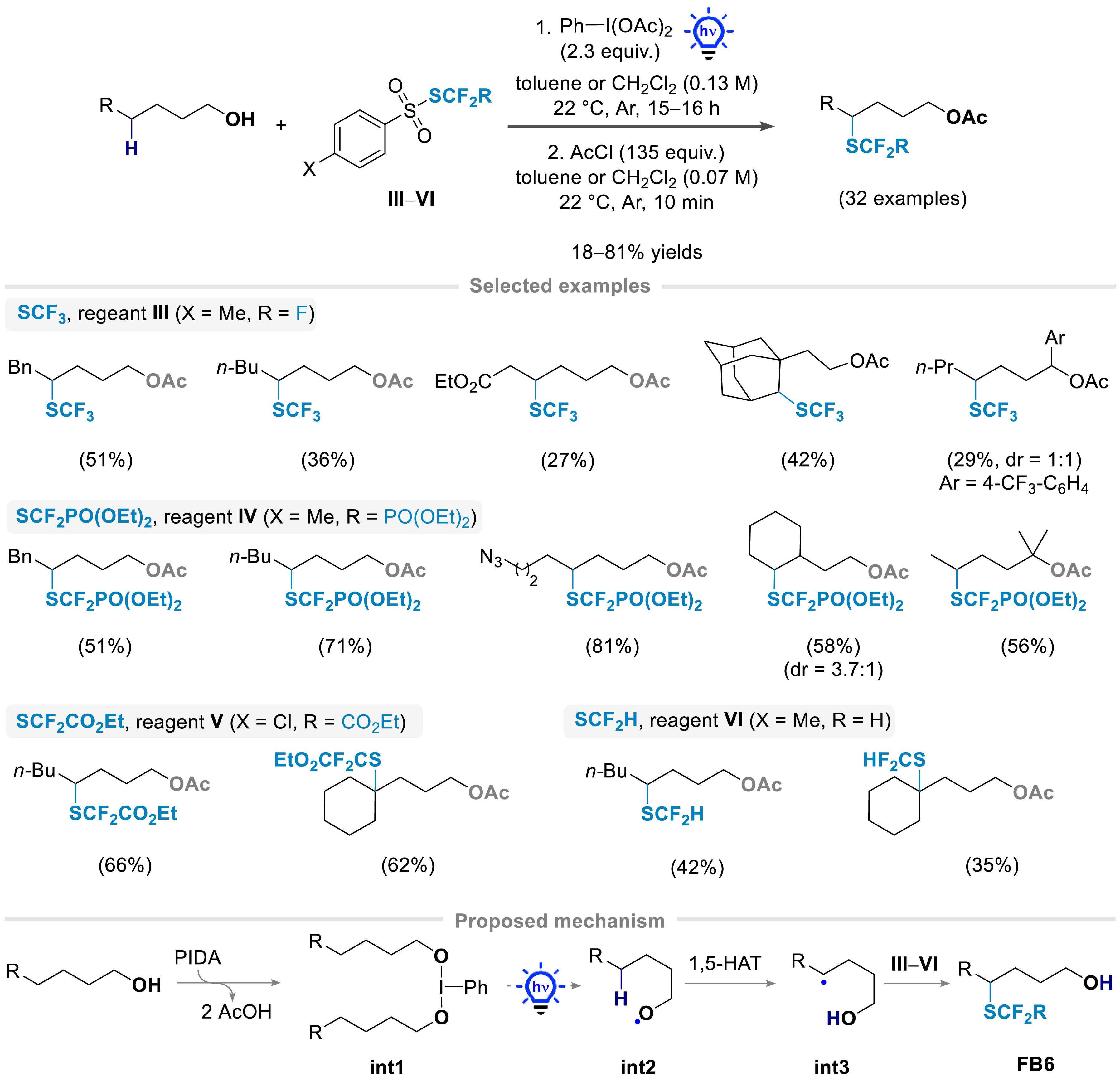
4. Conclusion
Over the years, major advances have been made in the functionalization of C(sp3) centers by using modern tools. Hence, methods based on transition metal catalysis, photocatalysis, and transition-metal-free processes were developed. With these contributions, the construction of C(sp3)–SR (R = CF3, CF2FG, CN) and C(sp3)–Cl at proximal and distal positions was possible, expanding the chemical space of compounds of interest for academia and industries.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work has been partially supported by University of Rouen Normandy, INSA Rouen Normandy, Centre National de la Recherche Scientifique (CNRS), European Regional Development Fund (ERDF), Labex SynOrg (ANR-11-LABX-0029), Carnot Institute I2C, XL-Chem Graduate School of Research (ANR-18-EURE-0020 XL CHEM), and Region Normandie. TB thanks the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 758710). FB thanks the European Union’s Horizon 2020 research and innovation program under Marie Skłodowska-Curie Grant Agreement Number 101034329, and the Region Normandie (WINNINGNormandy program) for funding. The French National Research Agency (ANR-21-CE07-0035-02 and ANR-22-CE92-0083) is gratefully acknowledged for generous financial support.