



 CC-BY 4.0
CC-BY 4.0
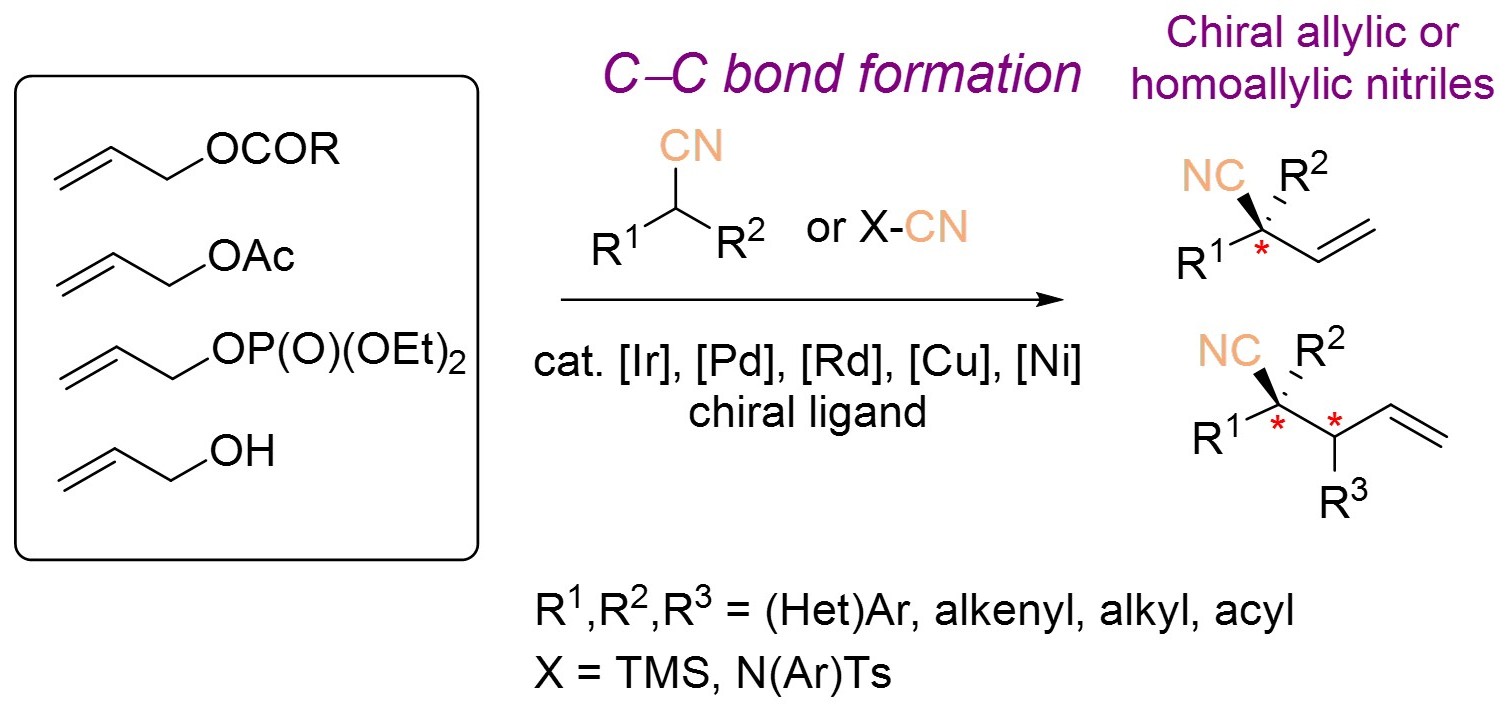
1. Introduction
Recent studies have highlighted the crucial role of three-dimensional architecture in bioactive molecules, and many drugs featuring a quaternary chiral center are derived from natural products (Figure 1) [1]. However, building structures with vicinal tetrasubstituted stereocenters still remains elusive due to steric repulsions occurring at the Csp3–Csp3 bond formation [2, 3, 4]. α-Chiral nitriles are common structural motifs in natural products, pharmaceuticals, and bioactive compounds. Moreover, the cyano group serves as a highly versatile chiral building block, readily convertible into aldehydes, ketones, carboxylic acids, amides, amines, and N-heterocycles. In this context, significant progress has been achieved in developing innovative strategies for the synthesis of Cα-tetrasubstituted or α-quaternary chiral nitriles with diverse architectures. This account summarizes recent advances in this area, with particular emphasis on the formation of homoallylic and allylic nitriles.
Examples of bioactive or natural compounds with quaternary stereogenic centers.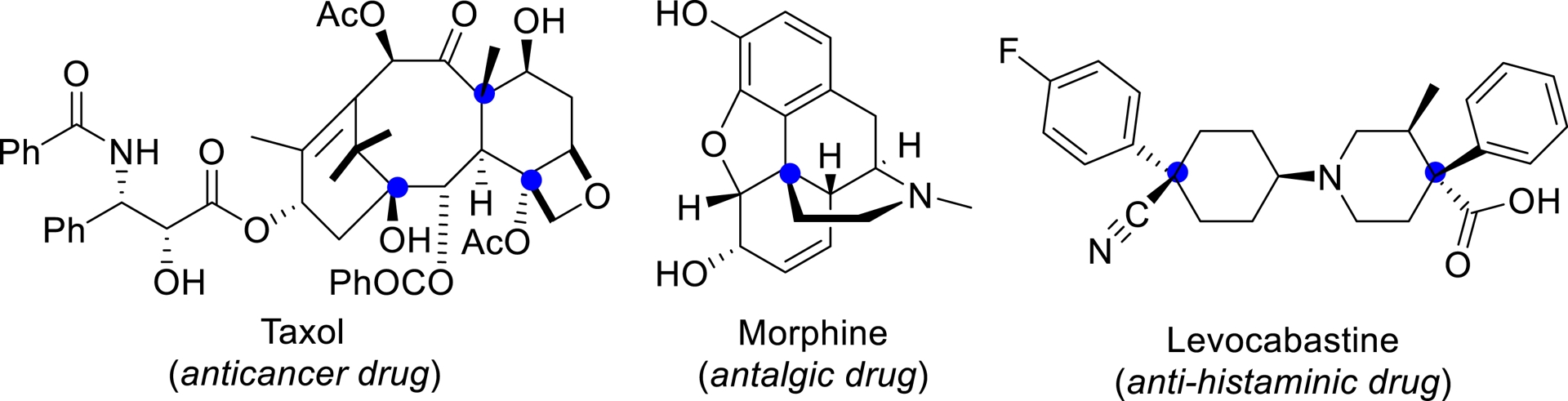
2. Asymmetric allylic alkylation
Since its introduction several decades ago, metal-catalyzed asymmetric allylic alkylation (AAA) has proven remarkably powerful in enabling the enantioselective synthesis of carbon–carbon bonds, making it a cornerstone in modern organic synthesis [5, 6, 7]. Extensive efforts have since been devoted to expanding the scope of nucleophiles, with notable advances achieved using carbon-based nucleophiles [8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24]. While α-carbanions derived from ketones and carboxylic acid derivatives have been effectively employed in Pd-catalyzed AAA [25, 26, 27, 28, 29, 30, 31], reports on the use of nitrile-derived α-carbanions remain scarce. This limitation likely stems from the intrinsic “hardness” of such carbanions and their tendency to interconvert between C- and N-metalated forms [15, 16, 17, 18, 19, 20, 21, 22, 23, 24]. As a result, several refined strategies have emerged that exploit electrophilic trapping of the N-metalated intermediate with silicon, affording axially chiral N-silyl ketene imines (SKIs) [32]. Despite these advances, the direct enantioselective alkylation of an α-metalated nitrile would represent a more straightforward and attractive approach. While diastereoselective alkylation [33] and enantiospecific acylation [34, 35, 36, 37] of such anions have been documented, achieving a truly enantioselective variant has proven elusive, particularly in the case of asymmetric allylation, which is the focus of this Section.
Branch-selective allylic substitution catalyzed by iridium complexes was first disclosed by Takeuchi and Kashio in 1997 (Scheme 1) [38]. Building on this pioneering study, the Takeuchi group reported the enantioselective synthesis of chiral homoallylic nitriles via iridium-catalyzed allylation of cyanoacetates, followed by Krapcho demethoxycarbonylation. This strategy provided a broad array of homoallylic nitriles in excellent enantiomeric excess (>95–99% ee). The resulting products serve as valuable chiral building blocks, as further diversification can be achieved from either the nitrile moiety or the terminal alkene.
Highly selective Ir-catalyzed allylic alkylation with a carbon nucleophile at the more substituted allylic terminus. Redrawn from Ref. [38].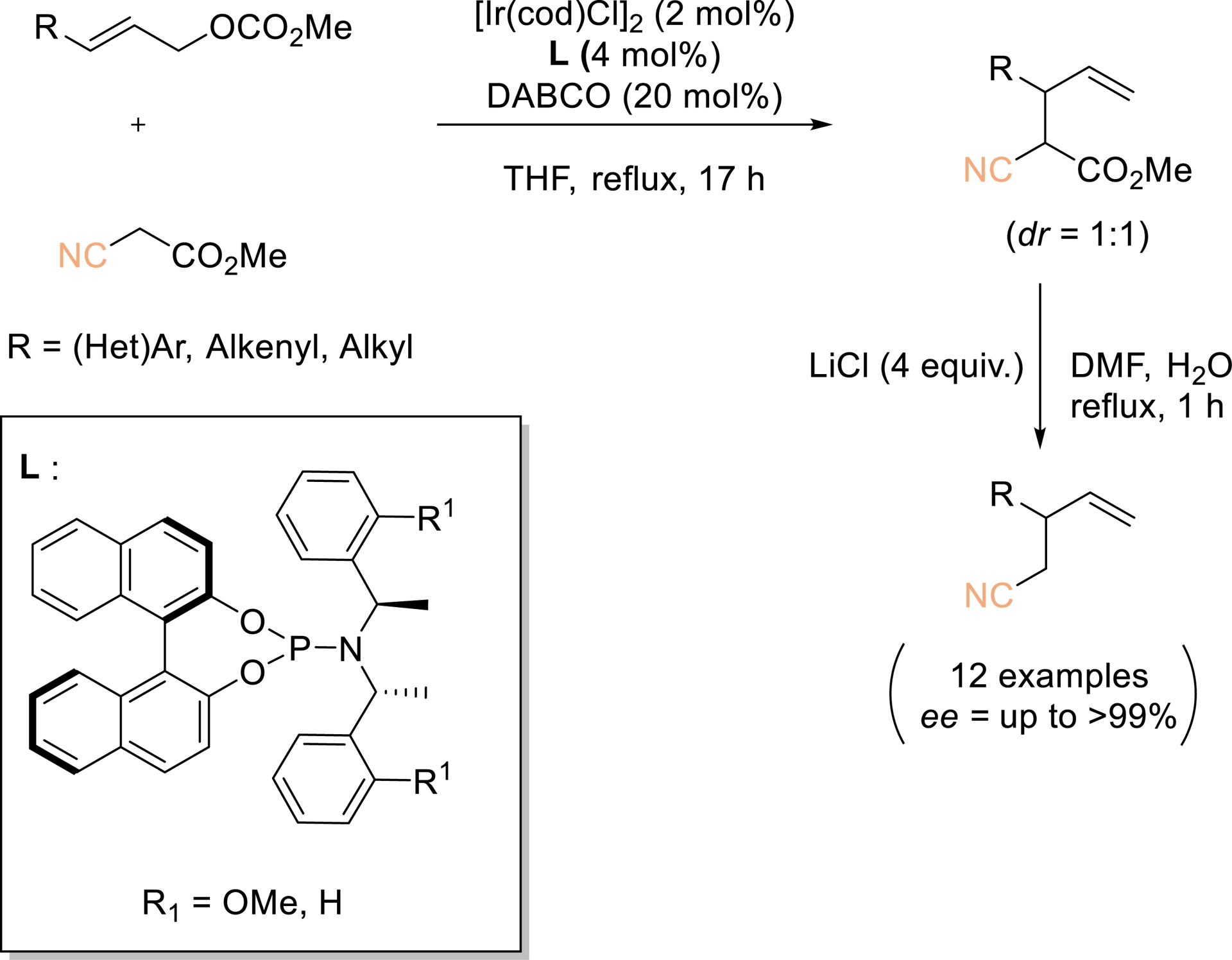
In 2005, Agbossou-Nidercorn demonstrated that Pd-catalyzed allylic substitution with aryl cyanoester pronucleophiles affords allylated products with significant enantioselectivity (ee up to 54%) (Scheme 2) [39]. Although the precise structure of the enolate involved in the addition step remains undefined, it seems to exert a critical influence on the stereocontrol. The noncyclic nature of the substrate likely accounts for the observed enantioselectivity, in line with precedents involving pronucleophiles that generate acyclic enolates. Among the chiral ligands examined, Trost-type “chiral pocket” ligands provided the highest selectivities. Notably, chiral alkylated cyanoesters bearing quaternary stereogenic centers constitute versatile building blocks for asymmetric synthesis.
Pd-catalyzed asymmetric allylic alkylation of prochiral aryl cyanoesters. Redrawn from Ref. [39].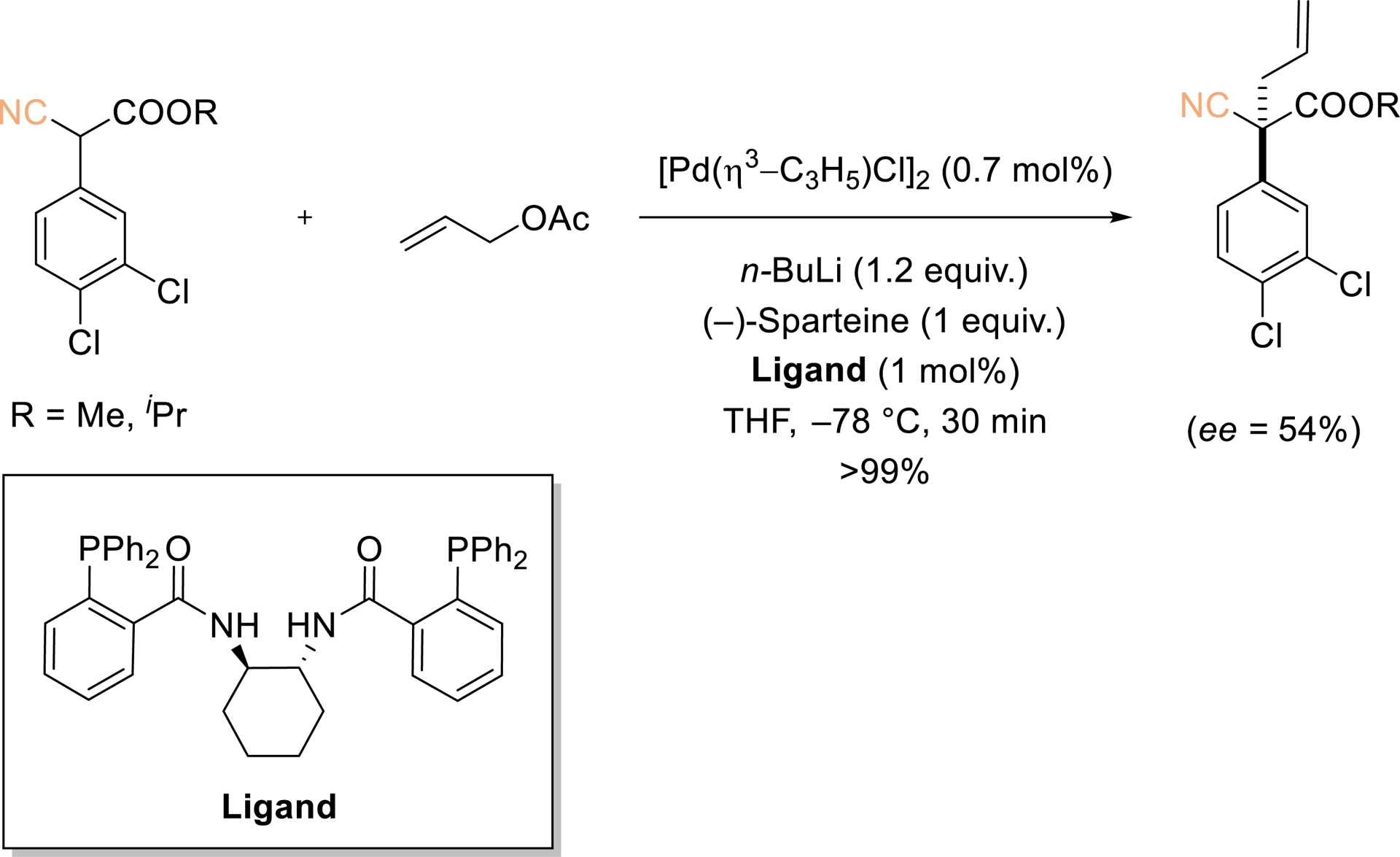
In this context, Sauthier also reported a nickel-catalyzed α-allylation of phenyl- and α-alkyl phenyl acetonitriles using allylic alcohols (Scheme 3) [40]. The reaction proceeded under neutral conditions, using nickel complexes generated in situ from Ni(cod)2 and dppf (1,1′-bis(diphenylphosphino)ferrocene).
Ni-catalyzed α-allylation of phenyl and α-alkyl phenyl acetonitrile with allylic alcohols. Adapted with permission from Ref. [40].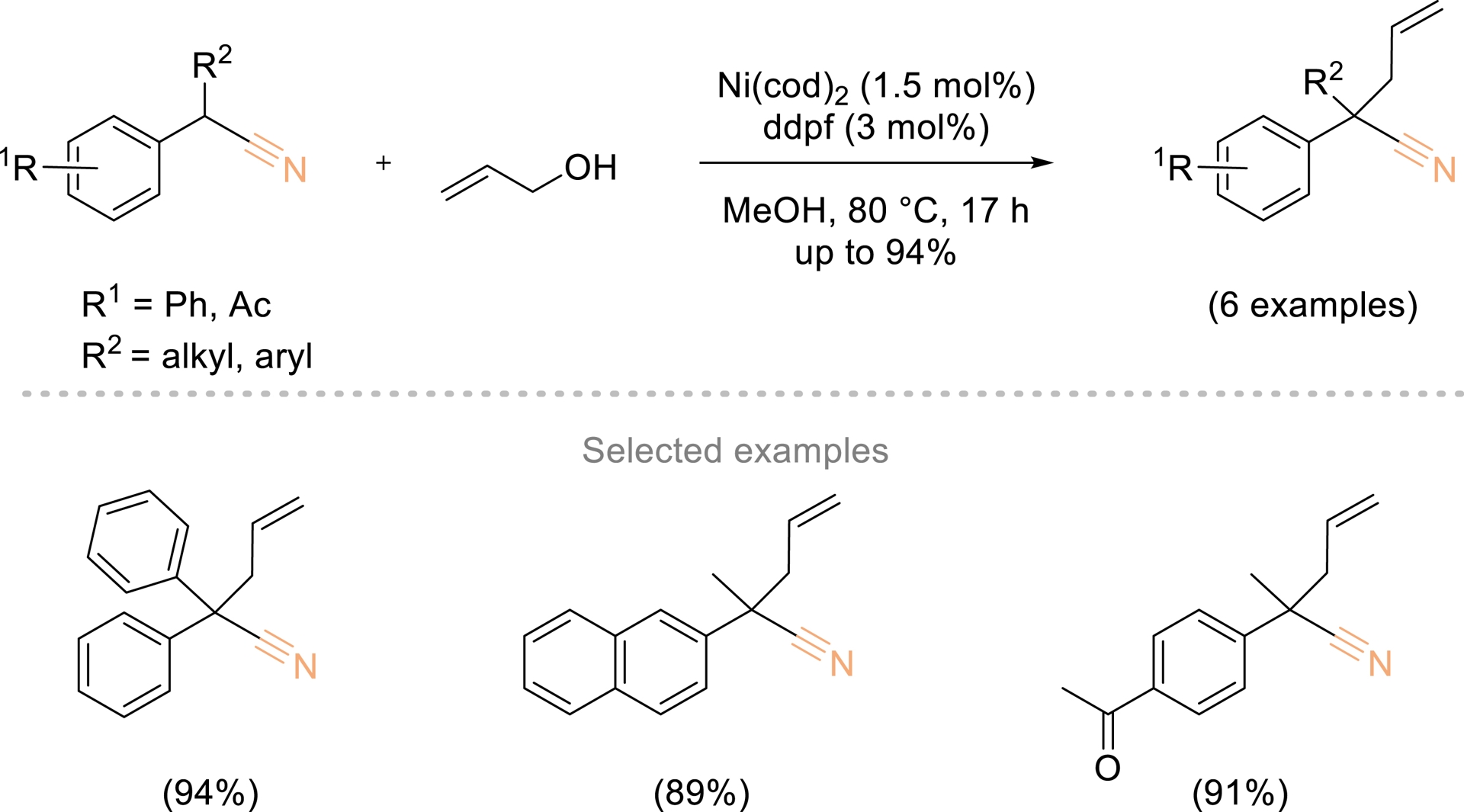
In 2015, Evans et al. described a direct and highly enantioselective rhodium-catalyzed allylic alkylation of allyl benzoate with α-substituted benzyl nitrile pronucleophiles (Scheme 4) [22, 41, 42]. This protocol provided a new approach toward the synthesis of acyclic quaternary carbon stereogenic centers and is the first example of direct asymmetric alkylation of an α-cyanocarbanion. The process is driven by the ability to control the C- versus N-metalated equilibrium with a crown ether. The synthetic utility of the nitrile products is amply demonstrated through conversion to various functional groups and the asymmetric synthesis of the calcium channel blocker (S)-verapamil in three steps and 55% overall yield.
(a) Enantioselective Rh-catalyzed allylic substitution. (b) Modular three-step synthesis of (S)-verapamil. Redrawn from Ref. [22, 41, 42].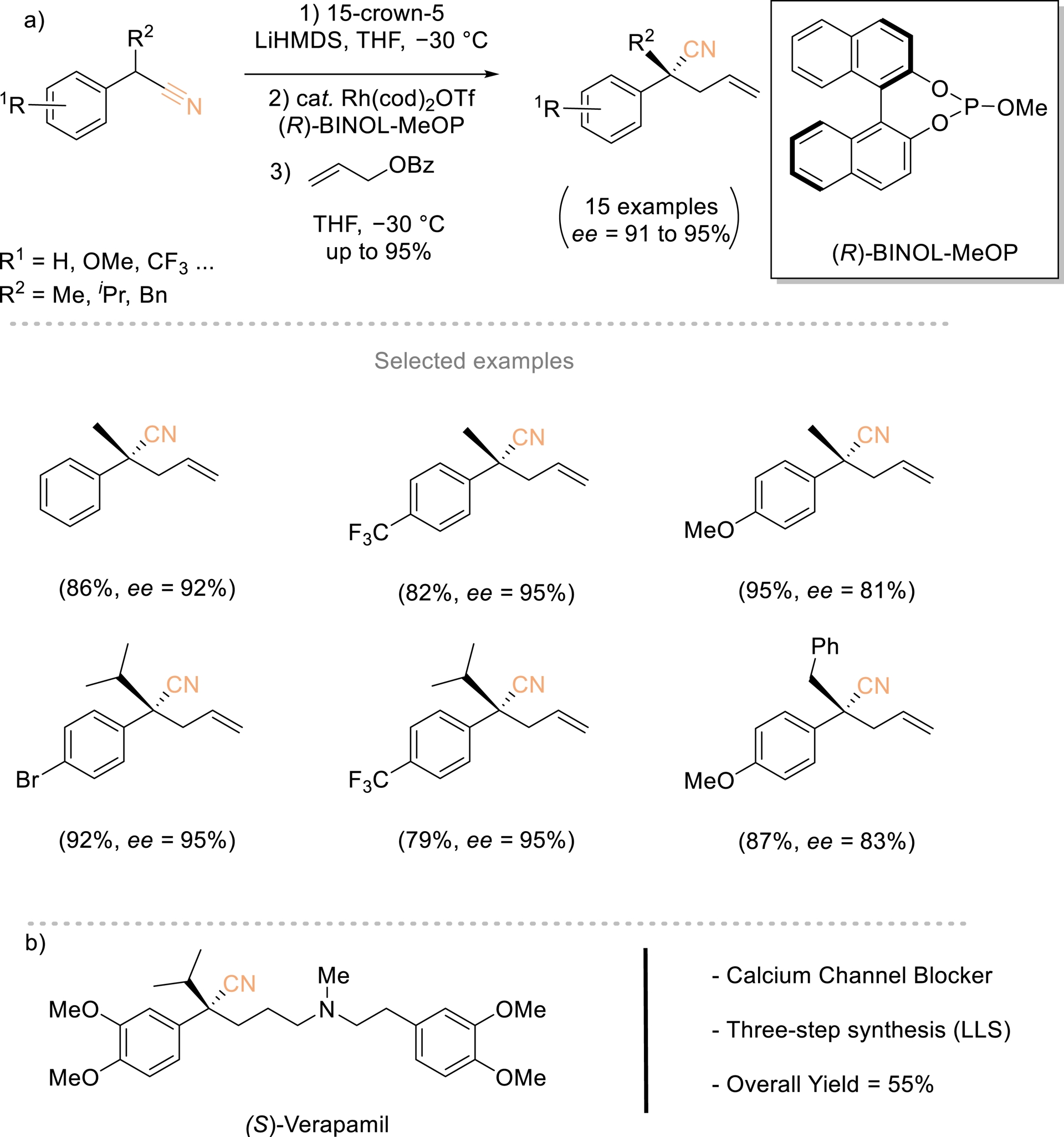
In 2017, Hou and Ding highlighted that the α-carbanion of acetonitrile (pKa ≈ 31 in DMSO) [43] was unreactive with allylic electrophiles [44], failing to yield the expected allylic-alkylation products, which instead underwent self-condensation to form β-enaminonitriles (Scheme 5). A novel nucleophilic species, the 3-imino nitrile carbanion, was subsequently generated in situ via the Thorpe reaction and employed in Pd-catalyzed asymmetric allylic alkylation of monosubstituted allyl reagents using the Pd/SIOCPHOX system. The resulting β-enaminonitriles isolated in high yields with high regio- and enantioselectivity constitute valuable building blocks for the synthesis of heterocycles, polymers, and pharmaceutical compounds. Regarding the reaction mechanism, the authors proposed that nitrile VI arises from the coupling of allyl reagent with nucleophile III, which is generated in situ through the attack of the acetonitrile α-carbanion I on a second acetonitrile molecule, followed by isomerization.
(a) New type of nucleophile in Pd-catalyzed AAA. (b) Proposed mechanism. Adapted with permission from Ref. [44].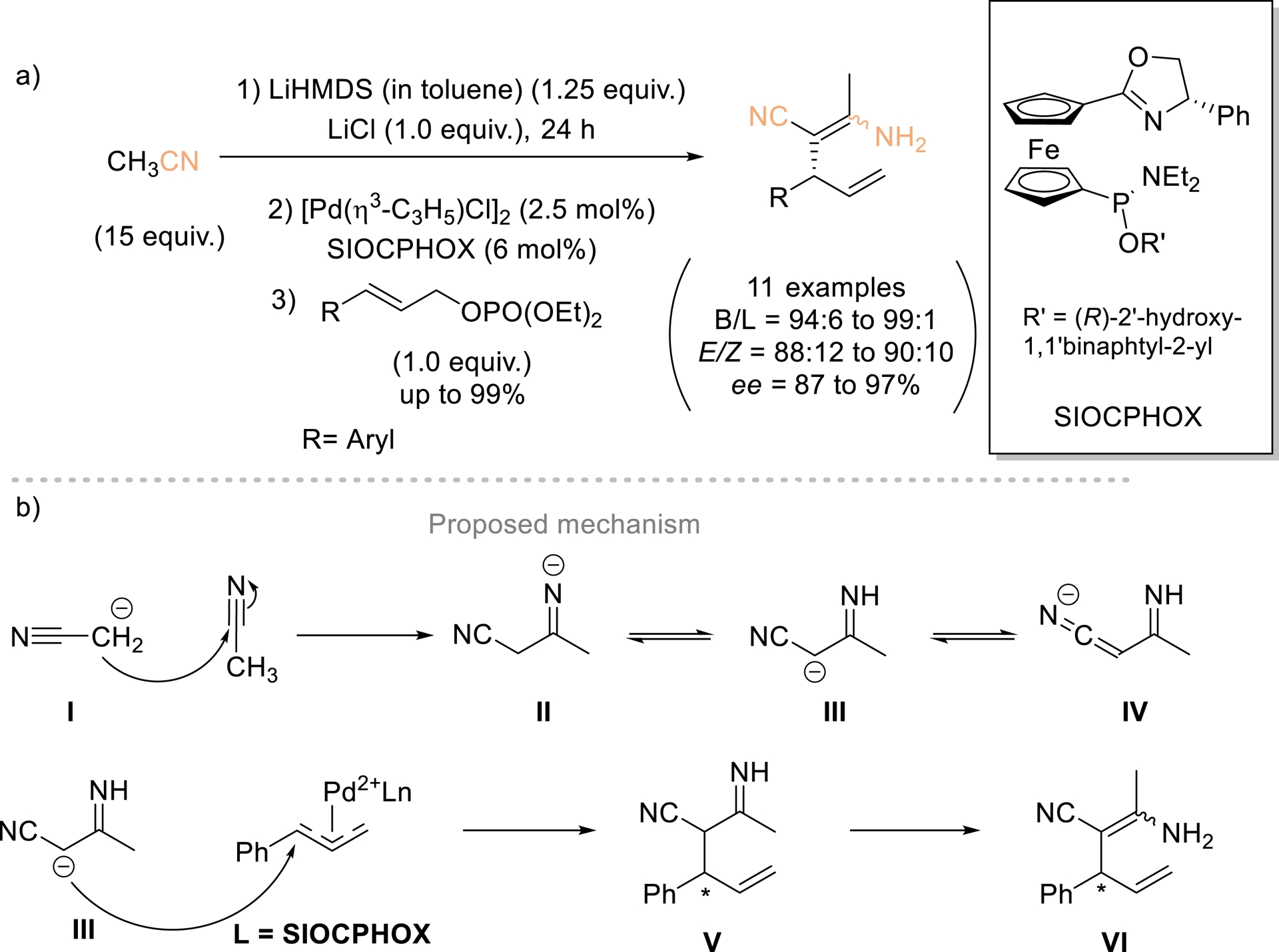
Cao and coworkers developed a method to overcome the side reactions associated with using nitrile α-carbanions as nucleophiles (Scheme 6). This strategy involved iridium-catalyzed enantioselective coupling between vinyl azides and allylic electrophiles [45]. Vinyl azides are cleverly employed as surrogates for acetonitrile carbanions, enabling the synthesis of γ,δ-unsaturated β-substituted nitriles with outstanding enantiomeric excess. These products can be efficiently converted to chiral nitrogen-containing building blocks and pharmaceutically relevant compounds. A mechanism has been proposed to account for the observed chemoselectivity of the coupling process (Scheme 6b). In the presence of a Lewis acid (e.g., BF3⋅OEt2 or a Zn(II) salt), the chiral Ir(I) complex activates allylic carbonate VII through oxidative addition, giving Ir(III) intermediate VIII and [LA–OtBu]−. An enantioselective C–C coupling between VIII and the vinyl azide then produces chiral iminodiazonium ion IX, which carries an α-2-hydroxypropan-2-yl group and then undergoes fragmentation to form X.
(a) Enantioselective Ir-catalyzed coupling reaction of vinyl azides and racemic allylic carbonates. (b) Proposed mechanism. Adapted with permission from Ref. [45].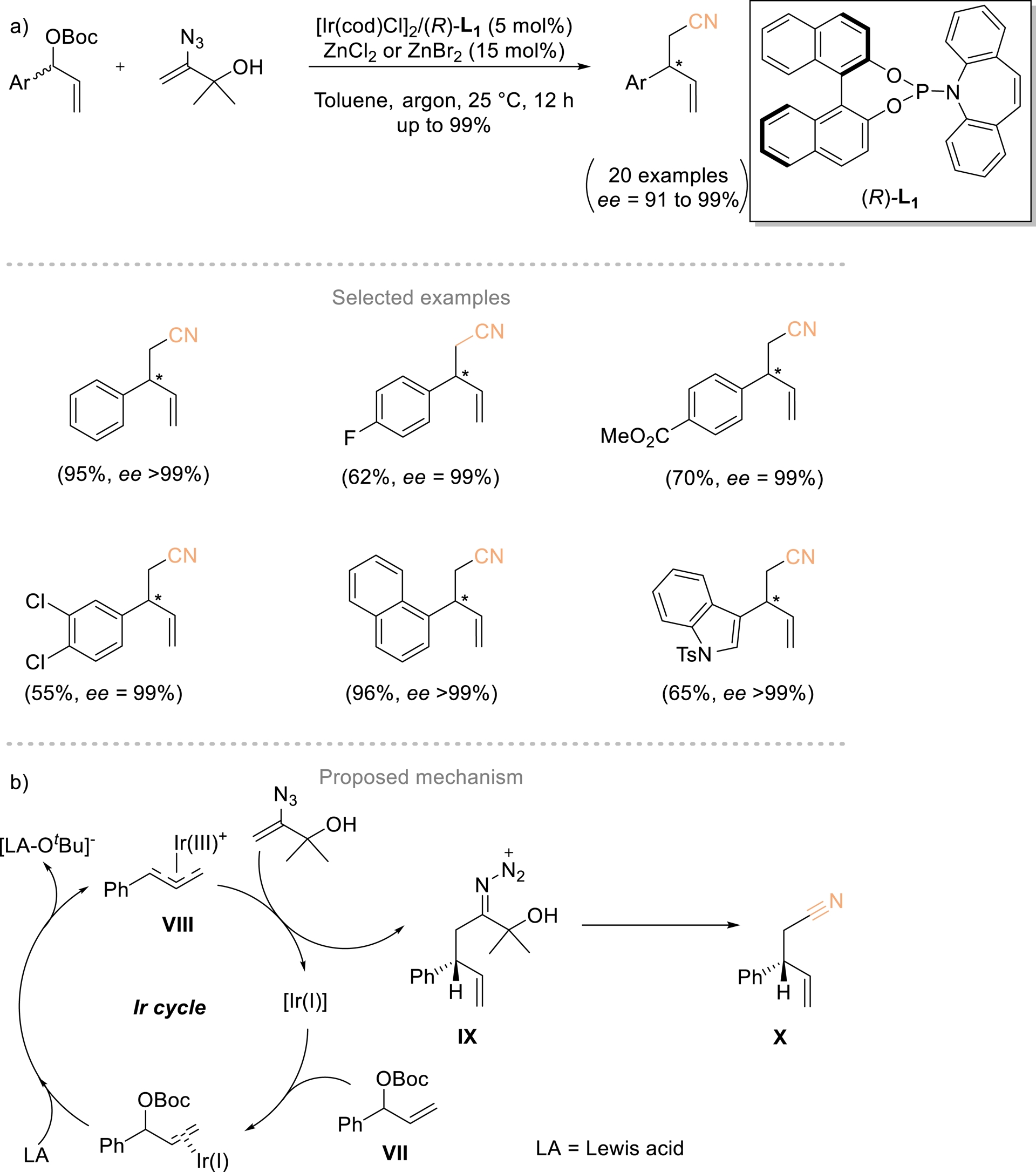
In 2022, a palladium(phosphinooxazoline)-catalyzed method for asymmetric allylic alkylation of α-aryl-α-fluoroacetonitriles was reported by Wolf and Sripada (Scheme 7) [46]. This reaction involves C–C bond formation and generates two contiguous chirality centers with moderate to good yields, high enantioselectivities, and up to 15:1 dr. The procedure is scalable, and it is worth noting that the brominated AAA products can be further derivatized using a Stille cross-coupling reaction without significant HF elimination.
Pd-catalyzed asymmetric allylic alkylation of α-aryl-α-fluoroacetonitriles. Adapted with permission from Ref. [46].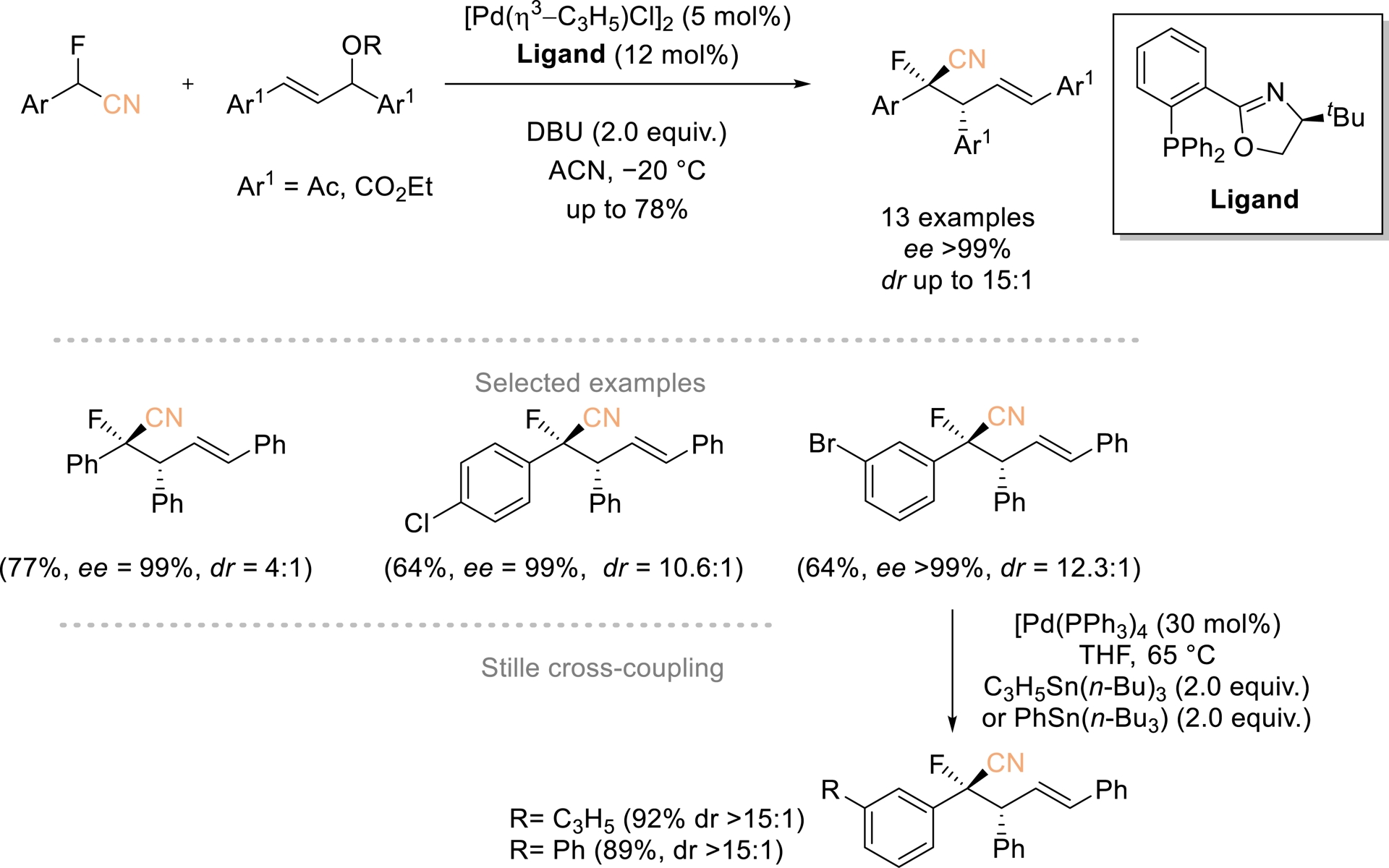
Ito, Sawamura, and Sudoh reported an innovative strategy involving dual catalysis, in which each transition metal complex (Pd and Rh) activates its own substrate (Scheme 8) [47]. The resulting intermediates undergo an enantioselective coupling to afford optically active products. The allylation of diethyl (1-cyanoethyl)phosphonate proceeded with excellent enantioselectivity, yielding an optically active phosphonic acid derivative (ee = 92%). Notably, this remains the only reported example of dual catalysis applied to nitriles. However, in this case, the catalytic system is restricted to the formation of a single quaternary stereogenic center.
Rh/Pd-Catalyzed allylic alkylation of activated nitriles. Adapted with permission from Ref. [47].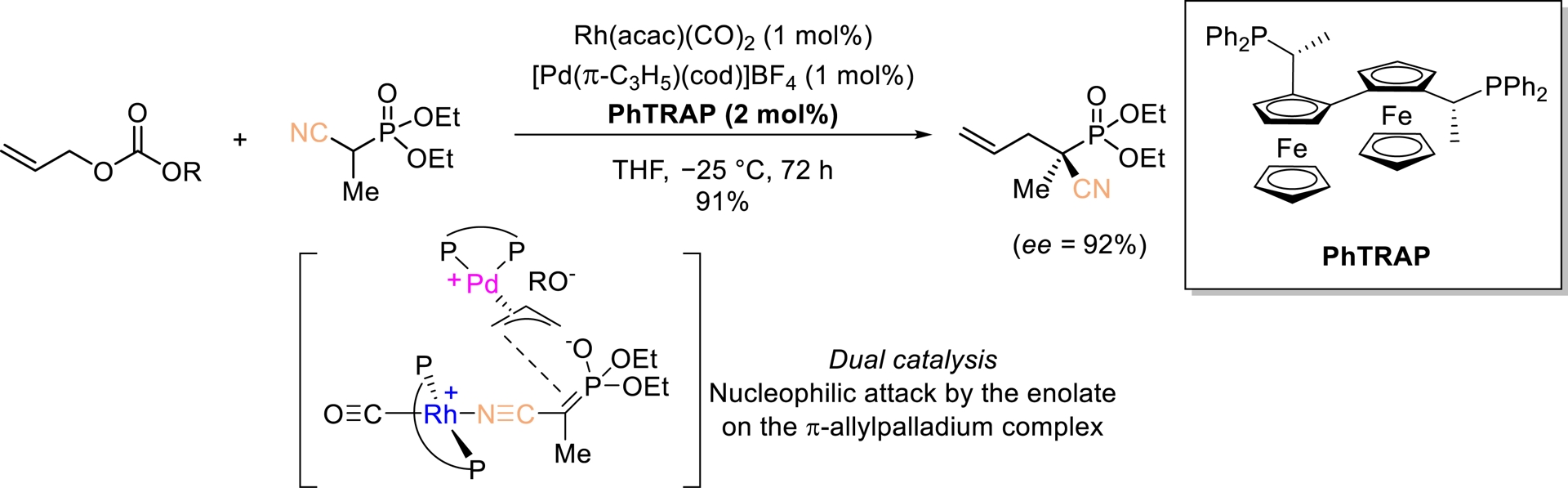
Gillaizeau and Nicolas et al. established an enantioselective strategy for constructing unprecedented homoallylic nitriles bearing vicinal stereogenic centers through an allylic alkylation process (Scheme 9) [48]. This transformation relied on a dual catalytic system in which a palladium catalyst controls the configuration of the allylic electrophile, while a copper catalyst governs the stereochemistry of the nucleophilic carbon via an N-MKI (metal ketenimine) active species XI. This strategy delivers nitrile-substituted products bearing vicinal tertiary and quaternary centers in up to 99% yield and 91% ee. All four stereoisomers are accessible, underscoring independent stereocontrol at both reactive sites (Scheme 9b). The reaction is readily scalable without compromising efficiency. Computational studies highlighted the pivotal role of the copper catalyst and attributed the limited diastereoselectivity to an orthogonal alignment of the ketenimine and allyl moieties within the key intermediate.
Dual Pd/Cu-catalyzed enantioselective synthesis of chiral homoallylic nitriles bearing vicinal stereogenic centers. Redrawn from Ref. [48].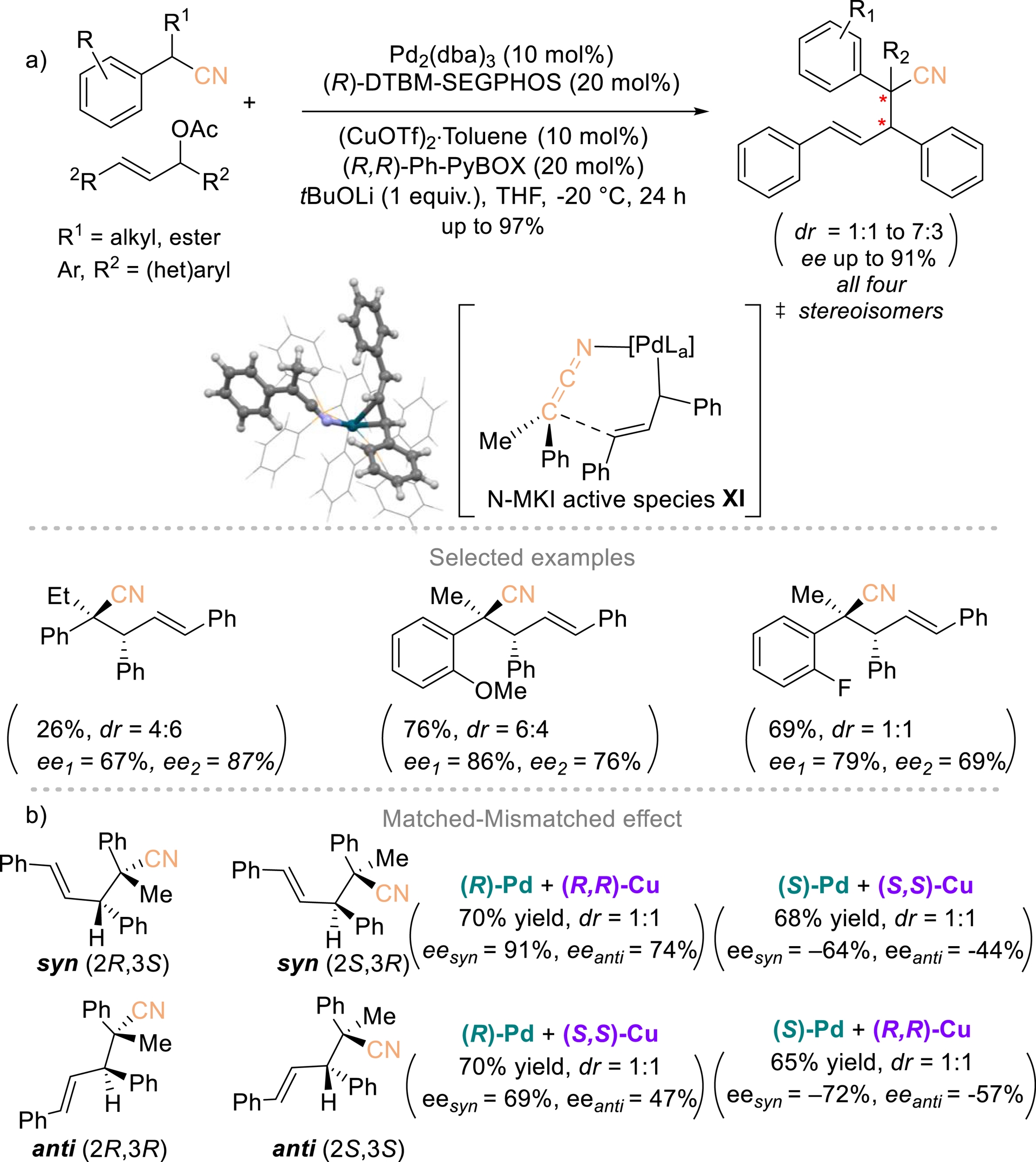
3. Metal-catalyzed conversion of allylic alcohols to nitrile
The transition-metal-catalyzed allylic substitution of diverse allylic substrates has emerged as a highly versatile transformation, particularly for constructing C–C bonds. This process typically relies on activated allylic precursors, such as acetates or carbonates, which promote the formation of η3–M complexes. The direct use of allylic alcohols is particularly attractive, as it avoids the generation of stoichiometric waste and eliminates the need for hydroxyl protection [49, 50]. In this regard, Tsuji and coworkers first established allylic cyanation using TMSCN as the cyanide donor and Pd(PPh3)4 as the catalyst [51]. Subsequent advances were made by Kawatsura et al., who employed CuI as the catalyst [52], and by Rousseaux et al., who developed a Ni(II)-based precatalyst in combination with Zn(CN)2 as the cyanide source [53]. Koert et al. also reported that rhodium-catalyzed allylic cyanation of 3-fluoroallylic trifluoroacetates with TMSCN in the presence of a bulky phosphite ligand provides 3-fluoroallylic nitriles with a broad range of substrates and excellent Z/E selectivity [54]. In dialkyl-substituted substrates, the fluorine atom governs the regioselectivity, favoring the formation of the distal product. Investigation of selected scalemic substrates indicated stereocenter inversion during cyanation, accompanied by minimal or no stereochemical erosion. Despite extensive studies on asymmetric allylic substitution, the enantioselective synthesis of allylic cyanides remains underdeveloped. In 1998, Tsuji reported the catalytic enantioselective cyanation of cyclic allylic carbonates with TMSCN using 5 mol% of the (S)-MeO–MOP–Pd complex, yielding only two chiral allylic cyanides with 19% and 63% ee, respectively (Scheme 10, Equation (1)) [55]. Later, Hou and coworkers demonstrated an asymmetric kinetic resolution of racemic 1,3-disubstituted unsymmetrical allylic carbonates by Pd-catalyzed allylic cyanation, affording allylic cyanides in 31–50% yields with 25–65% ee, alongside recovered chiral carbonates with an (S) configuration in 21–54% yields and with 24–99% ee (Scheme 10, Equation (2)) [31].
Pd-Catalyzed asymmetric allylic cyanation of allylic carbonates using TMSCN. Adapted with permission from Ref. [55].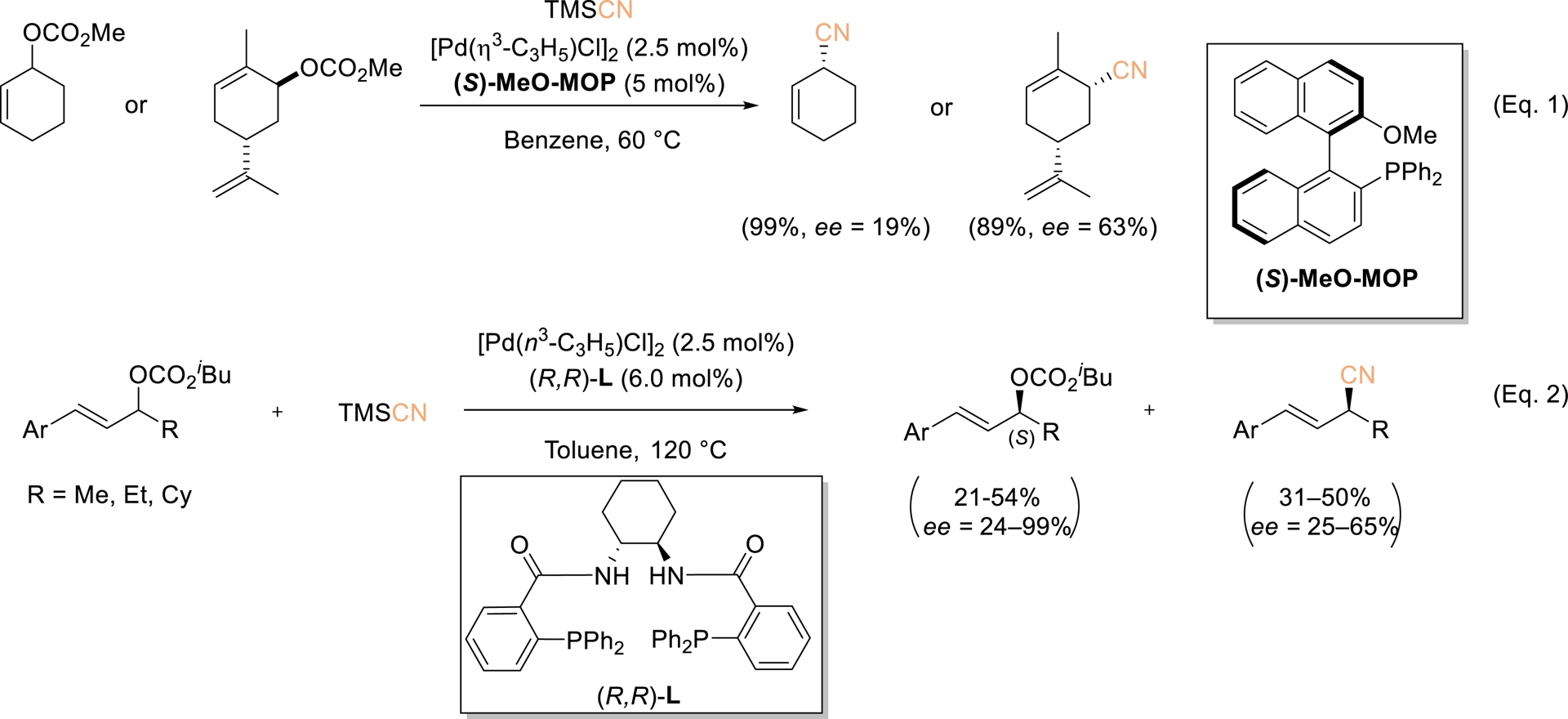
The Fang group described a one-pot conversion of allylic alcohols into chiral 1,3-dinitriles by merging allylic cyanation with asymmetric hydrocyanation through an auto-tandem catalysis strategy (Scheme 11) [56, 57]. Using 5 mol% of a nickel complex bearing a chiral bidentate phosphine ligand, a broad range of allylic alcohols reacted efficiently with acetone cyanohydrin as the cyanide donor to afford the corresponding enantioenriched 1,3-dinitriles in 37–89% yield and 65–94% ee. The proposed mechanism involves initial coordination of Ni(0)–L∗ species XII with the allylic alcohol to form a π-allyl–Ni(II)–OH intermediate XIV, which subsequently reacts with acetone cyanohydrin to generate intermediate XV. Reductive elimination delivers allyl nitrile XVI, which undergoes asymmetric hydrocyanation to furnish target dinitriles XX while regenerating active catalyst XII. These chiral dinitriles can be further transformed into piperidines XXI, a key step enabling the formal synthesis of the antidepressant (+)-paroxetine.
One-pot conversion of allylic alcohols to chiral 1,3-dinitriles. Adapted with permission from Ref. [57].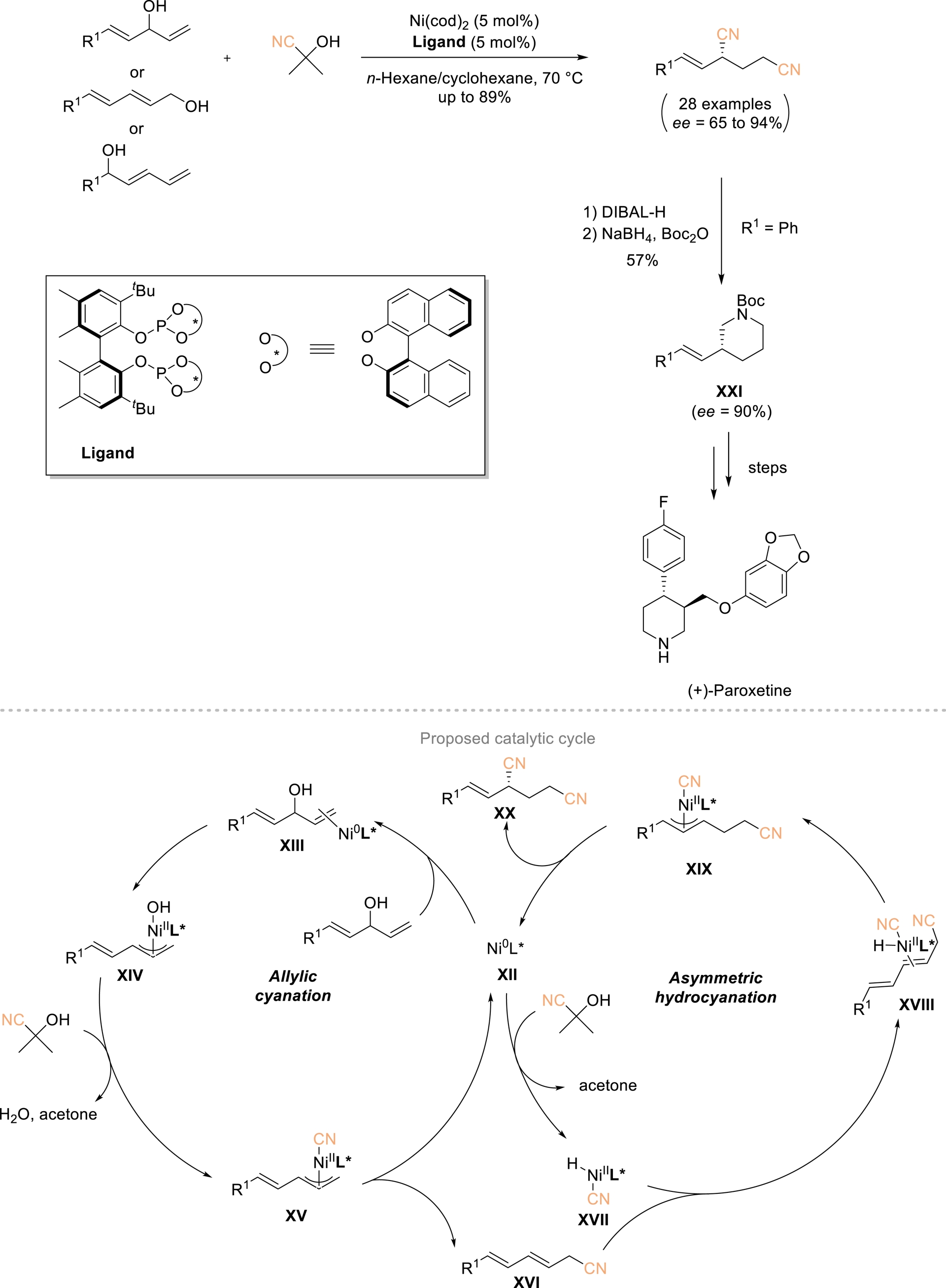
The same group reported a Ni-catalyzed asymmetric hydrocyanation of racemic allene- and methylene cyclopropanes (Scheme 12, Equation (1)). Using 10 mol% (R,R)-Ph-BPE with Ni(cod)2, it delivered enantioenriched allylic nitriles in up to 78% yield and 98% ee with excellent regioselectivity. Subsequently, a nickel catalyst based on a TADDOL-derived diphosphate (L1) enabled highly asymmetric hydrocyanation of disubstituted methylene cyclopropanes, providing chiral allylic nitriles in 82–99% ee (Scheme 12, Equation (2)) [58].
Ni-catalyzed asymmetric hydrocyanation of allene- and methylene cyclopropanes. Adapted with permission from Ref. [58].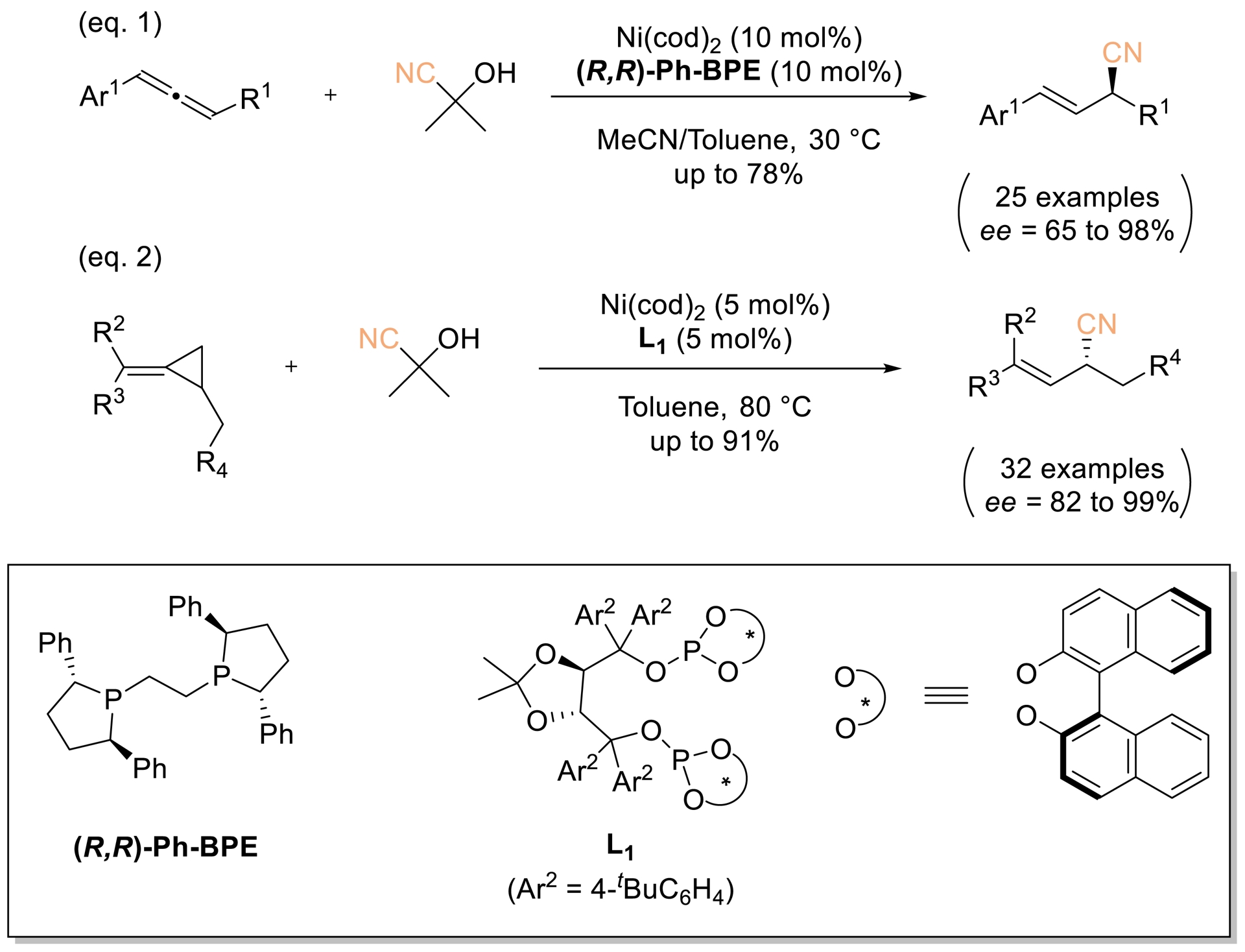
A nickel-catalyzed direct transformation of allylic alcohols with benzyl nitrile derivatives was also developed by Liu et al. in 2023, offering an efficient and racemic approach to access homoallylic nitriles featuring α-tertiary or quaternary carbon centers (Scheme 13) [59]. This protocol delivered good to excellent yields with high regioselectivity, broad substrate applicability, and high tolerance toward diverse functional groups. Notably, N,O-bis(trimethylsilyl)-acetamide (BSA) was identified as a key additive that significantly promotes the coupling, particularly in reactions involving alkyl-substituted allylic alcohols.
Ni-Catalyzed allylation of nitriles with allylic alcohols. Adapted with permission from Ref. [59].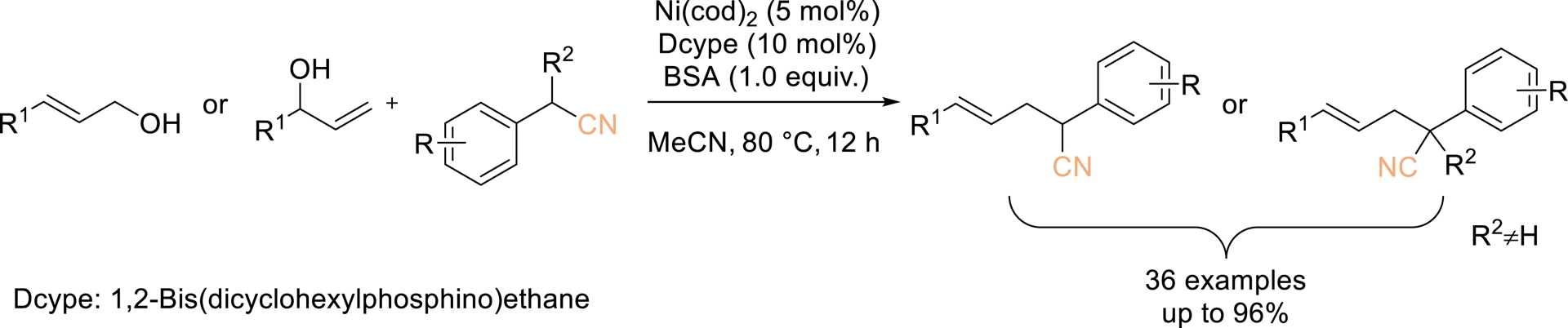
4. Metal-catalyzed cyanofunctionalization of unsaturated bonds
Copper has been shown to mediate cyanation of the π-bond in nitriles through C–CN activation [60]. Procter and co-workers developed a regio- and enantioselective Cu-catalyzed borocyanation of dienes using bis(pinacolato)diboron (B2pin2) and electrophilic cyanamides (Scheme 14) [61]. With a P,N-ligated copper complex (10 mol%) and Cu(I) thiophene-2-carboxylate (CuTC) (10 mol%), allylic nitriles bearing a Bpin substituent were obtained in up to 90% yield and 94% ee. The reaction is proposed to proceed through selective borylcupration of the terminal alkene, isomerization to (Z)-allyl copper intermediate XXIV, and subsequent cyanamide insertion. Despite high efficiency with simple dienes, this led to a decrease in reactivity and selectivity with multisubstituted substrates.
Enantio- and regioselective Cu-catalyzed borocyanation of 1-aryl-1,3-butadienes. Adapted with permission from Ref. [61].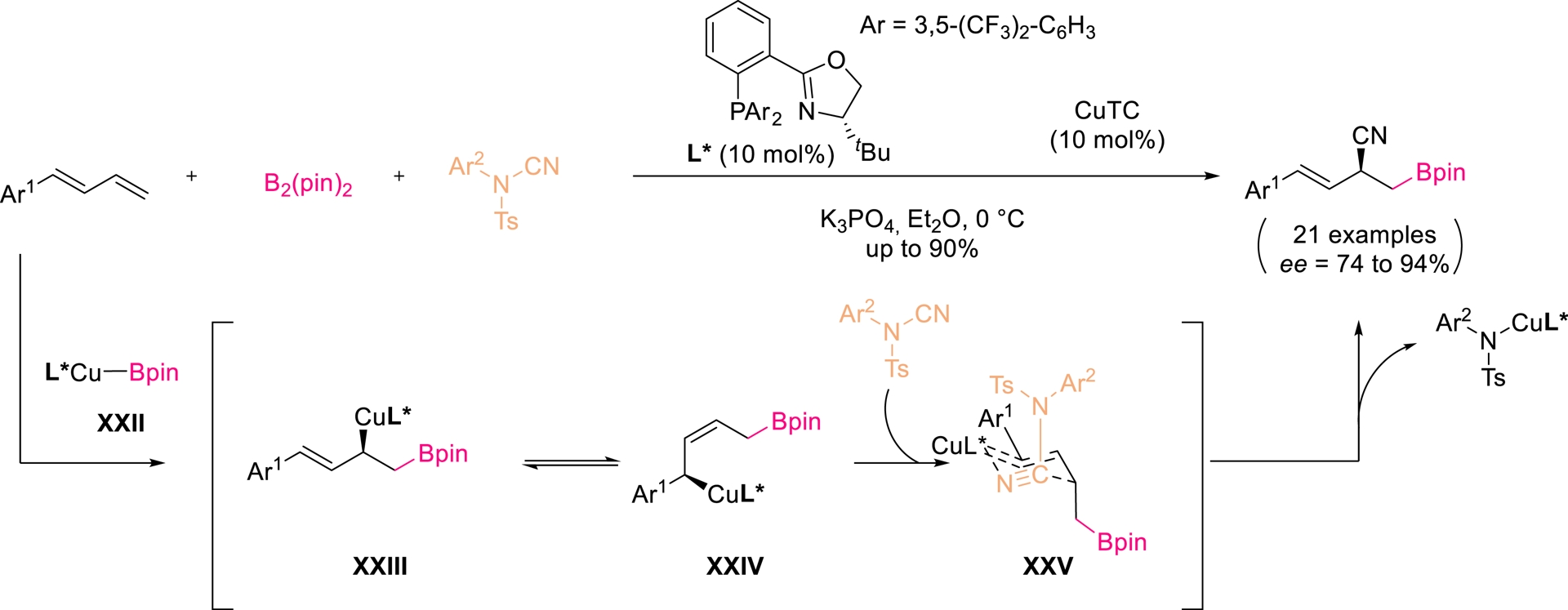
5. Site-selective C(sp3)–H cyanation of alkenes
While traditional asymmetric cyanation typically employs unsaturated substrates, recent advances have revealed that allylic C(sp3)–H bonds can also serve as radical precursors suitable for enantioselective cyanation. In 2019, Li et al. further addressed the challenge of trapping allylic radical with chiral Cu(II) cyanide species, achieving site-selective and enantioselective allylic C(sp3)–H cyanation of diverse di-, tri-, and tetrasubstituted alkenes via hydrogen atom transfer (HAT) pathways [62]. Using N-fluoroalkylsulfonamide (NFAS) as a precursor of the N-centered radical (NCR), the authors successfully realized allylic cyanation of trisubstituted alkenes bearing two distinct sets of allylic hydrogens. Under catalysis by 5 mol% of a chiral bisoxazoline L*/CuOAc complex, a diverse array of chiral allylic nitriles was obtained in yields of up to 91% and enantiomeric excesses as high as 99% (Scheme 15). Mechanistic studies suggested that the in situ—generated Cu(II)-bound NCR XXVI plays a decisive role in controlling site selectivity among multiple allylic C(sp3)–H bonds with similar properties. This method features wide substrate generality and excellent chemo-, regio-, and enantioselectivity. Its synthetic utility was underscored by diverse downstream derivatizations.
Enantioselective allylic cyanation of alkenes. Redrawn from Ref. [62].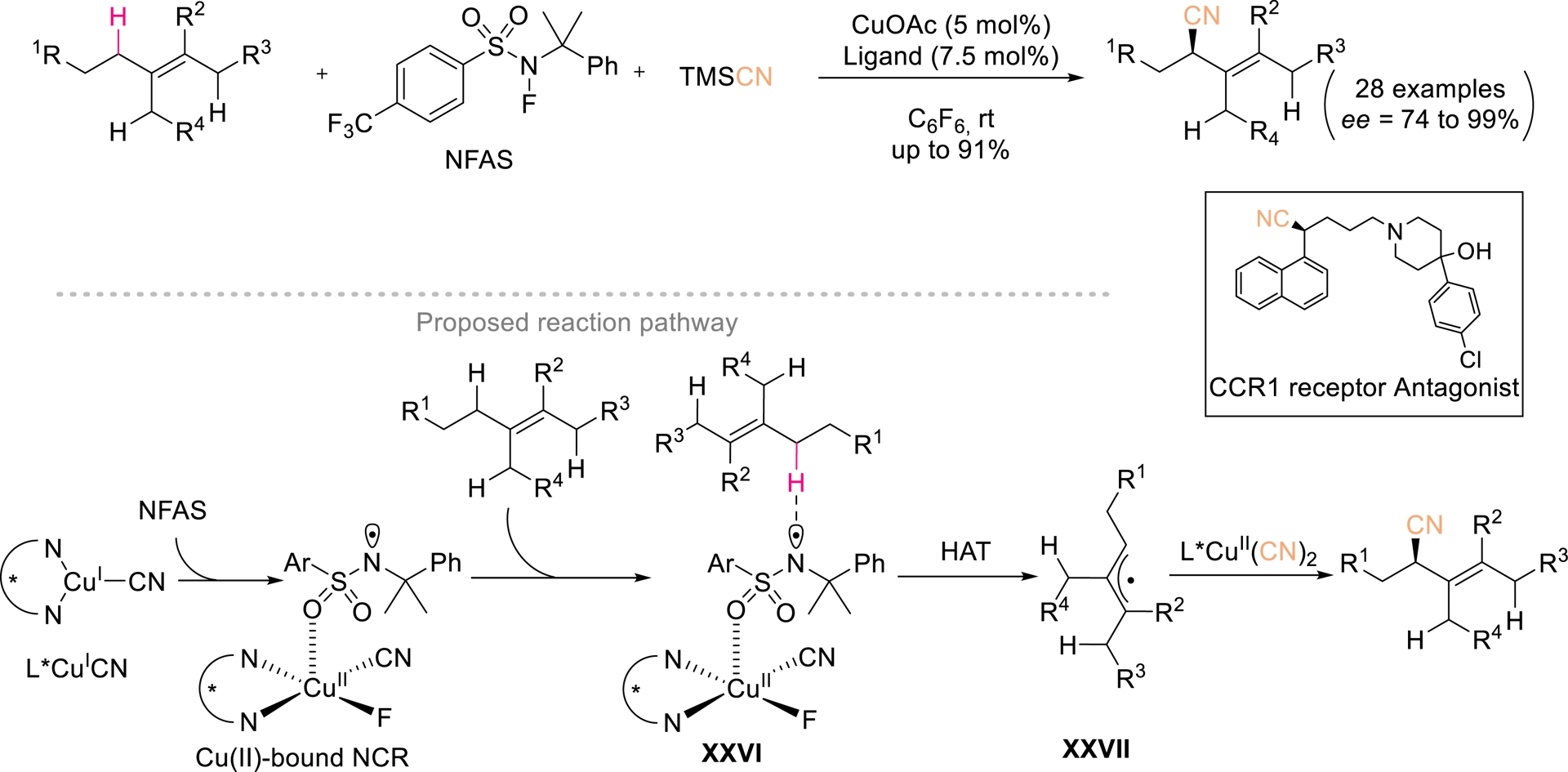
6. Intramolecular palladium-catalyzed decarboxylative allylic alkylation
Tan, Luan, and Ren outlined a practical strategy for the asymmetric synthesis of chiral acyclic nitriles bearing α-all-carbon quaternary stereocenters, employing a synergistic combination of palladium and phase-transfer catalysis on allyl 2-cyanoacetates under mild conditions (Scheme 16) [63]. This method provides an efficient and reliable route for the in-situ generation of tertiary α-cyano carbanions via an intramolecular palladium-catalyzed decarboxylative allylic alkylation. Moreover, it allows for exceptional enantioselective control of simple nitriles through ion-pairing interactions with chiral phase-transfer catalysts. The versatility of this approach was further highlighted by its scalability to gram-scale synthesis and its subsequent conversion into a range of chiral functionalized molecules containing acyclic all-carbon quaternary stereocenters. A plausible reaction mechanism was proposed (Scheme 16). The starting substrate is first activated by Pd(0), releasing CO2 and generating electrophilic [η3-π-ally-PdLn]+ species XXVIII and tertiary α-cyano carbanion XXIX, facilitated by Cs2CO3. Subsequently, tertiary α-cyano carbanion XXIX then associates with PN4 to form chiral ion pair XXX. Enantioselective nucleophilic attack by the α-cyano carbanion in XXX on the electrophilic species XXVIII furnishes the desired enantioenriched final homoallylic nitrile, while regenerating the palladium active catalyst for subsequent cycles.
Enantioselective construction of acyclic α-all-carbon quaternary nitriles via synergistic intramolecular Pd-catalyzed decarboxylative AAA and phase-transfer catalysis (PTC) from allyl 2-cyanoacetate substrates. Adapted with permission from Ref. [63].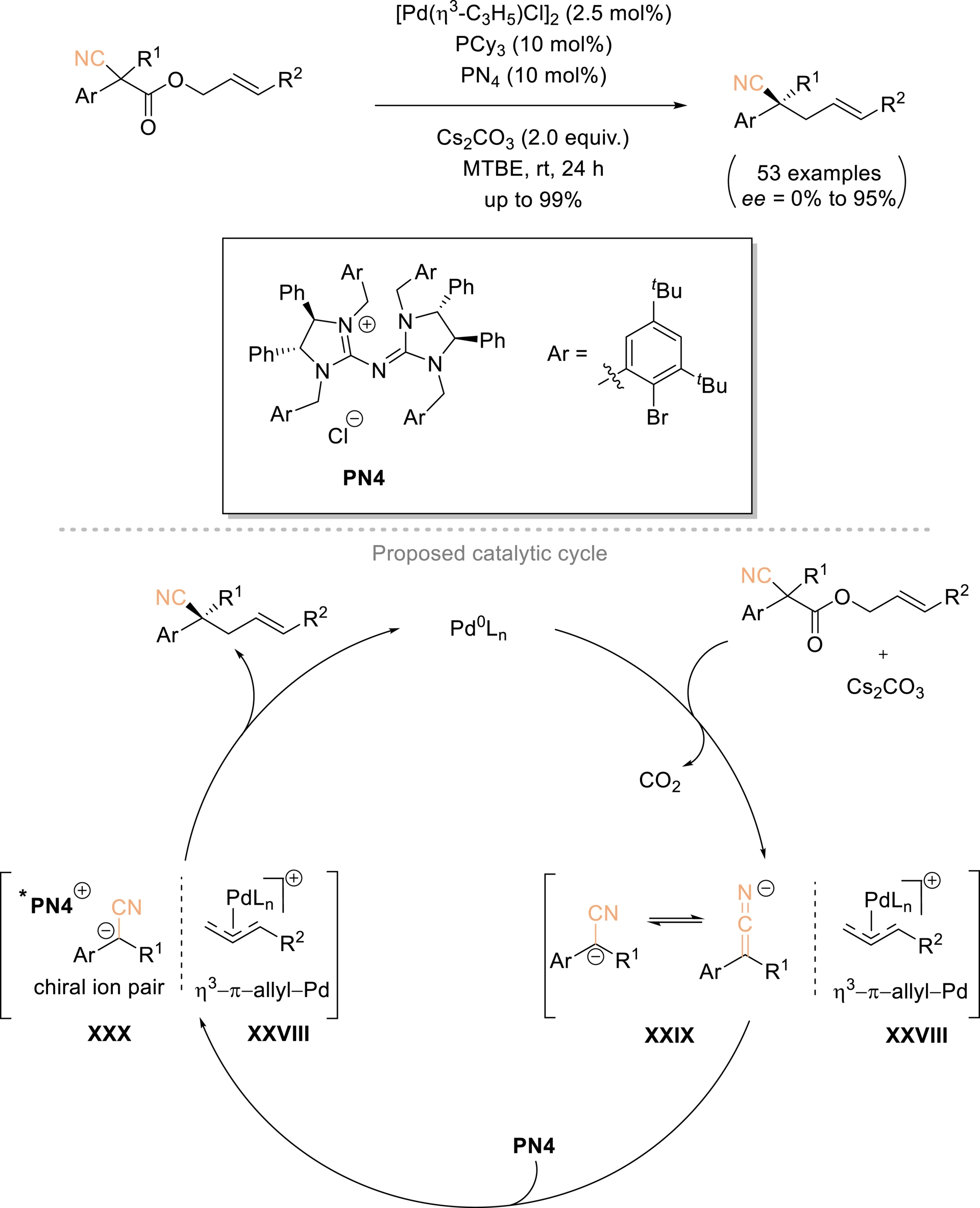
7. Asymmetric deacylative allylation
The use of readily available prochiral or racemic quaternary carbons to generate enantioenriched counterparts offers an efficient alternative to conventional syntheses from tertiary or planar substrates. Unlike desymmetrization, which modifies an existing substituent of limited reactivity, functional-group exchange introduces a new, structurally distinct motif. Achieving enantioconvergence in quaternary-to-quaternary transformations remains challenging, particularly for acyclic centers. Huang and coworkers reported a palladium-catalyzed deacylative allylation in which the acyl group of β-ketonitriles was stereoselectively replaced by an allyl fragment using simple alcohols as alkyl donors (Scheme 17) [64]. The transformation proceeded via a retro-Claisen-type elimination with alkoxide, and the absence of diastereoisomerism in the resulting ketenimine anion enabled highly selective asymmetric addition. The combination of α-substituents, retained nitrile, and introduced alkyl unit confers broad derivatization potential to the resulting enantioenriched quaternary stereocenters.
Enantioconvergent deacylative functionalization toward α-quaternary nitriles. Redrawn from Ref. [64].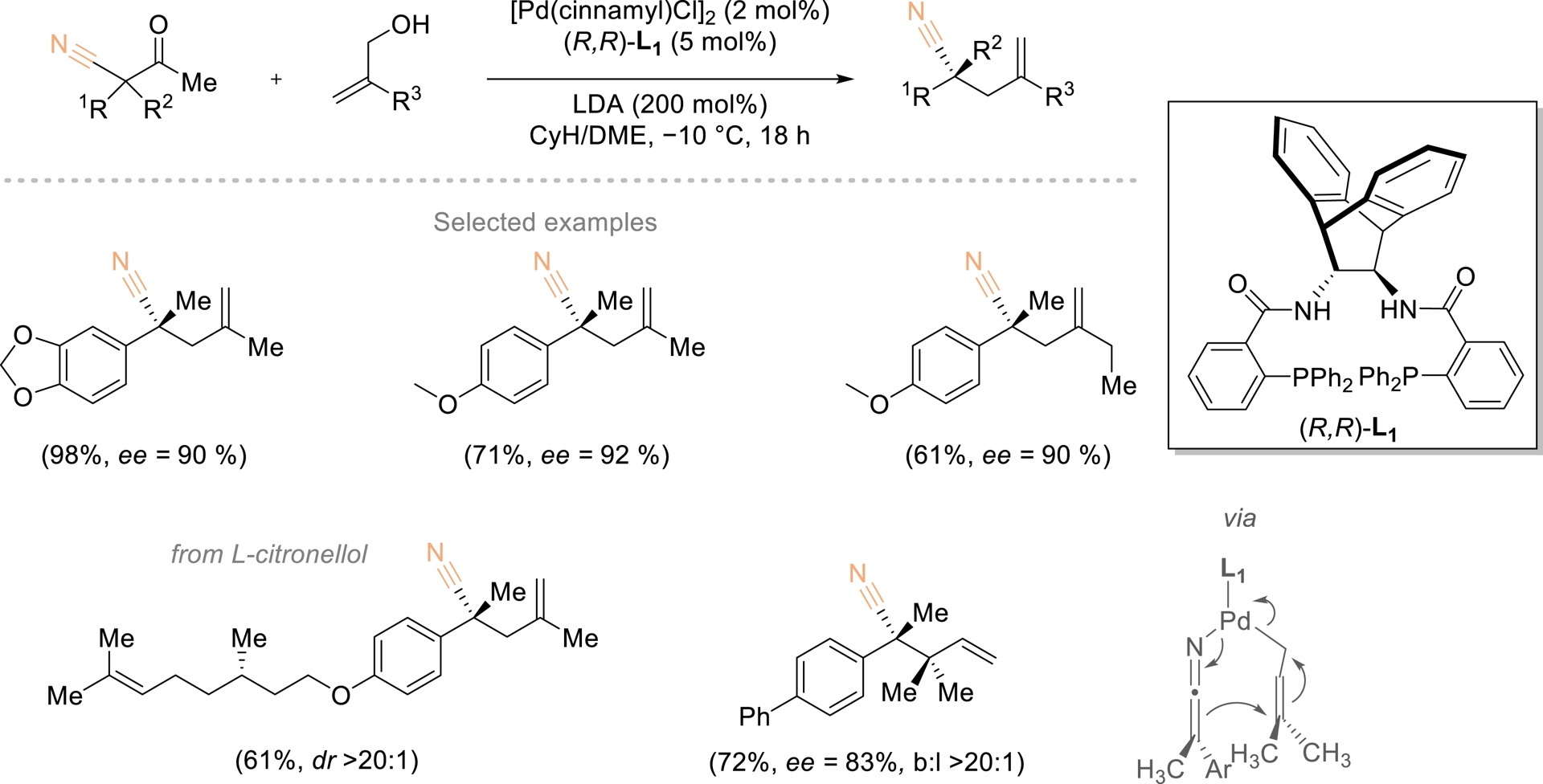
8. Conclusion
Over the years, significant progress has been made in the enantioselective synthesis of homoallylic and allylic nitriles through transition-metal-catalyzed allylation, enabling access to Cα-tetrasubstituted and α-quaternary chiral nitriles with diverse architectures. These advances have broadened the chemical space relevant to both academic and industrial research. Nevertheless, this account highlights the continuing need for enantioselective strategies capable of constructing chiral nitriles bearing vicinal tertiary or quaternary carbon centers.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was partially supported by University of Orléans, Région Centre-Val de Loire, and the French National Research Agency (ANR-20-CE07-0016-01 and ANR-23-CE07-0014-04).