



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Spirotetronates are a remarkable class of macrocyclic natural products isolated predominantly from actinomycetes, a phylum of gram-positive bacteria. Since the discovery of chlorothricin in 1969, the first isolated spirotetronate [1] (Figure 1), numerous related compounds have been structurally characterized [2, 3] such as spirohexenolide A and B [4], abyssomicin C [5], quartromicins [6], versipelostatin [7], streptaspironates A–C [8], glenthmycins A–M [9], and others [10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]. These compounds have attracted substantial interest due to their diverse and potent biological activities, including antibacterial, antiviral, and antitumoral properties. Their fascinating structures and significant pharmacological potential have made spirotetronates central to ongoing synthetic and pharmaceutical research.
Structure of chlorothricin and its aglycon chlorothricolide.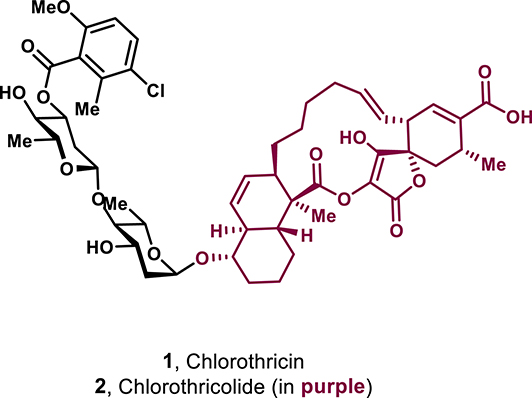
Structurally, spirotetronates feature a characteristic tetronic acid (4-hydroxy-[5H]furan-2-one) moiety that is spiro-linked to a cyclohexene ring at the C-4 position, a distinctive motif within this family (Figure 1). The spirotetronate core consists of a five-membered lactone ring with a high degree of oxidation and unsaturation. This structural unit is typically substituted at the C-2 position by an acyl group or, in rare cases, by an acyloxy chain, such as in chlorothricolide (Figure 1). Adjacent to the spiro-linkage at the C-5 position of the cyclohexene ring is another stereocenter, which can lead to two diastereoisomeric arrangements: the endo configuration, where the higher alkyl chain is cis relative to C-3 of the spirotetronate (highlighted in blue), and the exo configuration, where these groups are trans (highlighted in red). Both diastereomers have been observed in spirotetronates, posing a key synthetic challenge in achieving diastereoselective access to the desired configuration (Figure 2A).
(A) Structure of the two different diastereomers. (B) Spirotetronates harnessing a quaternary stereocenter at C5.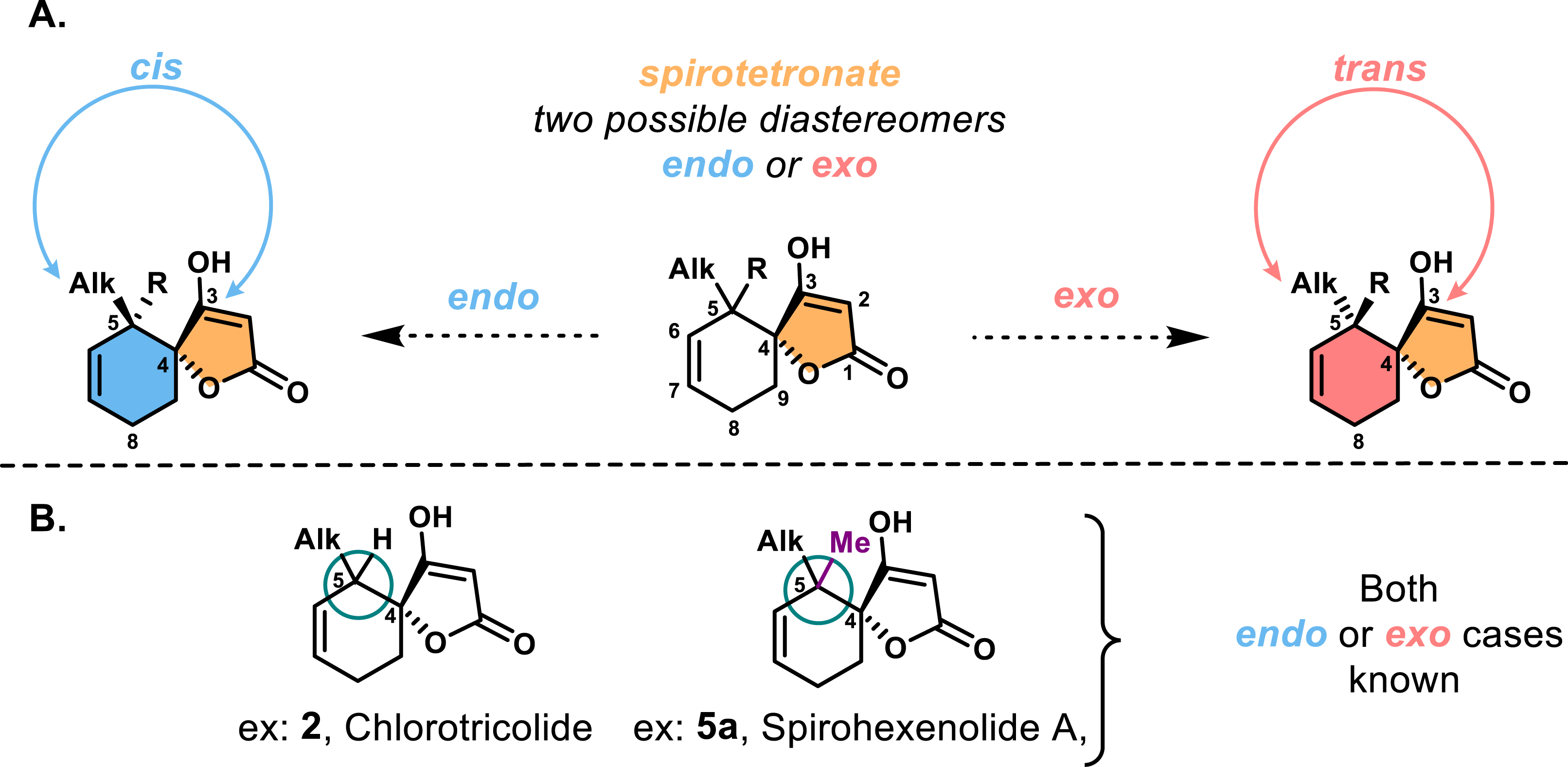
This stereocenter at the C-5 position of the cyclohexene ring can be quaternary, which is a significant structural feature in some spirotetronates (Figure 2B). In these cases, a methyl group replaces a hydrogen, as seen in compounds such as in spirohexenolides 5a and 5b, maklamicin 6, and quartromicins 9–14. This modification dramatically increases the complexity of the synthesis by introducing steric hindrance in the form of adjacent quaternary stereocenters. The presence of these bulky neopentylic carbons restricts functionalization and adds strain to the cyclohexene ring, making these molecules even more challenging targets for synthetic chemists.
In addition to the tetronic acid core and the stereocenters at C-5, spirotetronates exhibit diverse modifications at the C-7 and C-8 positions of the cyclohexene ring. These substituents vary from simple methyl groups, as seen in okilactomycin 4 or spirohexenolides 5a–b, to more complex, oxidized alkyl chains in compounds like maklamicin 6 and chlorothricolide 2 (Figure 3). The oxidation state and size of these side chains contribute to the structural diversity of spirotetronates and influence their biological activities. In some cases, such as abyssomicin C (compound 3), the cyclohexene ring is further oxidized to form vicinal diols.
Diverse structures of Class I and Class II spirotetronates.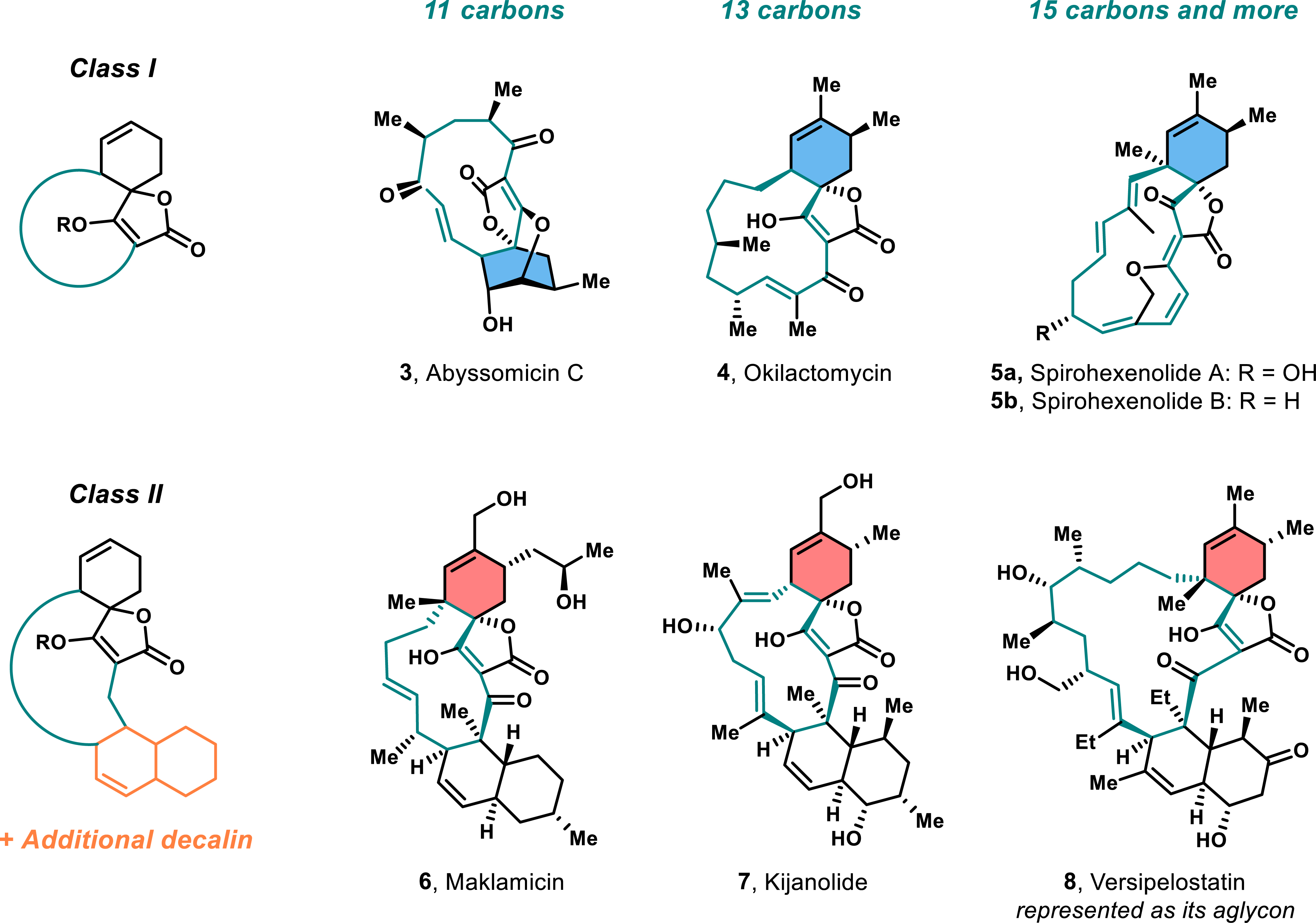
A key feature of spirotetronates is their macrocyclic structure, which links the tetronic acid to the cyclohexene ring (C2–C5). The size and substitution of the macrocycle vary, resulting in three subclasses: small spirotetronates with 11-carbon macrocycles, medium-sized with 13 carbons, and larger structures with 15 carbons or more. These macrocycles can range from simple structures to those bearing conjugated alkenes, alcohols, or ketones. Additionally, some members of the spirotetronate family contain a trans-decalin ring system within the macrocycle, further increasing structural complexity (Figure 3). Based on these features, spirotetronates are divided into two main classes: Class I, containing a single spirotetronate moiety in the macrocycle, and Class II, which incorporates a decalin ring system in addition to the spirotetronate.
Perhaps one of the most intriguing subclasses within the spirotetronates is the quartromicin family, which lies outside of the conventional Class I and Class II designations. Unlike the typical members of the spirotetronate family, quartromicins (first isolated in 1991 from a strain of Amycolatopsis orientalis) [6, 21] contain four spirotetronate units within their scaffold (Figure 4). These compounds exhibit strong metal-ion-binding affinity and are a 32-carbon macrocycle, which contributes to their highly symmetrical C2 axial structure.
General structure of quartromicins and their structural characteristics.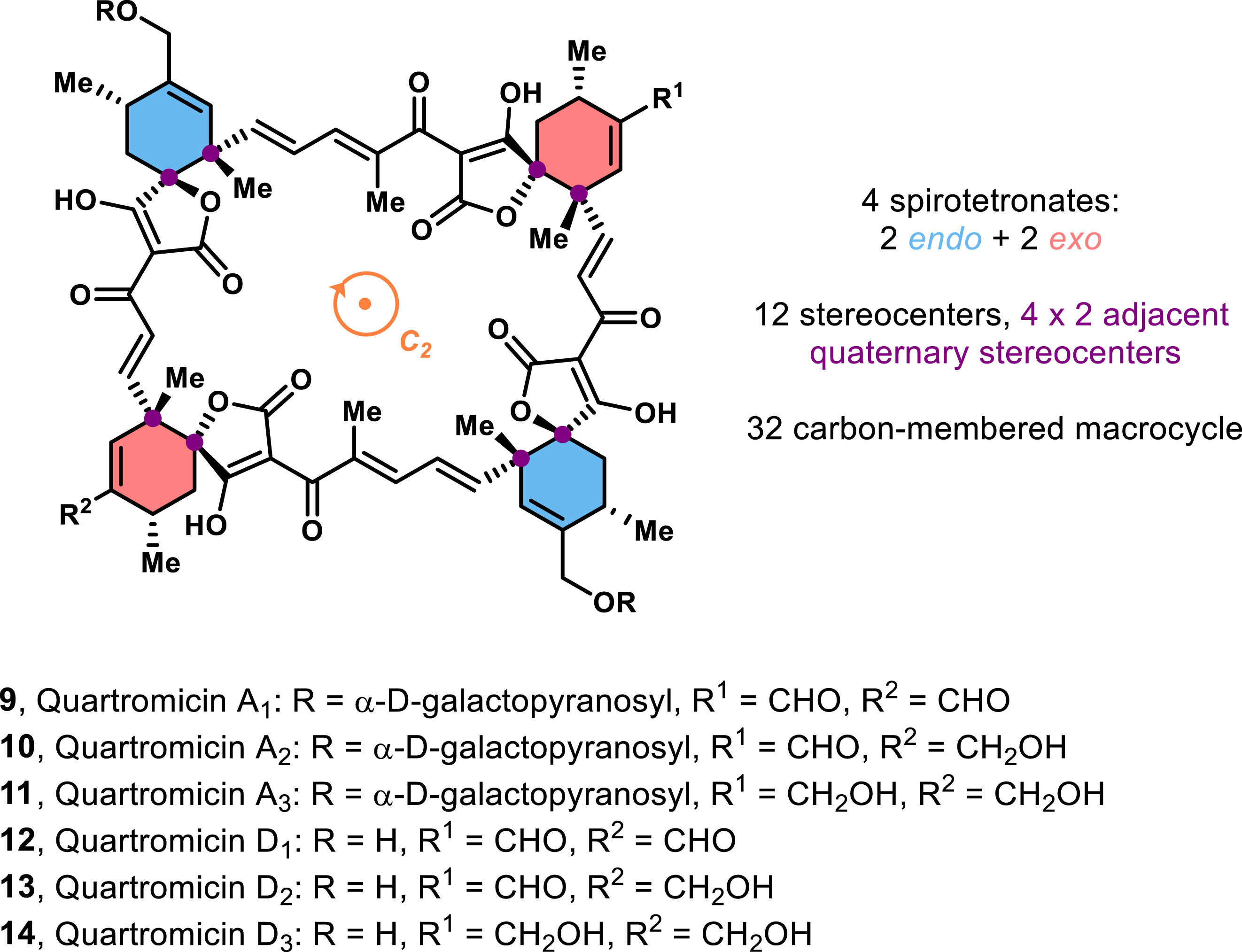
Spirotetronates, with their fascinating structures, have captured significant interest due to their diverse biological activities [2, 3, 22]. For example, chlorothricin 1 inhibits pyruvate carboxylase, an enzyme crucial for bacterial metabolism, making it an effective antibacterial agent against gram-positive bacteria [23]. The abyssomicin family, known for inhibiting the growth of Mycobacterium tuberculosis and Staphylococcus aureus, has also been extensively studied [24]. Abyssomicin C specifically targets the chorismate pathway, which is vital for the production of aromatic amino acids in microorganisms [25]. This compound works by acting as a double Michael acceptor, irreversibly modifying a cysteine residue in an enzyme involved in this pathway, showcasing the importance of its enone group and strained macrocyclic structure. In contrast, quartromicins are recognized for their potent antiviral effects, including against HIV and HSV-1 [21, 26]. Although their exact mechanisms are still under investigation, their interactions with metal ions and their complex macrocyclic structures suggest they might employ novel modes of action.
The intricate structures and significant biological activities of spirotetronates have fascinated synthetic chemists for decades. Previous reviews have covered their biological activities, biosynthesis, and total syntheses in detail, often focusing on the macrocycle and decalin components [2, 3, 22, 27, 28]. However, they often overlook the construction of the spirotetronate core—a crucial element with significant stereoselective challenges. The synthesis of this spiro-linked tetronic acid, in particular, remains complex, requiring precise stereocontrol and functionalization. This review aims to explore the methods developed for constructing spirotetronates, highlighting both successful strategies and ongoing challenges. By focusing on the stereochemical and synthetic difficulties, we seek to provide a thorough understanding of the progress made in synthesizing this intriguing class of natural products.
2. Synthetic strategies to access spirotetronates
We will discuss the key disconnections investigated up to date and their drawbacks as well as their benefits. However, except when deemed useful, we will not discuss the construction of the macrocycle and the decalin moieties, as these have been reviewed and studied elsewhere and are not the purpose of this review [2, 3, 22, 27, 28, 29, 30, 31, 32, 33, 34, 35].
2.1. Seminal works established the Diels–Alder reaction as a preferred strategy
One of the earliest synthetic studies on spirotetronates was conducted by Ireland and co-workers in 1979, focusing on the synthesis of chlorothricin (2), the aglycon of chlorothricolide [36]. This molecule features an exo-type spirotetronate moiety, a functionalized decalin, and a macrocyclic structure with a single unsaturation. Based on a retrosynthetic analysis, the authors assumed that an Ireland–Claisen rearrangement could be key in forming the macrocyclic backbone and therefore the first requirement would be to build the advanced spirotetronate intermediate 15 (Scheme 1).
Retrosynthetic analysis to access chlorothricolide 2 by Ireland and co-workers.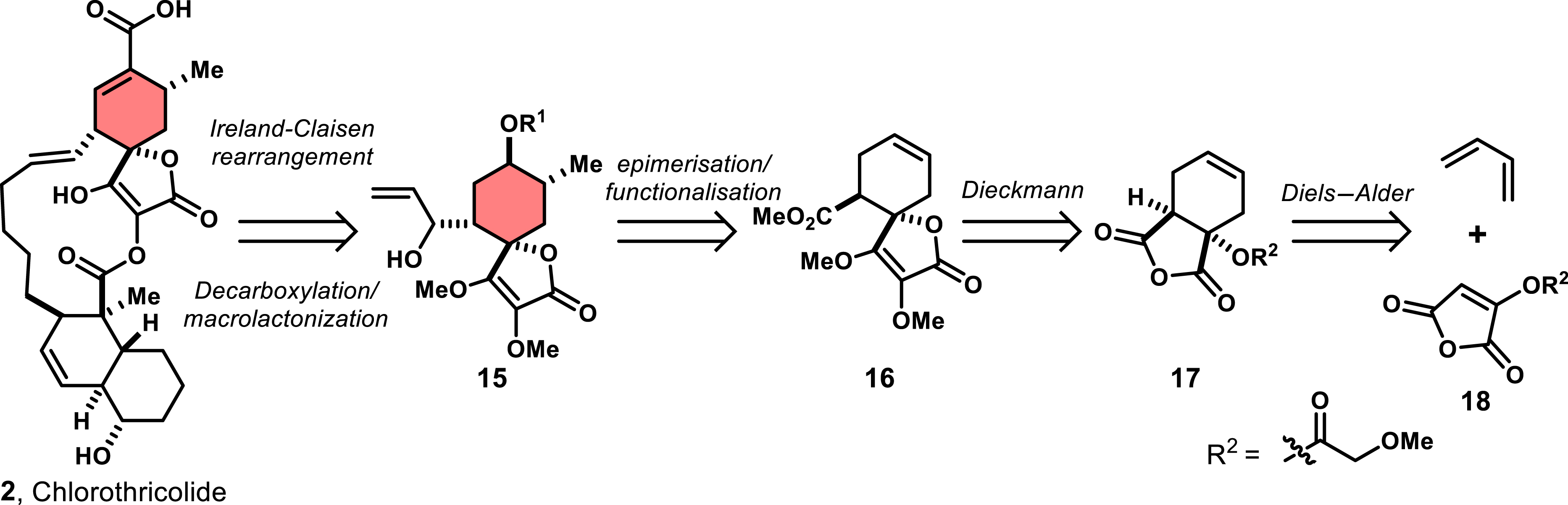
For constructing such a spirotetronate moiety, the authors proposed a Diels–Alder reaction between maleic anhydride derivative 18 and butadiene. The maleic anhydride was chosen not only for its dienophilic properties but also because it enabled an intramolecular Dieckmann condensation to form the tetronic acid moiety.
Although efficient, the Diels–Alder reaction between 18 and butadiene produced an unfunctionalized diastereo- and regioisomeric adduct of the desired spirotetronate backbone, requiring therefore post-manipulation of the cyclohexene ring. Scheme 2 outlines the high-pressure, high-temperature reaction of 18 with butadiene, leading to cycloadduct 17 in 89% yield (Scheme 2). Subsequent Dieckmann cyclization and methylation produced 16 in 60% yield. An epimerization event using sodium methoxide resulted in the desired diastereomer 19. Following a series of redox manipulations and the addition of vinyl manganese iodide, the authors obtained intermediate 20 (79% yield over three steps).
Construction of the unfunctionalized spirotetronate moiety.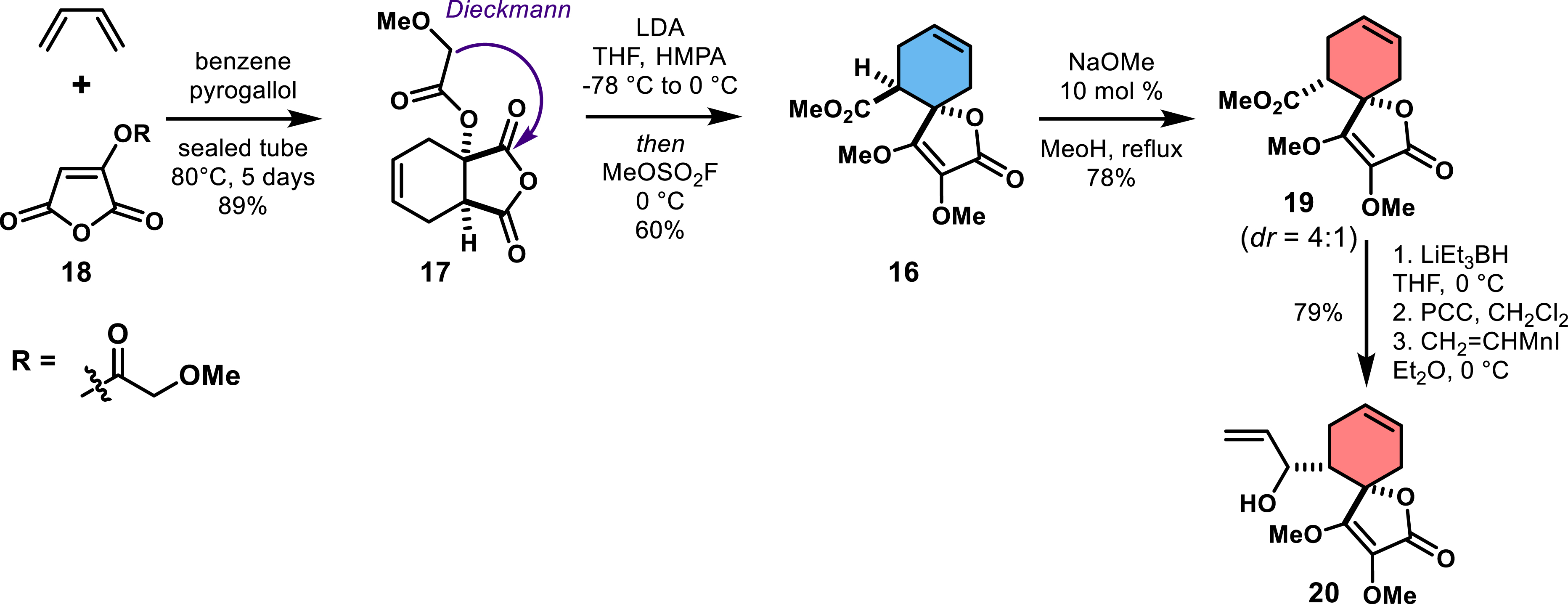
The functionalization of the cyclohexene ring in 19 required incorporation of a methyl and a carboxylate group, and regioselective formation of an endocyclic double bond. The approach, started from silyl-protected alcohol 21 and led to epoxide 22, which was transformed to intermediate 23 (Scheme 3A). Further transformations produced intermediate 24, which allowed further functionalization of the spirotetronate. Despite significant progress, Ireland’s group was unable to complete the synthesis of chlorothricin due to difficulties in the final stages, particularly in further functionalizing the cyclohexene ring. This first synthetic access to a functionalized spirotetronate was notable but burdened by many protecting-group manipulations, redox steps, and moderate selectivities. Attempts to improve the strategy by using an already pre-functionalized butadiene analogue as a reactive dienophile in the Diels–Alder reaction, namely activated silyl enol ether 25, was largely unsuccessful due to poor regioselectivity (26/27 = 1:1; 67% overall yield) observed during cycloaddition (Scheme 3B). Nevertheless, this research laid the groundwork for future synthetic efforts by demonstrating the utility of a cycloaddition and a Dieckmann condensation for constructing the bicyclic tetronic acid ring, a key feature in spirotetronate synthesis.
(A) Post-functionalization of the cyclohexene ring. (B) Reactivity of 18 toward substituted dienes.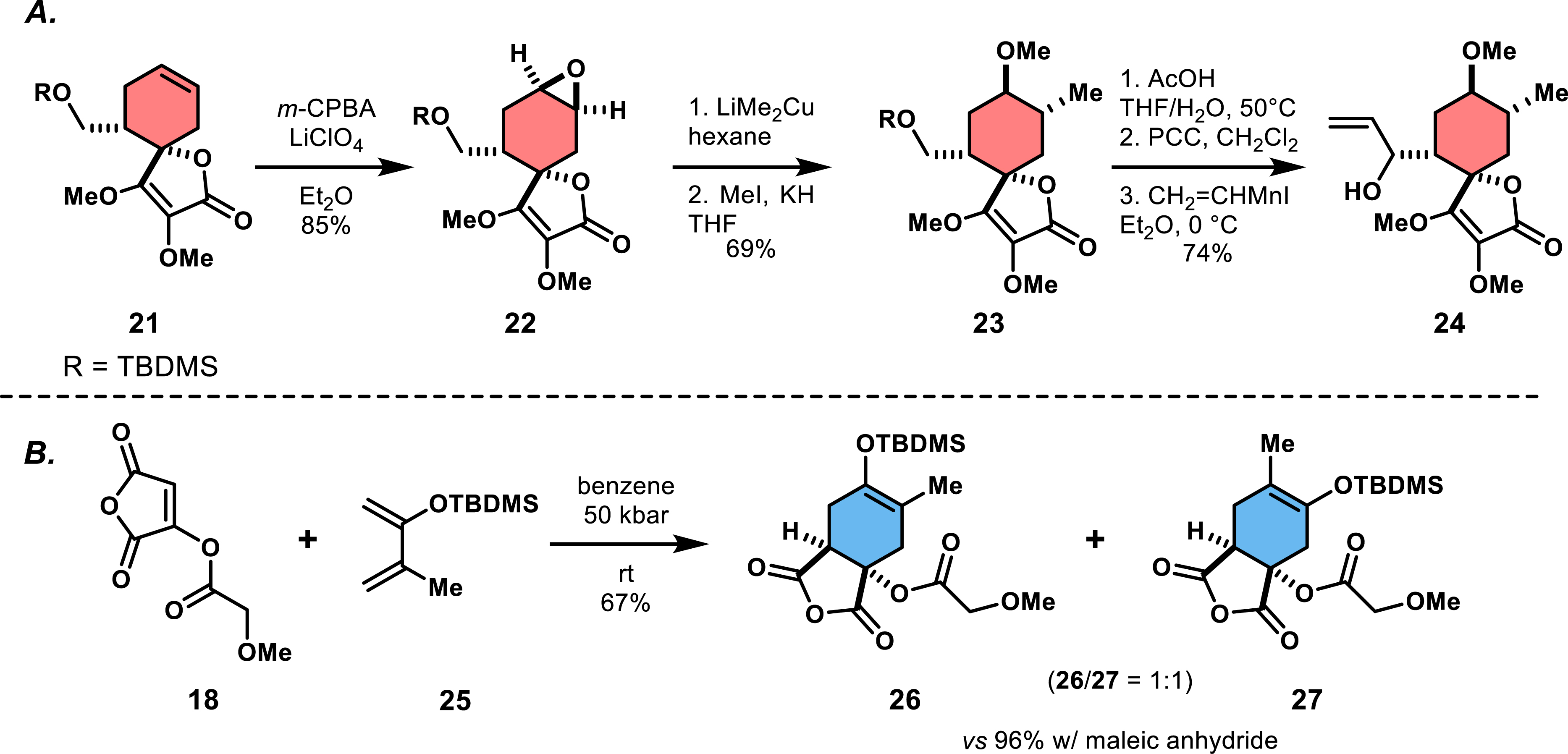
Yoshii’s group adopted a related Diels–Alder/Dieckmann cyclization route during their effort toward the synthesis of tetronolide 28, a structurally related spirotetronate with anticancer and antibacterial activity [37]. The synthesis relied on the construction of a cyclohexene fused lactone that could subsequently undergo a Dieckmann condensation to reveal an advance spirotetronate (Scheme 4A). In a model study, it was demonstrated that tetrahydrophthalide (obtained from maleic anhydride/butadiene cycloaddition and reduction) could be hydroxylated and converted to spirotetronate 32a (Scheme 4B). In the second approach, diverging from Ireland and co-workers’ reported work, 2-hydroxyacrylate derivative 34 rather than maleic anhydride was used as the coupling partner in the Diels–Alder reaction and opposed to symmetrical diene 35, avoiding regioselectivity issues. This allowed the progress toward a more functionalized spirotetronate (Scheme 4C).
Retrosynthetic analysis toward tetronolide by Yoshii and co-workers.
Yoshii et al.’s second strategy began with diene 35 and acrylate 34, which upon Diels–Alder cycloaddition and heating in methanol delivered bicyclic lactone 36 in 61% yield (Scheme 5). After several steps, including protection/deprotection, acylation, and redox manipulations, the Dieckmann condensation led to the desired regioisomeric cyclohexene spirotetronate 41. Next, oxidation of the terminal alcohol to the aldehyde allows the epimerization event to take place, leading to 42 as a 5:1 mixture of exo/endo diastereomers. Further post-transformations were then conducted to install the required side chain on the cyclohexene [38]. This sequence, although more efficient than Ireland and co-workers’ approach, still required 17 steps with an overall yield of less than 2%, due in part to extensive protecting group manipulations [39].
Synthetic access to spirotetronate precursor 70 by a Diels–Alder strategy with an acrylate and a symmetrical diene.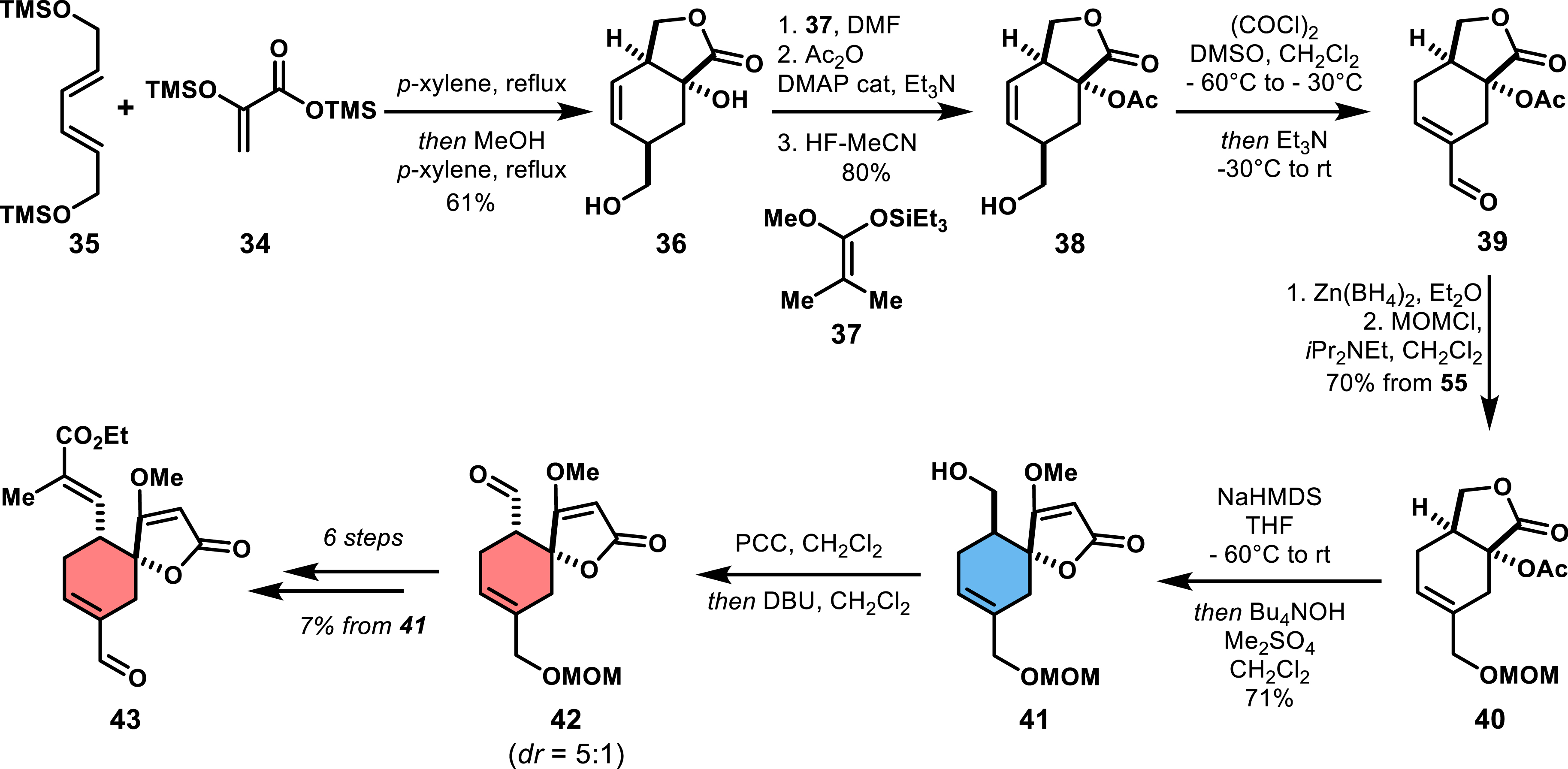
Despite these challenges, Yoshii et al.’s work represented the first use of an acrylate derivative in a Diels–Alder reaction to access spirotetronates. It was found to be particularly well suited to spirotetronate synthesis, allowing the installation of both carboxyl and acyloxy groups, critical to the tetronate ring formation.
Both Ireland et al. and Yoshii et al. studies underscore the centrality of the Diels–Alder reaction and Dieckmann condensation in spirotetronate synthesis despite the challenges in achieving regio- and stereoselective control. Within such a strategy, the use of an acrylate derivative in a Diels–Alder reaction was largely pursued in subsequent work. Since costly post-functionalization remained a bottleneck, efforts undertaken subsequently have increasingly focused on designing more functionalized dienes and dienophiles to simplify the overall synthetic routes. The biosynthetic insights gained over the past decade have further reinforced the suitability of the Diels–Alder strategy, providing a pathway for future advancements in the synthesis of this complex class of natural products.
2.2. Acrylates and acrolein derivatives as privileged dienophiles in the synthesis of spirotetronates
As mentioned above, the utilization of deactivated alkenes such as α-acyloxy acrolein 44 and acrylate 45 and substituted dienes in [4+2]-cycloadditions has proven to be a significant strategy in the synthesis of spirotetronates (Scheme 6). These dienophiles lead to intermediates containing essential functional groups required for subsequent Dieckmann condensation, while also establishing the requisite endocyclic double bond and desired substituents.
Diastereoselective outcome of a Diels–Alder reaction using an acrolein or an acrylate as the dienophile.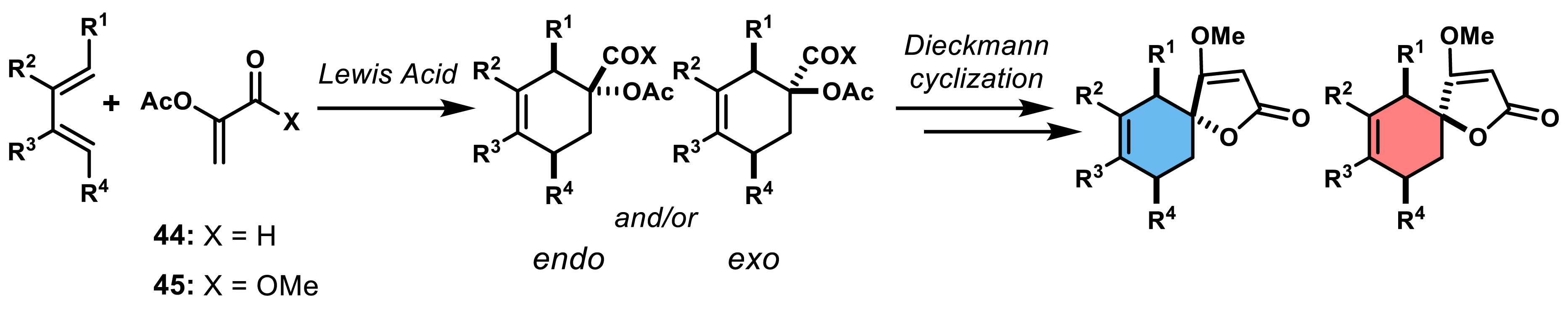
Acrolein derivatives and α-substituted acrylates are characterized by their enhanced reactivity as dienophile partners, attributed to the presence of electron-withdrawing groups. Their reactivity under thermal conditions has been well documented with simple cyclopentadiene or activated dienes analogous to Danishefsky’s diene [40, 41, 42]. However, the interaction of these dienophiles with inactivated, highly substituted, in particular α,α′ di-substituted acyclic dienes, was scarcely explored until Roush et al. reported the use of Lewis acids to promote such reactivity in 1997 [43]. Their findings demonstrated that aluminum(III) or tin(IV) based Lewis acids could facilitate [4+2]-cycloadditions between hindered, inactivated acyclic dienes and α-acyloxy acrolein, yielding good to excellent selectivity. As shown in Scheme 7, diene 46 reacted with 44 upon exposure to stoichiometric amounts of SnCl4 at −78 °C, leading to cycloadduct 47 bearing two vicinal quaternary carbons with excellent regio- and diastereoselectivity in favor of the endo diastereomer.
Lewis acid–promoted Diels–Alder reaction with hindered acyclic dienes.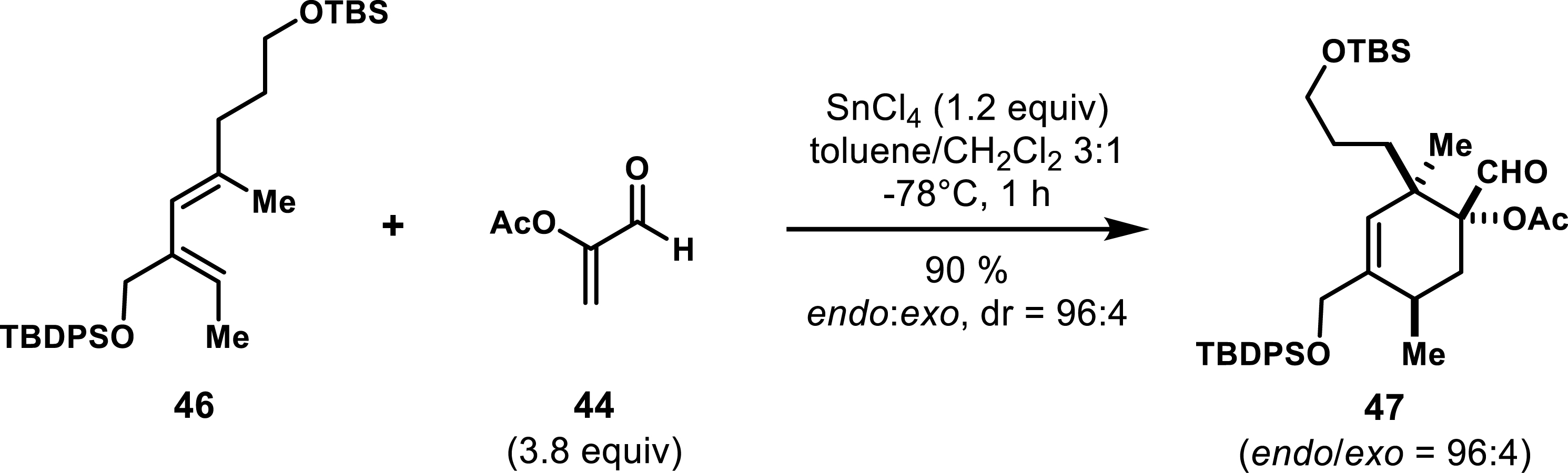
Further evaluations of various dienophiles, including α-bromoacrolein and methacrolein, indicated that MeAlCl2 could promote these reactions effectively even if longer reaction times were observed for certain dienophiles. Importantly, the application of MeAlCl2 in these cycloadditions resulted in cycloadducts exhibiting complete regioselectivity and good to excellent diastereoselectivities (endo/exo < 85:15) in favor of the endo cycloadduct. It should be mentioned that the α-substituent of the acrylate residue has a strong influence on the outcome of the reaction; indeed, the use of acryloyl and methacryloyl chlorides leads to the exo adduct or poor diastereoselectivity. An example is illustrated in Scheme 8, where exo-selective cycloaddition achieved from compound 48 was followed by a methanol/pyridine treatment to facilitate the formation of trans-fused lactones 50. The successful application of Lewis acids not only demonstrated the ability to achieve stereoselective outcomes that are often challenging under thermal conditions but also highlighted their utility in promoting the Diels–Alder reaction involving acrolein derivatives and acyclic dienes.
Diastereoselective outcome between diene 1.66 and acryloyl chloride.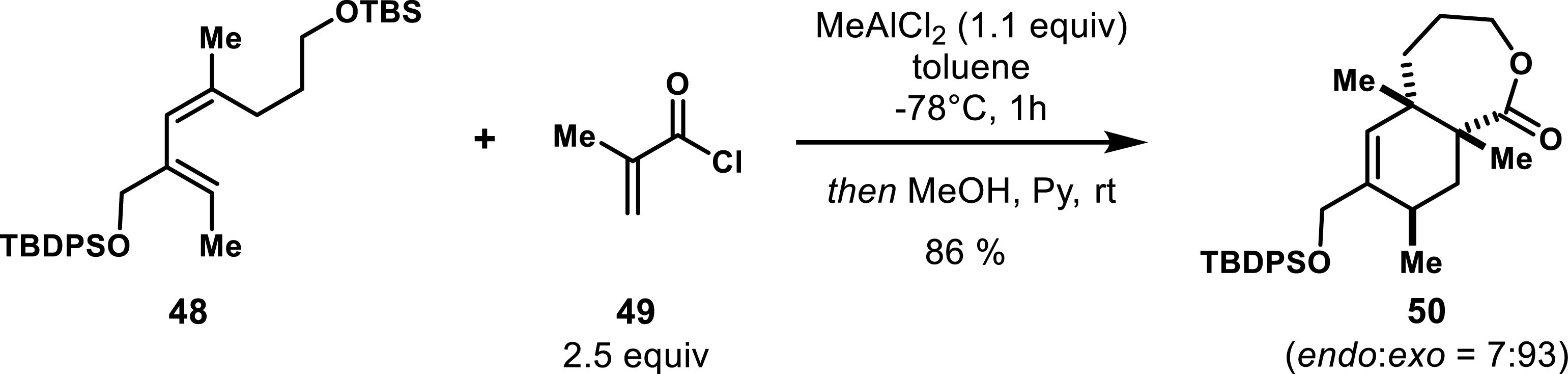
In 2002, Roush’s group employed this strategy to devise a diastereoselective synthesis of both exo and endo spirotetronate fragments of quartromicins [44]. Capitalizing on the C2 axial symmetry of quartromicins, they reasoned that the tetramer could result from an incremental twofold dimerization of endo and exo spirotetronates (Scheme 9). Specifically, this strategy could be accessible from a coupling reaction between tetronates 51 and 52, which should be feasible by Diels–Alder cycloadditions between 46 and an appropriate acrolein derivative. The reaction with acrolein 44, upon activation by a Lewis acid, would lead to the endo spirotetronate. For the exo spirotetronate, they relied on an endo-selective Diels–Alder reaction of α-bromoacrolein and 46, followed by a subsequent inversion of configuration of the quaternary stereocenter described below [45].
Retrosynthetic analysis to access quartromicins by Roush and co-workers.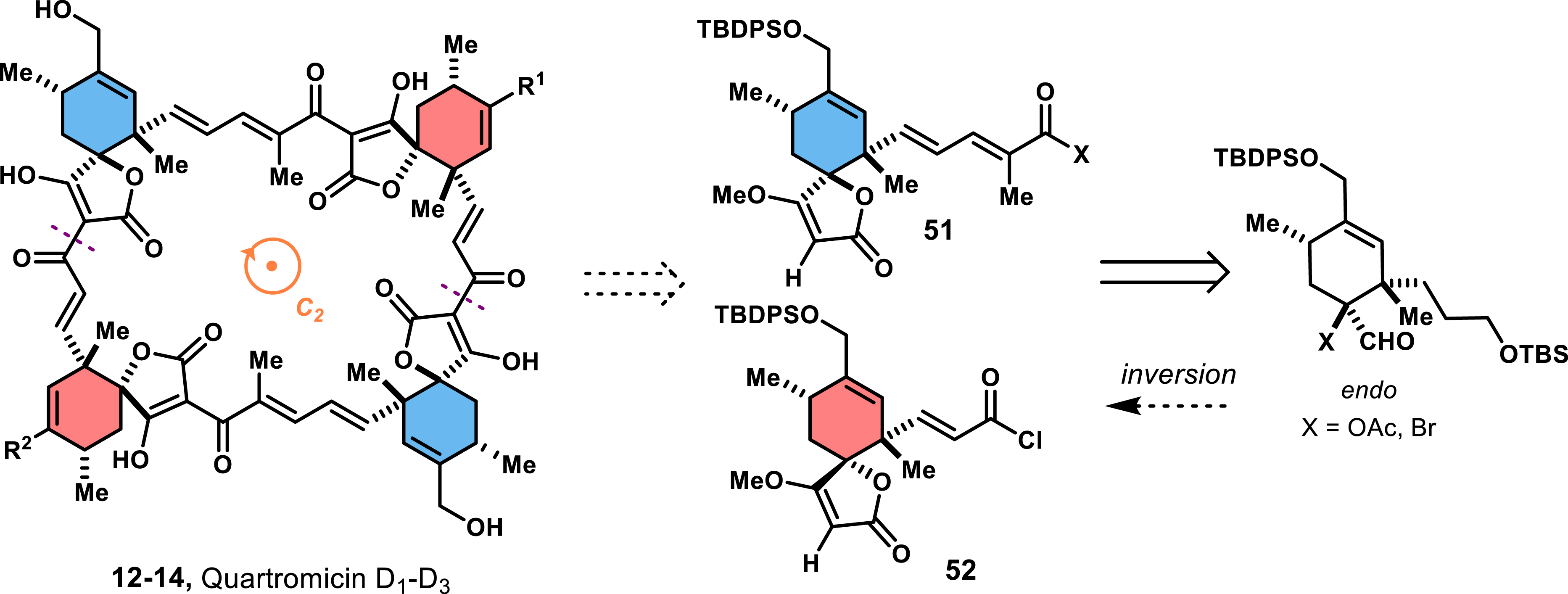
Indeed, the reaction of 46 with α-bromoacrolein in the presence of MeAlCl2 produced cycloadduct 53 as the single endo diastereomer (Scheme 10). Then, a series of five synthetic steps enabled the conversion of cycloadduct 53 to the spiroepoxide 54, which was obtained in 72% yield. This involved the reduction of aldehyde 53 to the corresponding alcohol, followed by an alkoxide-mediated inversion of the quaternary stereocenter. Although spiroepoxide 54 displayed reluctance to react with oxygen-based nucleophiles, it was successfully opened by sodium thiophenolate, yielding sulfoxide 55 upon oxidation with m-CPBA. The subsequent reaction with Ac2O and NaOAc produced the desired intermediate 56 via Pummerer rearrangement and acylation of the tertiary alcohol. After further oxidation of the aldehyde using KMnO4 and conversion to the methyl ester with TMSCHN2, the TBS-protected hydroxyl group was deprotected using pyridinium p-toluenesulfonate. This allowed for further functionalization to obtain the α,β-unsaturated ester 58, ultimately leading to the classical Dieckmann condensation that produced spirotetronate 59 in good yield.
Inversion of a quaternary stereocenter to access exo spirotetronate 78.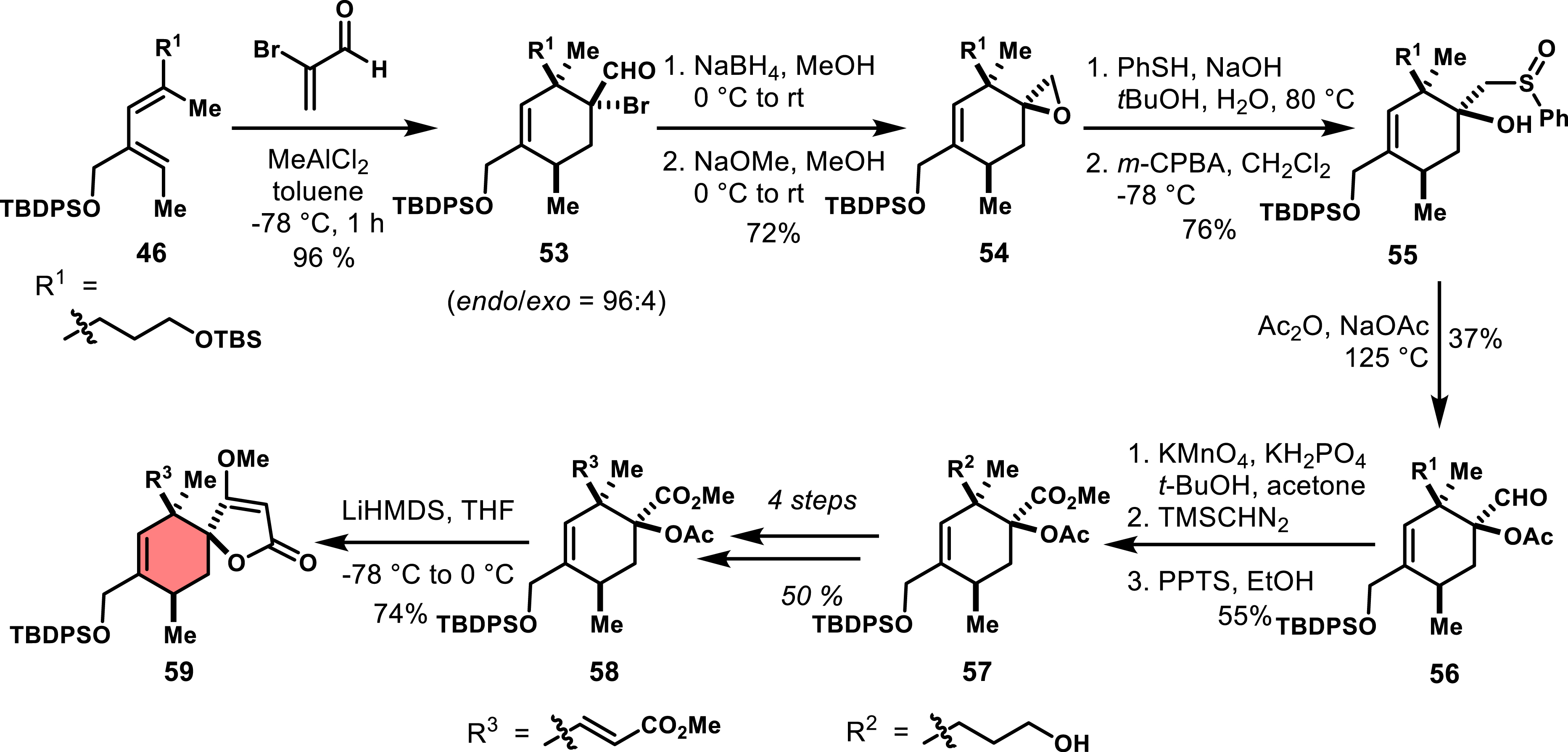
For the construction of the endo spirotetronate, diene 46 was made to react with α-acyloxy acrolein to generate cycloadduct 60, followed by functional group interconversion and redox manipulations to access the desired endo product in 10 steps (24% yield; Scheme 11). These synthetic sequences highlight the versatility of acrylates and acroleins as privileged dienophiles, underscoring their significance in the efficient construction of complex spirocyclic architectures relevant to biologically active compounds. Further advance in the synthesis was undertaken [46], but the total synthesis of quartromicins remained incomplete.
Synthesis of the endo spirotetronate moiety en route to quartromicins.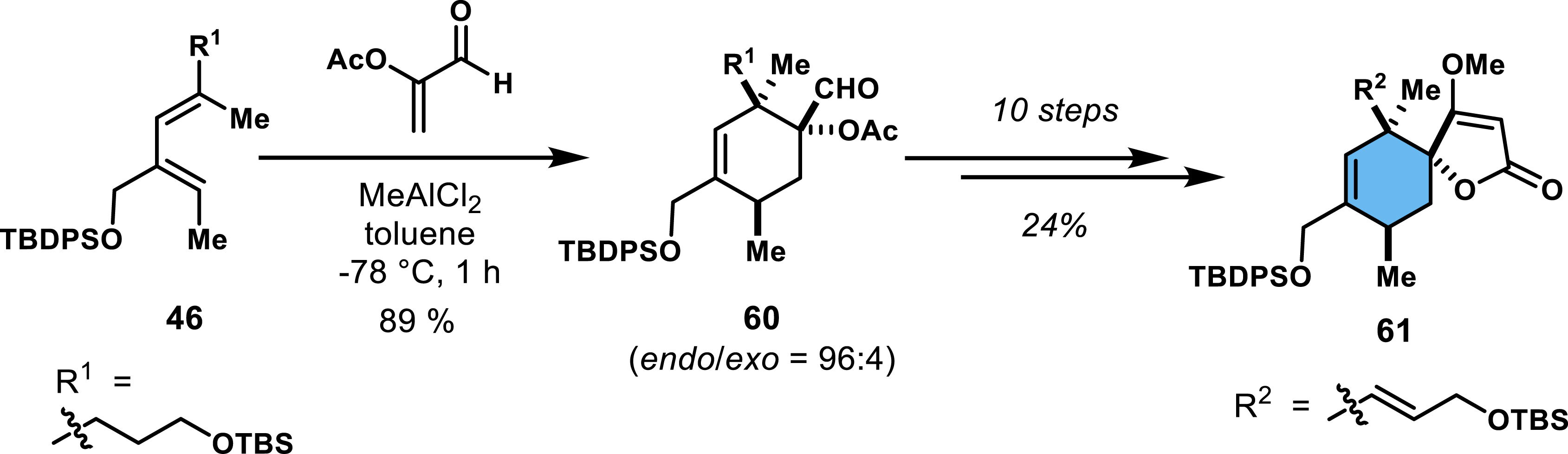
Lewis acid–promoted cycloadditions of functionalized dienes with acrolein derivatives or acrylates have proven effective for synthesizing various endo diastereocontrolled spirotetronates. Several studies [47, 48, 49] have highlighted this approach, particularly in the context of abyssomicin C. Two independent studies published in 2005 reported the synthesis of the spirotetronate moiety using diene 62, which contains an unprotected allylic alcohol (Scheme 12). The reaction with α-acyloxyacrolein 44 produced cycloadduct 63 as the sole endo diastereomer [48]. The fused lactol was subsequently oxidized to lactone 64 and engaged in a Dieckmann condensation that first required a TBSCl protection of the terminal alcohol to prevent recyclization to the lactone. Attempts to achieve enantioselectivity through aluminum/BINOL complexes during the cycloaddition were unsuccessful. In the second study, methyl acrylate served as the dienophile, leading to the fused lactone 66, necessitating an additional hydroxylation step to introduce the acetate group required for the spirotetronate formation [49]. Both studies ended with the formation of compounds 65 or its deprotected variant 68, without completing the total synthesis of abyssomicin C.
Synthetic access to the spirotetronate moiety of abyssomicin C.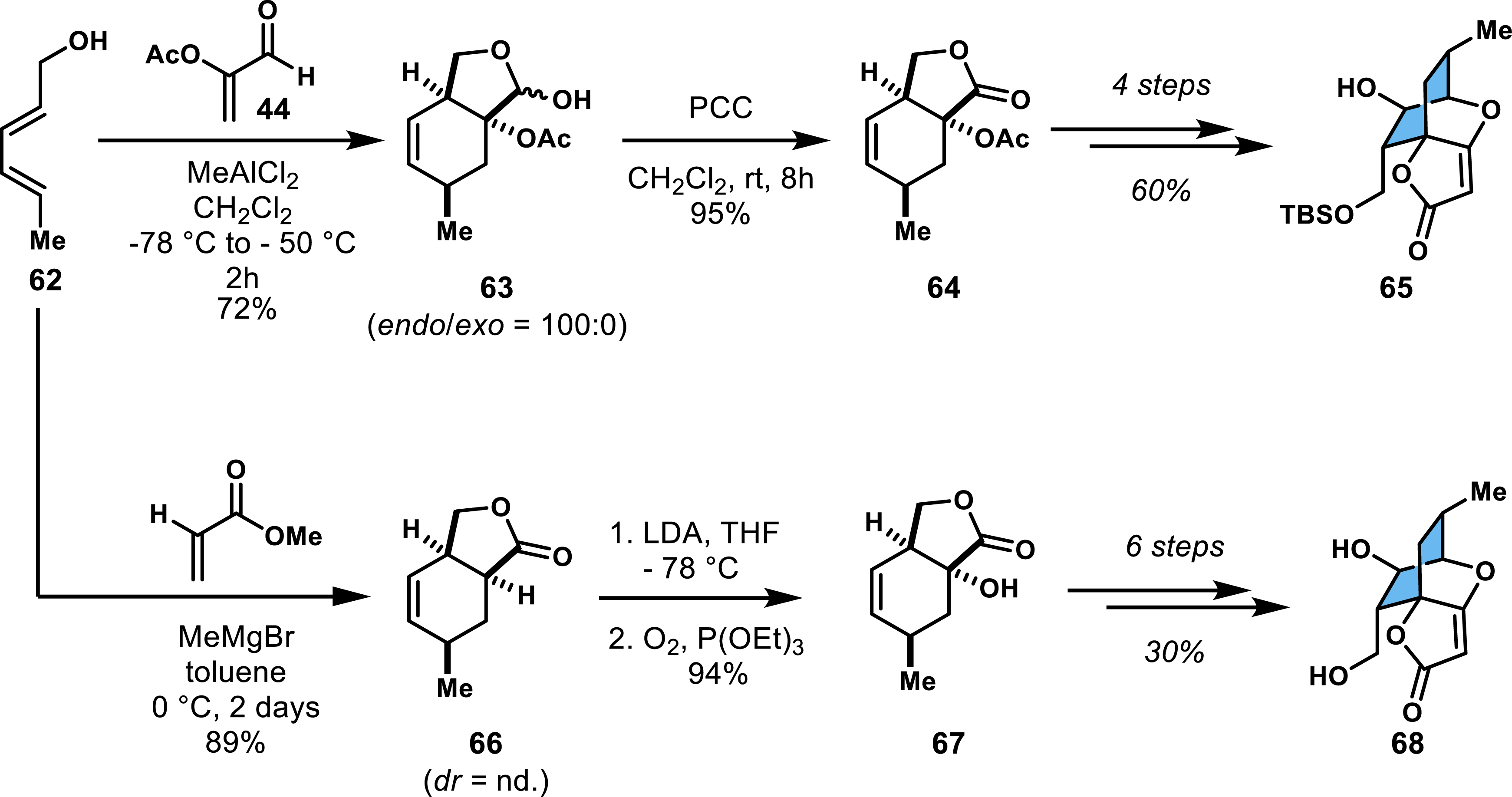
Burkart’s group mimicked this method to access the core structure of spirohexenolides 5a and 5b, previously isolated compounds exhibiting antitumoral activity [4]. They achieved a regio- and diastereoselective cycloaddition between a hindered triene and α-acyloxyacrolein 44 [50], but encountered the undesired deacylation of the tertiary alcohol during aldehyde oxidation, necessitating additional protection/deprotection steps and reducing the overall synthetic efficiency.
Nicolaou and co-workers implemented this strategy in a diastereoselective process during their synthesis of enantioenriched spirotetronate moiety of abyssomicin C [51, 52]. Engaging enantioenriched diene 69 bearing a chiral secondary alkoxy side chain with methyl acrylate in a Diels–Alder reaction, they managed to isolate lactone 70 as a single diastereoisomer in 80% yield (Scheme 13). This magnesium-promoted reaction used 2-(pyrrolidin-1-yl)phenol as a ligand, which enhanced the reaction rate by facilitating a favorable spatial arrangement between the magnesium ions and the dienophile. The cycloadduct underwent further transformations, including α-hydroxy ester formation, lithium–sulfur exchange, epoxidation, and Dieckmann cyclization, ultimately yielding endo spirotetronate intermediate 73. Final macrocyclization was achieved through ring-closing metathesis (RCM) using a Hoveyda–Grubbs catalyst, culminating in the successful synthesis of abyssomicin C and its atropisomer alongside numerous analogues, helping in understanding the structure–activity relationship.
Enantioselective access to the spirotetronate moiety of abyssomicin C.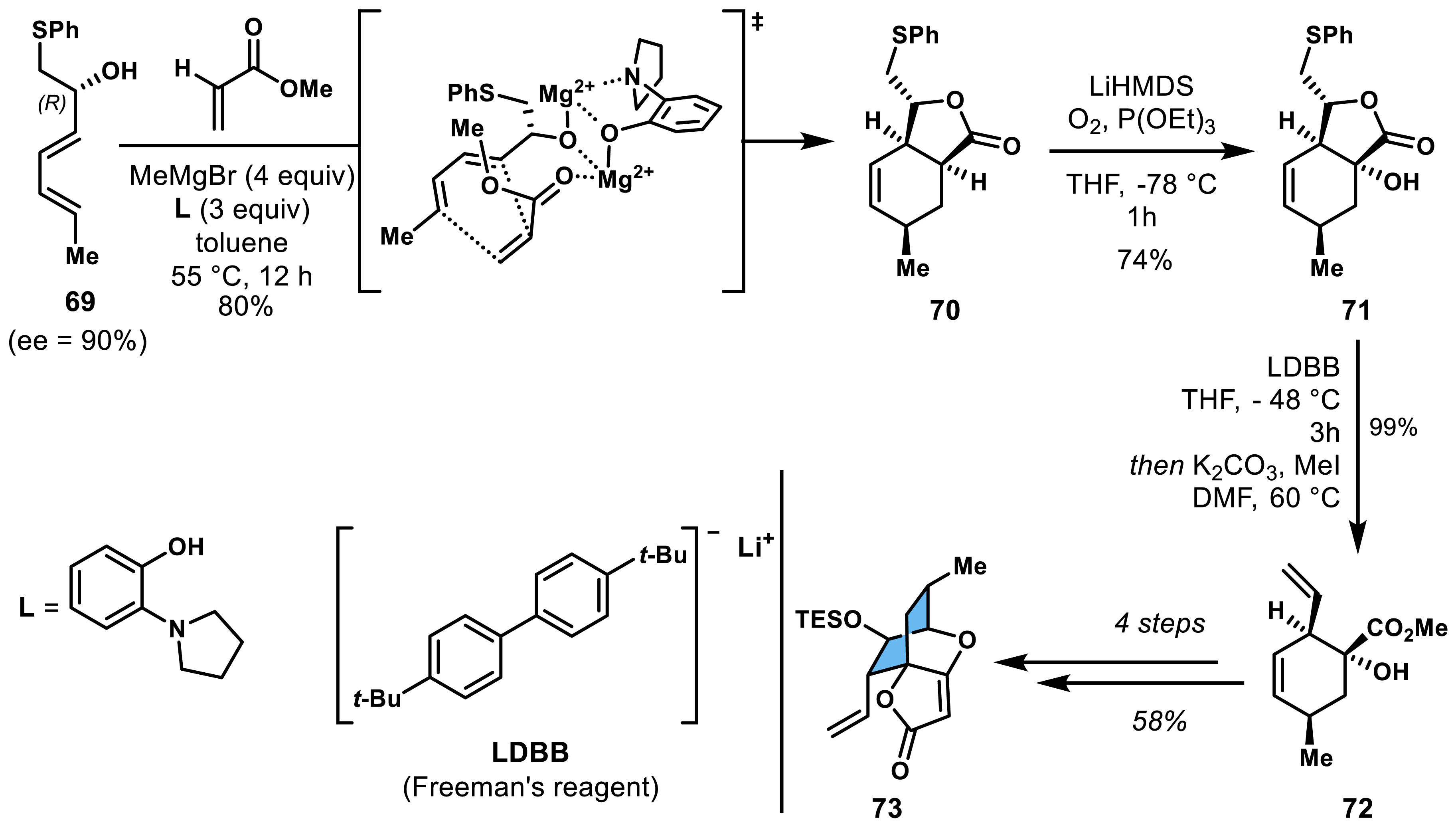
Exploration of chiral dienophiles to induce diastereoselectivity and obtained enantioenriched cycloadducts have also been reported. Roush et al. demonstrated that 2-alkyl-5-methylene-1,3-dioxolan-4-ones participate in Diels–Alder reactions involving cyclobutene via an exo transition state [53, 54, 55, 56], leading essentially to one diastereomer with an exo configuration. In particular, 2-(tert-butyl)-5-methylene-1,3-dioxolan-4-one 74 with its bulky tert-butyl substituent predominantly forms product 75 from exo-TS-75, with selectivity attributed to the constrained s-cis conformation of the dienophile and steric interactions influencing the transition states (Scheme 14).
Diastereoselective Diels–Alder cycloaddition of (R)-74 with cyclopentadiene.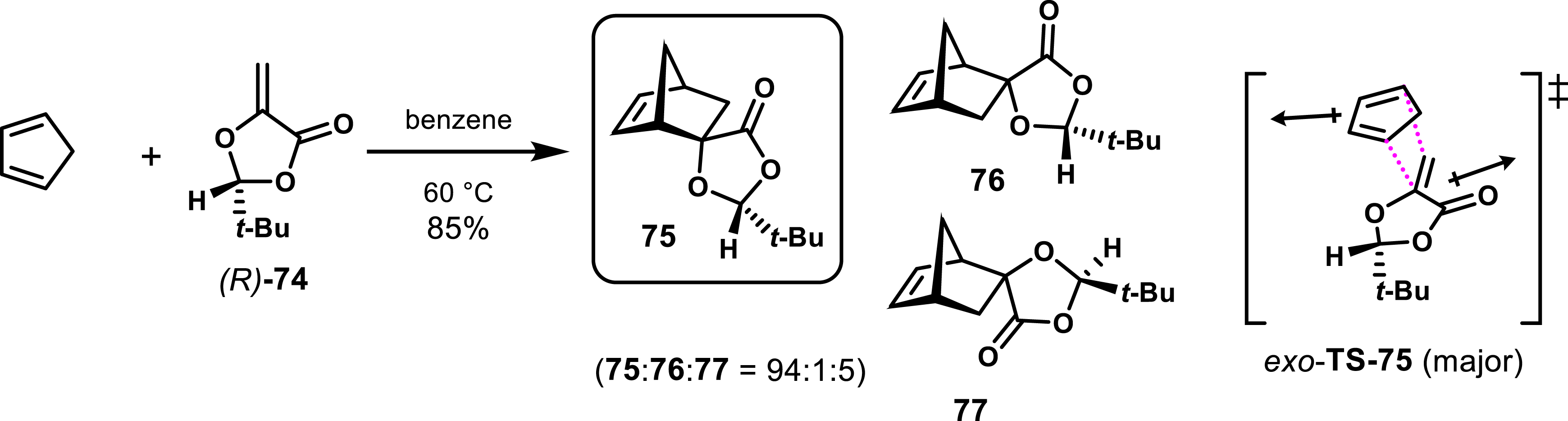
Utilizing this diastereoselective exo-selective approach, Roush and co-workers implemented an intramolecular Diels–Alder (IMDA) strategy to access diverse diastereomers of spirotetronates relevant to quartromicins [57]. The synthesis of the sulfone precursor 79 was achieved in six steps from a known vinyl iodide 78, yielding an overall 50% yield. The IMDA reaction conducted at 210 °C generated cycloadducts 80, 81, and 82, albeit with low yields and poor stereoselectivity (Scheme 15). The absolute configurations of products 80 and 82 were confirmed through crystallographic analysis, while nOe correlations determined the relative configuration of 81 [58, 59]. Ring opening of the lactone was achieved using ethanedithiol to reveal the key α-hydroxy ester motif subsequently used to build the tetronate. This IMDA strategy has proven effective in the total synthesis of chlorothricolide [60, 61], tetronolide [62, 63], and kijanolide [64, 65], showcasing the potential of spirotetronate derivatives.
Diastereoselective outcome of the intramolecular Diels–Alder reaction applied to 79.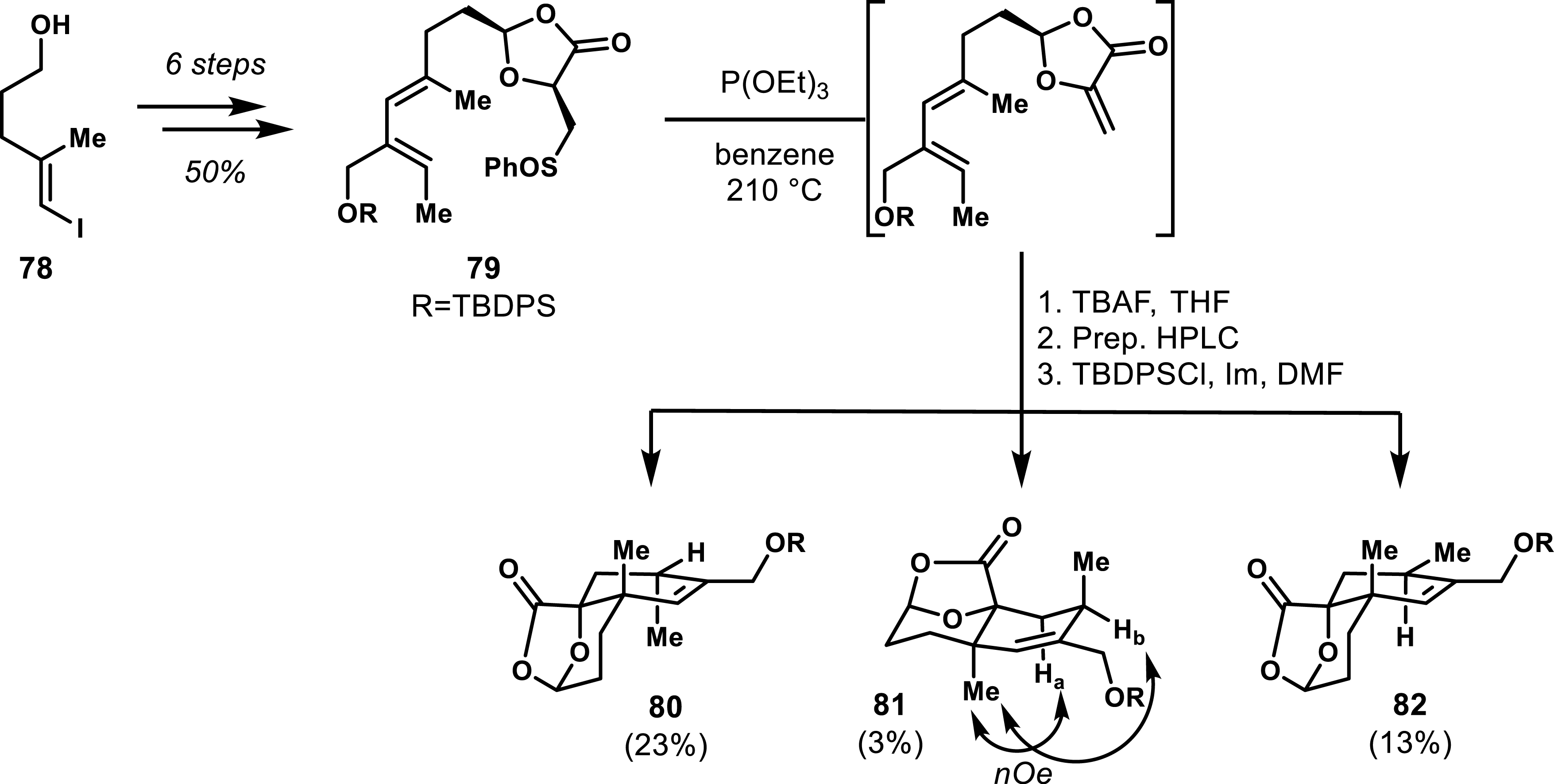
Although 5-methylene-1,3-dioxolan-4-one derivatives were demonstrated to be competent partners in Diels–Alder reactions, their reactivity remained low, and even under quite forcing conditions, furnished low yield and limited selectivity. To improve the outcome, Roush’s group explored the reactivity of 1,3-oxazolidine-4-one analogues 83a–b under Lewis acid activation to facilitate an intermolecular Diels–Alder reaction. However, the reaction between dienophile 74 and diene 46 exhibited extensive decomposition at cryogenic temperatures in the presence of a Lewis acid (Scheme 16). An aza analogue of 1,3-oxazolidine-4-one, in the form of oxazolidine 83, was more robust as a dienophile and underwent the cycloaddition smoothly. Unfortunately, efforts to subsequently cleave the lactam were unsuccessful [66].
Reactivity issues posed by the chiral auxiliaries developed by Roush et al.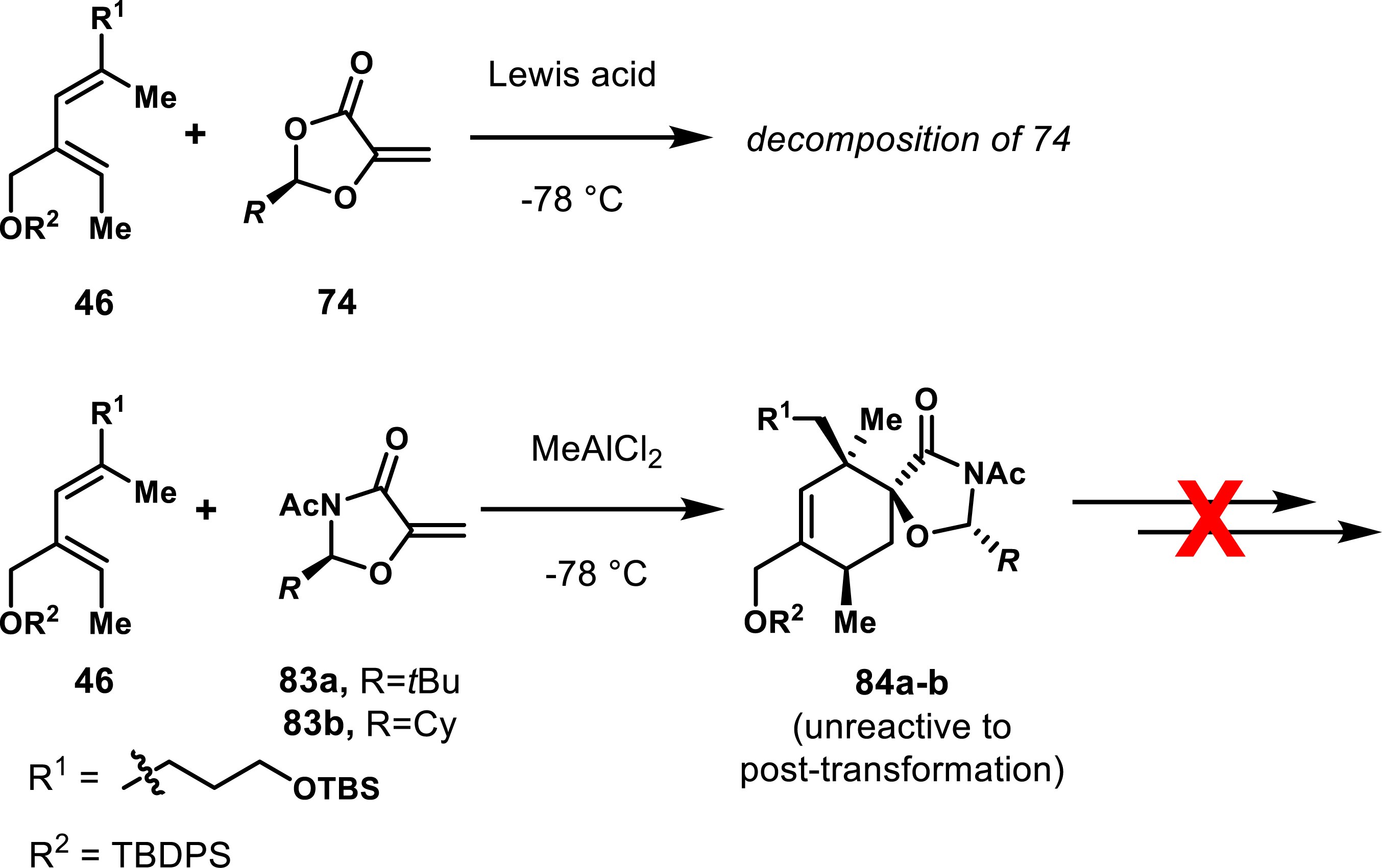
To improve the strategy without struggling with low yields and decomposition and for subsequent handling, Roush’s group turned its attention toward bicyclic oxazolidinone based dienophiles 85a–c, each featuring substituents such as methyl, isopropyl, or benzyl groups, designed to enantioselectively access the spirotetronate moieties of quartromicins (Scheme 17) [66]. These chiral building blocks proved to be effective in the anticipated Lewis acid–activated Diels–Alder reaction even if the reaction was specially slow (5 days). Among the dienophiles tested, 85a provided the most promising results leading to a 5:1 ratio with a total yield of 70% for the exo adduct. Subsequent transformations involved the cleavage of the chiral auxiliary using lithium aluminum hydride followed by oxidation of the primary alcohol with DMSO, yielding the enantioenriched precursors 88a and 89a. Despite the advancements achieved with these bicyclic oxazolidinone dienophiles, their relatively low diastereoselectivity in the Diels–Alder reaction (exo vs endo ratio) highlights the limitations of this approach. Although other chiral auxiliaries based on acrylates and acrolein derivatives have been explored for spirotetronate synthesis, they generally encounter similar challenges related to post-functionalization [67, 68].
Bicyclic chiral oxazolidinone enables access to the exo and the endo fragments of quartromicins.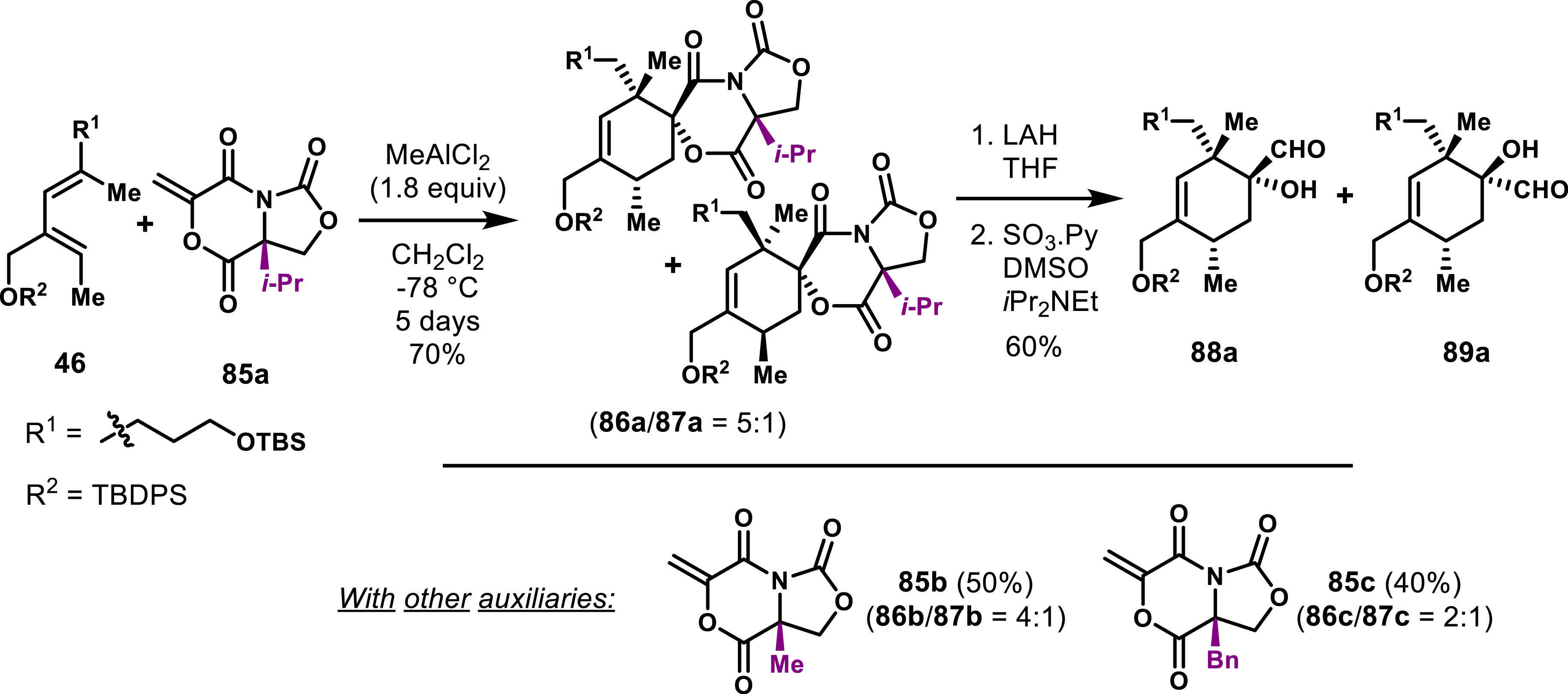
Roush and co-workers also examined a Lewis acid–promoted Diels–Alder reaction using N-acryloyl camphorsultam 90 and diene 46, achieving a diastereomeric ratio of exo/endo 7:1 (Scheme 18A) [44]. Despite this stereoselectivity, the approach did not provide direct access to the required quaternary hydroxy carbon, thereby diminishing its relevance. Post-manipulations nevertheless allowed building suitable spirotetronate precursors. Subsequent attempts to utilize α-substituted N-acryloyl camphorsultams were unsuccessful, prompting further investigations.
(A) Use of camphorsultam-based auxiliary to access both endo and exo spirotetronates of quartromicins by Roush et al. (B) Scheidt et al. strategy displaying a N-acryloyl oxazolidinone.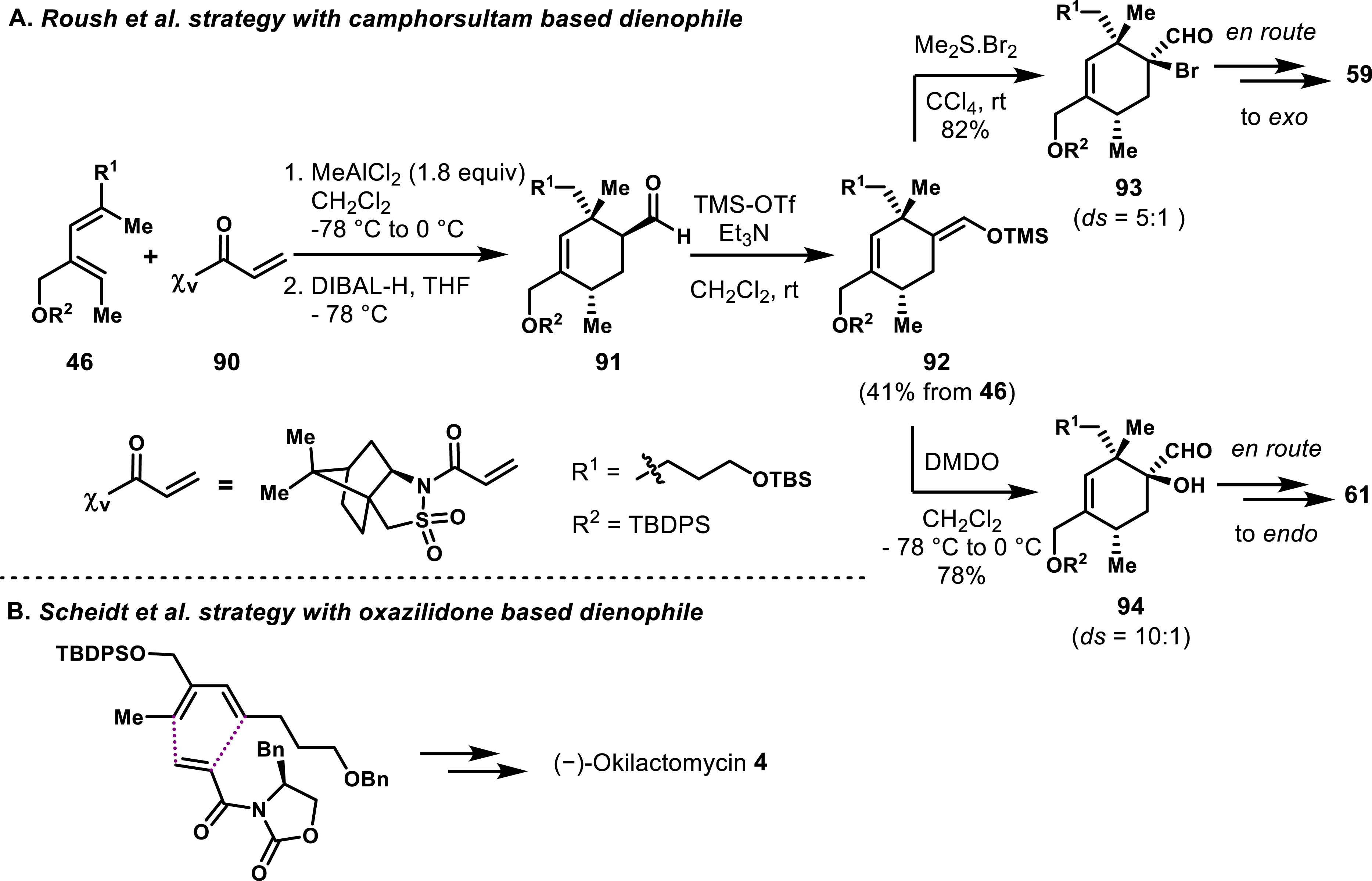
In a related approach, Scheidt and co-workers successfully utilized a chiral N-acryloyl oxazolidinone [69] in the synthesis of the endo spirotetronate of (-)-okilactomycin, demonstrating that this strategy could be extended to complex natural products (Scheme 18B) [70].
Given the challenges with chiral dienophiles, it would be advantageous to achieve enantioenriched intermediates from achiral substrates by controlling the enantioselectivity of the Diels–Alder reaction through the use of chiral promoters or catalysts. In the synthesis of spirotetronates, this approach remains underexplored, with the only successful example reported by Marshall and Xie in 1992 [45]. They leveraged an enantioselective Diels–Alder reaction using reactive dienes and acrolein derivatives, promoted by the Corey–Bakshi–Shibata (CBS) catalyst, a boron-based oxazaborolidine. They conducted the reaction between functionalized triene 95, synthesized in eight steps from glycolaldehyde, and α-bromoacrolein, using one equivalent of the CBS catalyst 96. This reaction led to the formation of the endo diastereomer 97 with moderate enantiocontrol (ee = 72%) but impressive regioselectivity (Scheme 19). The choice of bromo-substituted acrolein was driven by its reactivity toward dienes when activated by the oxazaborolidine catalyst and the requirement to invert the quaternary stereocenter to access the exo spirotetronate. Although the authors did not provide a detailed mechanism, a plausible transition state based on Corey and Loh’s earlier work was proposed in Scheme 19 [71]. The key inversion of the stereocenter followed a sequence similar to that previously outlined by Roush et al., involving an epoxide intermediate, sulfide ring opening, oxidation of the sulfoxide, and a Pummerer rearrangement. Following this, a classical acylation of the tertiary alcohol and a subsequent Dieckmann cyclization were used to form the spirocyclic intermediate 98 in nine steps from 97 (overall yield 68%). Despite the moderate enantiomeric excess and the stoichiometric use of the oxazaborolidine catalyst, this regio- and diastereoselective cycloaddition represents a rare example of spirotetronate synthesis not relying on chiral dienophiles.
Enantioselective Diels–Alder reaction with achiral substrates with the use of CBS catalyst.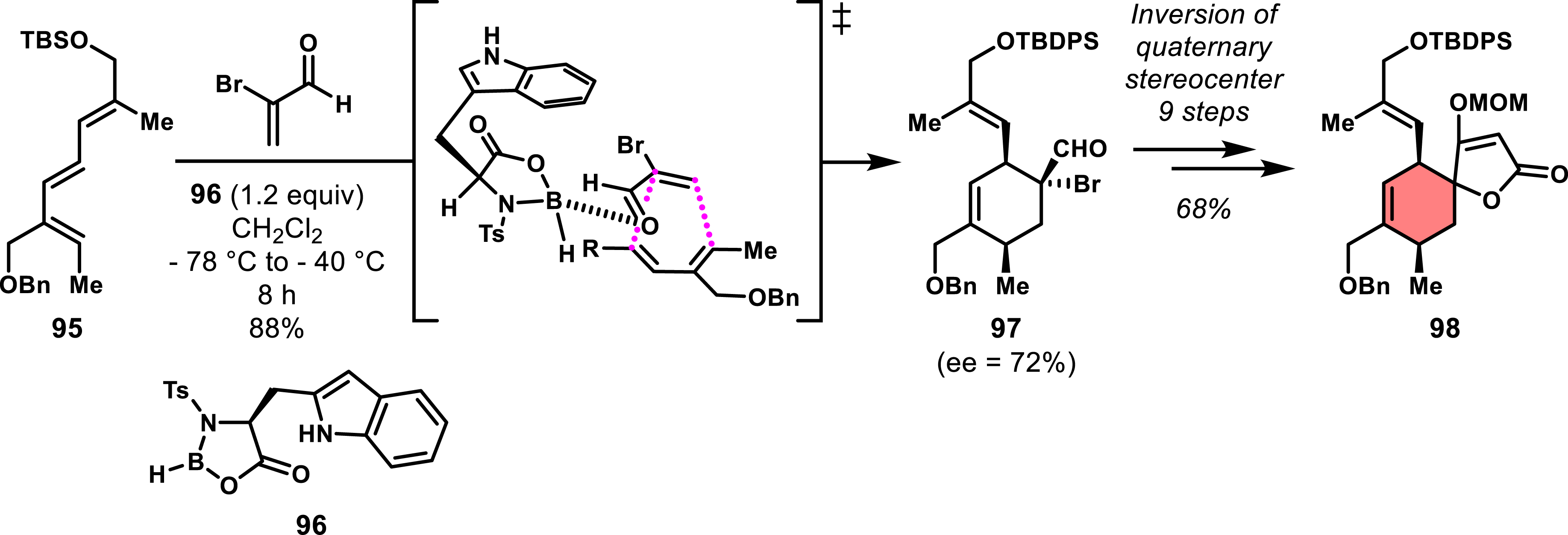
2.3. Emergence and use of tetronate-derived dienophile in Diels–Alder reactions
The use of Diels–Alder reactions to access spirotetronates has been predominantly based on acrolein derivatives or acrylates, both chiral and achiral. However, some studies have explored alternative dienophiles, such as 5-methylenetetronate 100 (Scheme 20), which bypass the need for post-functionalization steps required for Dieckmann condensation [72]. Yoshii’s group successfully synthesized 100 in just three steps starting from 4-methoxyfuran-2(5H)-one. They were the first to apply this dienophile in an intermolecular Diels–Alder reaction with triene 99 at high temperatures (180 °C in 1,2-dichlorobenzene), obtaining a modest diastereoselectivity (dr = 1:3) and yield (43%). The reaction favored the undesired endo diastereomer 102, considering that targeted kijanolide contains an exo-type spirotetronate. Despite the low yield and selectivity, the reaction showcased the ability of tetronate dienophiles to form spirocycles in a single step. Following this work, the same group explored α-acyloxy derivatives of 100, synthesized in two steps from 100, to access the spirotetronate moiety of chlorothricolide [73]. Although the reaction conditions were less severe (140–150 °C in chlorobenzene), the outcome was again a diastereomeric mixture favoring the endo-type spirotetronate along with regioisomeric products. These results highlight the limitations of 5-methylenetetronate in terms of regio- and diastereoselectivity, yet its use circumvents extensive post-reaction functionalization.
First example of intermolecular Diels–Alder reaction using a tetronate dienophile.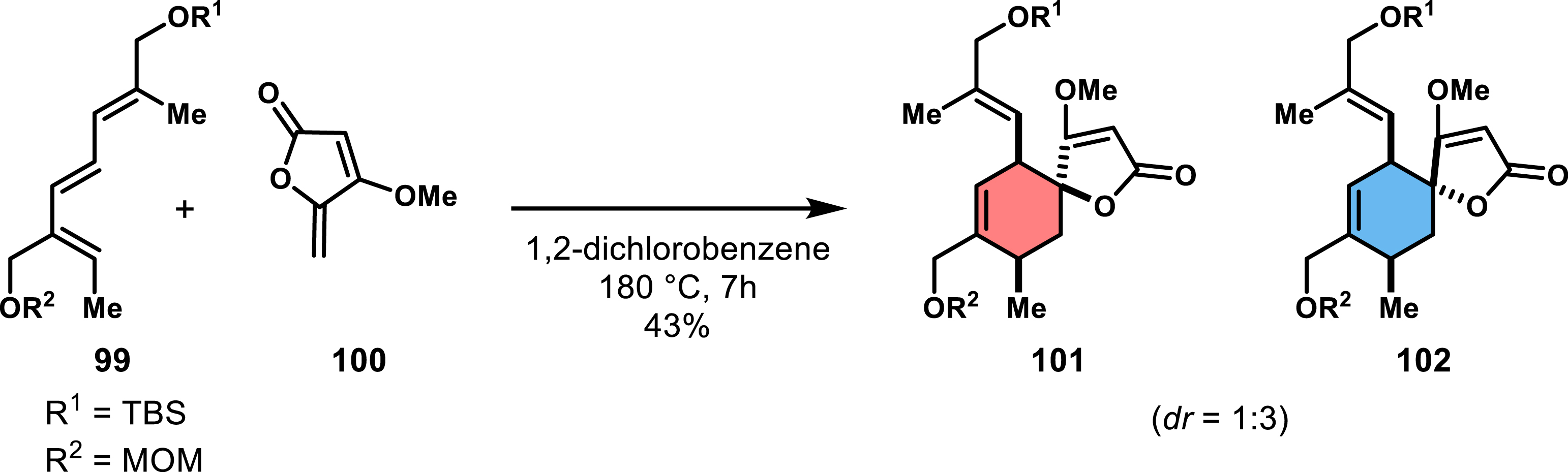
The use of tetronate-derived dienophile as a synthetic precursor to spirotetronate was illustrated by Sorensen and co-workers (2005) [74], who achieved the enantioselective and diastereoselective synthesis of abyssomicin C. Sorensen et al.’s approach relied on a biomimetic IMDA reaction that constructs the spirotetronate and the macrocyclic ring in a single step (Scheme 21). The synthesis began with meso-anhydride 103, which was accessible from diethyl methylmalonate in three steps on a large scale. After several key transformations, including stereoselective hydrolysis and aldolization with trans,trans-2,4-hexadienal, aldehyde 104 was obtained in nine steps from 103 with a 22% overall yield. A critical step in the sequence was the addition of deprotonated tetronate 105 to aldehyde 104, followed by oxidation of the resulting secondary alcohol using the Dess–Martin periodinane, generating intermediate 106. Initially, the authors attempted to eliminate the protected alcohol and initiate the Diels–Alder reaction by using scandium(III) triflate, but this approach produced an unstable polyene intermediate. They subsequently found that treating 106 with lanthanum(III) triflate triggered both polyene formation and intramolecular cycloaddition in one step, furnishing the key intermediate 107 in 50% yield. Notably, this step was completely diastereoselective, a significant improvement over previous methods employing tetronate dienophiles. The Diels–Alder reaction proceeded through a favorable transition state (TS-107) as confirmed by computational studies. Further transformations, including epoxidation of the cyclohexene ring and nucleophilic attack of the deprotected tetronic acid, completed the synthesis of abyssomicin C. This strategy not only provided the first enantioselective synthesis of abyssomicin C but also closely mirrored the natural biosynthetic pathway, which involves a similar IMDA reaction with a tetronate dienophile. In addition to abyssomicin C, this method has been extended to the synthesis of abyssomicin C analogues and okilactomycin, further demonstrating the versatility of the IMDA reaction with tetronate dienophiles [75, 76]. These efforts have significantly expanded the scope of spirotetronate synthesis, offering a powerful tool for accessing complex natural products with spirocyclic cores.
Enantio- and diastereoselective access to abyssomicin C via a biomimetic strategy by Sorensen and co-workers.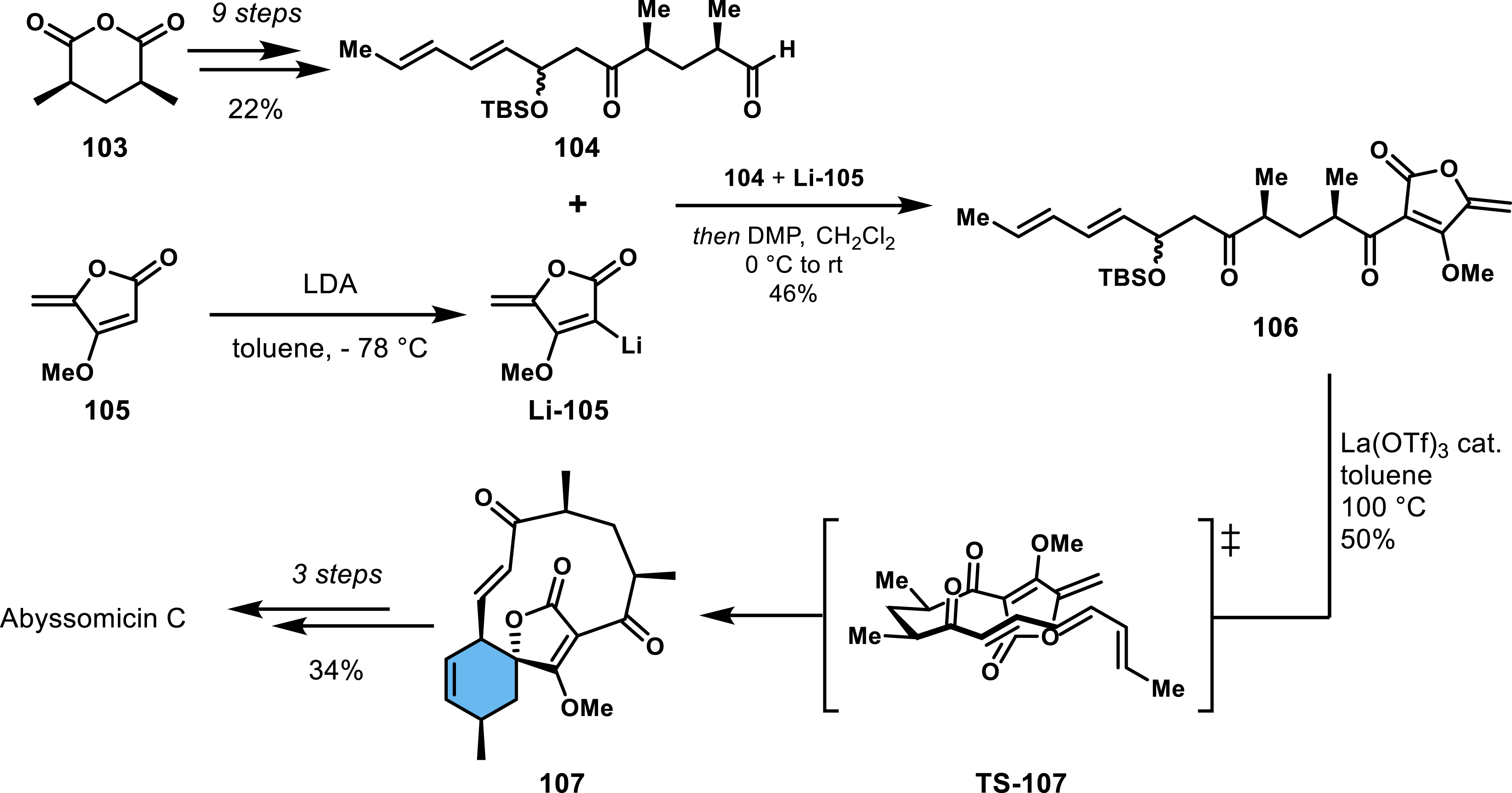
3. Other strategies
As illustrated previously, the main strategies to access spirotetronates rely on two key disconnections: a Diels–Alder reaction to build the cyclohexene ring and a Dieckmann condensation to form the tetronate unit. However, alternative retrosynthetic disconnections have been explored to address the synthesis of this challenging spirotetronate motive.
3.1. Alkylation of cyclic ketones
The conversion of ketone with β-lithiated acrylates has been reported as an alternative to the Dieckmann condensation in the context of spirotetronate natural compound synthesis. For instance, selective β-lithiation of β-methoxyacrylate 108 under kinetic control and subsequent addition to cyclohexanone produced unfunctionalized spirotetronate 109 (Scheme 22) [77, 78]. Schmidt and Hirsenkorn used this method in the synthesis of chlorothricolide [79, 80], and showed that β-ethylthioacrylate improved yields compared to methoxyacrylate, while both nucleophiles provided the same diastereoselectivity. The resulting sulfide 111 could be further converted into a methoxy derivative and α-hydroxylated to yield intermediate 112, useful in chlorothricolide synthesis.
Alkylation of cyclohexanone with a β-lithiated acrylate.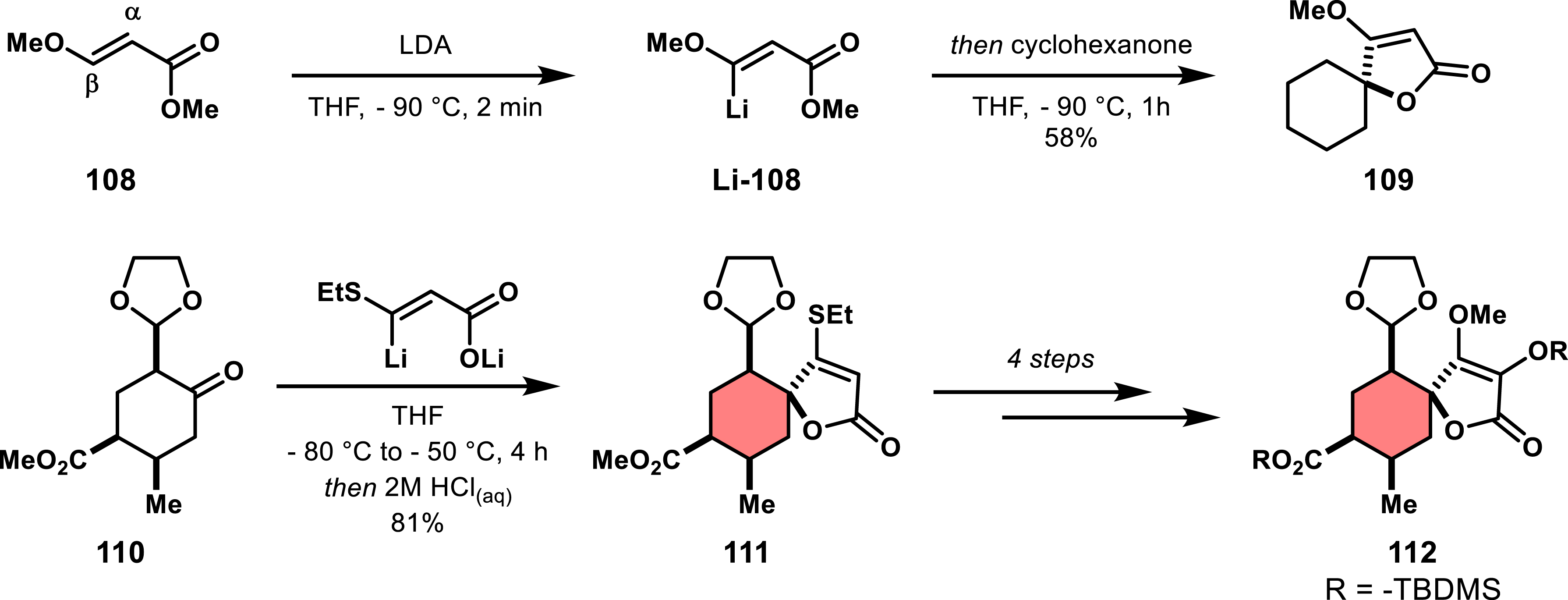
Yoshii and co-workers also took advantage of this strategy in several studies toward kijanolide and tetronolide [81, 82]. They found that using a cerium(III) derivative of Li-108 allowed better compatibility with enolizable ketones due to the nucleophilicity of organocerium compounds, which are poor bases [83]. Their synthesis of tetronolide started with enantioenriched cyclohexanol 113 [84] and proceeded to cyclohexanone 114 in three steps with 80% yield. Addition of β-dichlorocerium salt of 108 provided the spirotetronate intermediate 115 as the exo diastereomer (Scheme 23).
Enantioselective access to an exo spirotetronate by alkylation of a cyclohexanone with a cerium salt.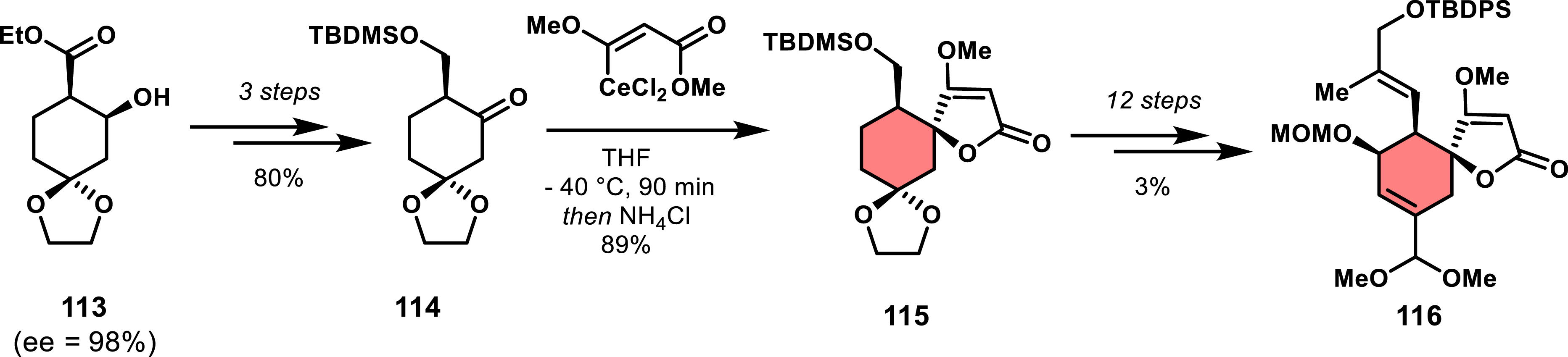
Other notable efforts include the total synthesis of (-)-atrop-abyssomicin C by Bihelovic and Saicic, relying on a gold-catalyzed step to form the bridged spirotetronate (Scheme 24) [85]. This approach involved the addition of a lithium derivative of ethyl orthopropiolate to a cyclic ketone that occurred with high diastereoselectivity, providing compound 122 in 75% yield [86]. A gold-catalyzed hydroxyl addition to the alkyne was subsequently performed, leading to a tricyclic (Z)-3-alkoxyacrylate. Photochemical (Z)–(E) isomerization did then offer the suitable conformation for tetronate formation.
Synthetic access to an intermediate in the synthesis of (-)-atrop-abyssomicin by Saicic et al.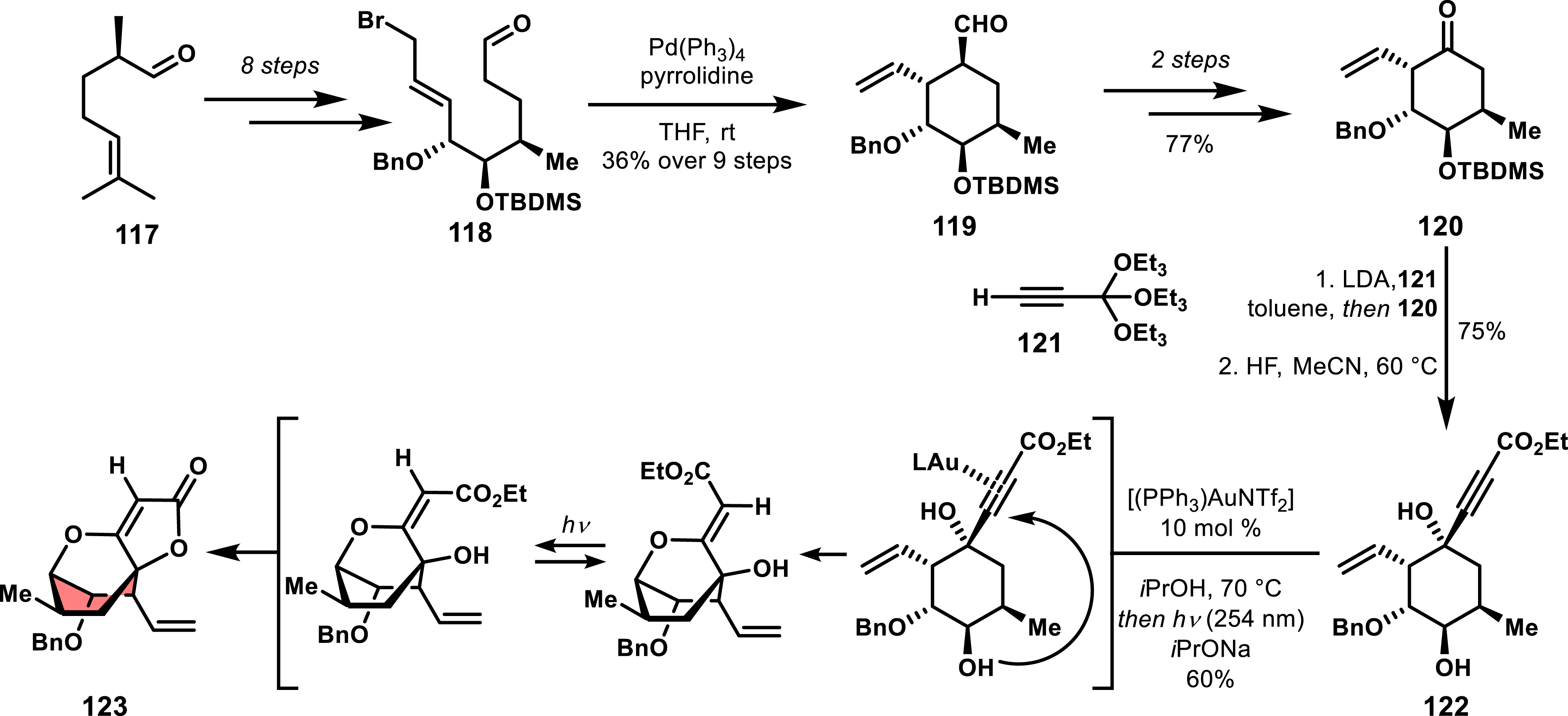
Interestingly in Bihelovic and Saicic’s work, the synthesis of the functionalized cyclohexanone precursor was not established by a cycloaddition strategy (see vide infra) but was achieved through a dual organo/metal-catalyzed Tsuji–Trost cyclization of linear allyl–aldehyde 118 [87]. This key cyclization involves a transient enamine formation that undergoes a cyclative π-allyl palladium displacement. The resulting cyclic aldehyde was next transformed through an oxidation into desired cyclohexanone 120.
3.2. Alternatives to Diels–Alder reaction for cyclohexene ring construction
The Dieckmann condensation of 1-acetoxycyclohexene-1-carboxylate derivatives was largely used when targeting spirotetronates, and the Diels–Alder reaction has been found to be particularly well suited to prepare these precursors. Selected ring-forming approaches, bypassing cycloaddition strategies, were nevertheless also reported.
Roush’s work related to quartromicins is a notable example of such an alternative strategy, wherein enantioselective access to a functionalized cyclohexene precursor is achieved through an intramolecular aldol reaction [88]. The synthesis involves trialdehyde 125 (obtained in 11 steps), which undergoes an intramolecular aldol reaction, transiently forming a cyclohexanol that is trapped by the third aldehyde leading to bicyclic lactol 126 in 54% yield. Elimination of the hydroxy after several steps furnished an entry into the unsaturated cyclohexene that was finally converted through a Dieckmann condensation to spirotetronate (Scheme 25).
Enantioselective synthesis of an endo spirotetronate via an intramolecular aldolization step.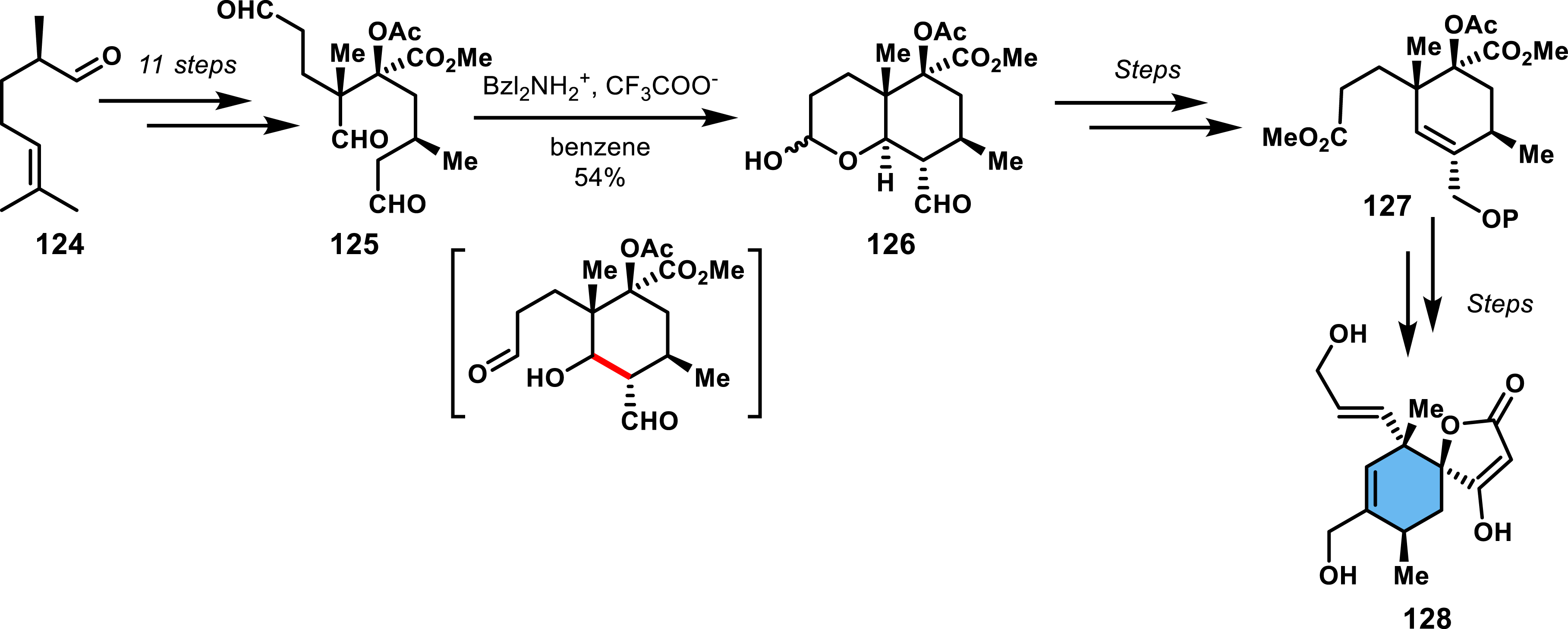
Retrosynthetic disconnections employing RCM have been explored in a few reports, demonstrating their utility in synthesizing functionalized cyclohexenes. In the context of spirotetronates, RCM requires the formation of a β-unsaturated ester, typically synthesized via a Claisen rearrangement. An example of this strategy is found in the work of Bedel and co-workers, who used an Ireland–Claisen/metathesis sequence to generate precursors for quartromicins [89]. More recently, an enantioselective total synthesis of spirohexenolides A and B (5a and 5b) utilized the same strategy, further illustrating the power of this approach [90]. In this synthesis, enantiopure ester 129 undergoes an Ireland–Claisen rearrangement with high yield and diastereoselectivity, driven by a chair-like transition state (Scheme 26). Two adjacent quaternary centers are formed in this step. The subsequent RCM proceeded efficiently, achieving an 88% yield using the Hoveyda–Grubbs second-generation catalyst (HG-II). The succeeding steps included a Dieckmann cyclization to form spirotetronate 132, another RCM to construct the macrocyclic backbone, and an elimination to introduce the unsaturation into the macrocycle [91, 92, 93]1 . An alternative RCM-based strategy targeting versipelostatin involved the formation of a cyclohexenone, followed by the stereoselective addition of a deprotonated alkyne to the ketone [94].
Ireland–Claisen rearrangement followed by an RCM step en route to spirohexenolides.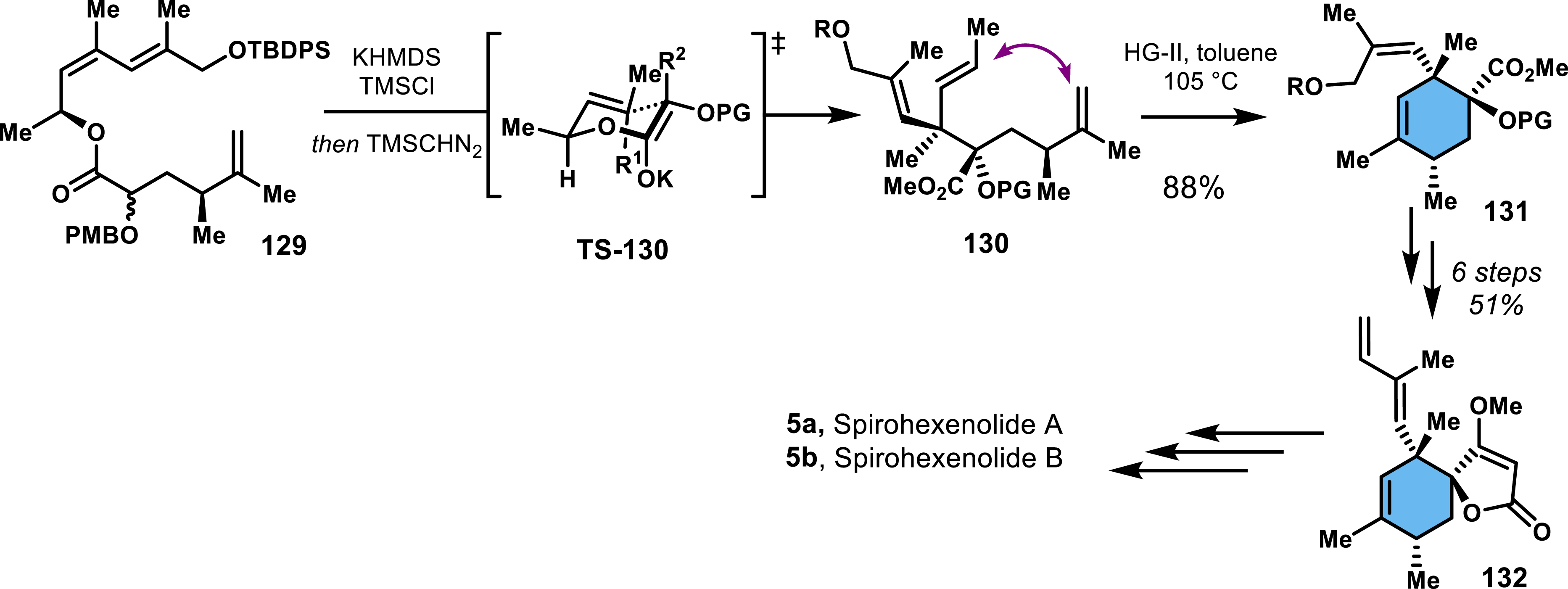
4. Conclusion
Among the strategies discussed, the Diels–Alder cycloaddition between a diene and an acrolein derivative stands out as the most versatile and widely studied approach for spirotetronate synthesis. The ease with which functionalized dienes can be accessed through reactions like Wittig olefination and vinyl lithium addition, combined with the utility of substituted acroleins, makes this method highly efficient for constructing spirotetronate frameworks. However, enantioselective control remains a key challenge. Although asymmetric variants have been developed, they often require pre- and post-functionalization steps to manipulate chiral auxiliaries, adding complexity to the overall synthesis. The reported enantioselective Diels–Alder reaction achieving 72% enantiomeric excess using achiral substrates demonstrates progress, but further refinement is needed to increase both efficiency and stereocontrol.
The diastereoselective IMDA variants, particularly those leveraging biomimetic approaches with tetronate-type dienophiles, show promise for applications. Meanwhile, alternative strategies like the addition of acrylates to cyclohexanone and the use of RCM have shown potential but require further optimization, particularly in improving the selectivity and overall step economy of the synthesis. The recent successes in spirohexenolide synthesis highlight the potential of these methods when effectively applied, but they also underscore the significant challenges related to step count, diastereoselectivities, and enantioselectivities.
Looking ahead, future research should focus on improving the enantioselective control in Diels–Alder reactions by exploring new chiral catalysts or reagents that can enhance selectivity without necessitating multiple auxiliary steps. Additionally, integrating biomimetic strategies with catalytic approaches may offer new avenues to streamline the synthesis of spirotetronates, particularly for complex natural products like spirohexenolides. Further investigation into RCM and the development of novel organocatalytic systems could provide more efficient routes to construct these intricate molecular architectures. Ultimately, the goal will be to reduce the synthetic burden while achieving greater precision in stereoselectivity, enabling broader access to spirotetronate-based natural products and their analogues.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.