

1 Introduction
Organosilicon compounds are versatile and powerful reagents in the field of organic synthesis [1–3]. Moreover, silanes have attracted considerable attention not only as biologically acceptable analogues of natural products [4–7], but also because silicon acts as a directing atom that increases [8–10] or reverses selectivities usually observed for the carbon analogues [11,12]. Recently, organosilanes have been extensively studied in cross-coupling reactions catalysed by palladium [13–15]. For example, Tietze took advantage of the presence of a silicon group to direct the last step of a domino-Heck double cyclisation process [16]. On the other hand, Hirao et al. reported first, nearly two decades ago, that 1- or 3-trimethylsilylallyl acetates in the presence of a palladium catalyst could react regio- and stereoselectively with stabilized carbon nucleophiles giving the corresponding vinylsilanes as the only product [17].
We report here some aspects of the influence of silicon in palladium-catalysed reactions. Interestingly, parts 2.1, 2.2 and 3.1 describe the reactivity of silylated vinyloxiranes with the silicon either in the γ or the α position to the vinyl function. Silicon is shown also to bring chemoselectivity in the reactivity of silylated butene diols and some interesting examples of synthetic applications are selected to illustrate the silicon effects in small substrates engaged in palladium-catalysed transformations.
2 Palladium-catalysed rearrangement of silylated vinyloxiranes
At the beginning of this project concerning the development of vinyloxiranes 1, we intended to synthesize unsaturated amino acids 2 and this immediately raised the problem of the existence of three electrophilic carbons on the vinyloxiranes 1 that might react with a nitrogen nucleophilic species (Scheme 1).
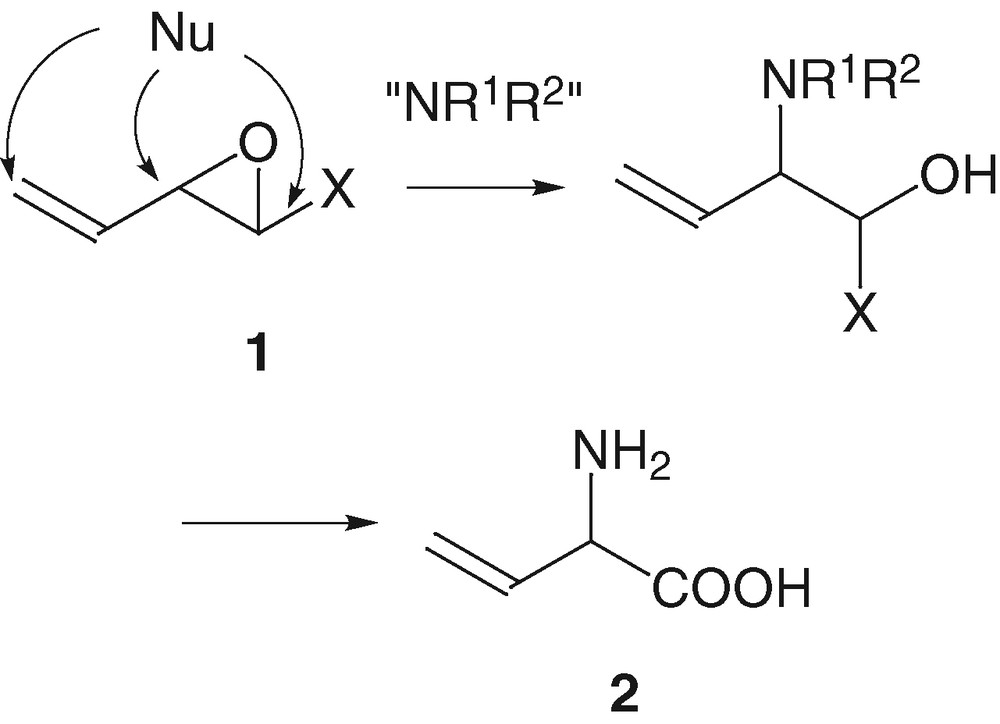
Trost [18] and Tsuji [19] had already shown that the regioselectivity of the palladium-catalysed nucleophilic addition on vinyloxiranes depended on the experimental conditions: the solvent, the ligands of the palladium, the temperature and the nature of the nucleophile. The choice of these conditions gave a 1,2- or a 1,4- selective addition of the nucleophilic species respectively on the proximal or the distal carbon centre of the π-allyl complex. Therefore, we decided to achieve the synthesis of the target compounds 2 via a palladium-catalysed key-step as well as to start with a vinyloxirane substituted by a silicon atom that might control the stereochemistry of the nucleophilic addition and also its regioselectivity since it might stabilize the positive charge on the β-carbon and favour the attack on the proximal carbon (Scheme 2).
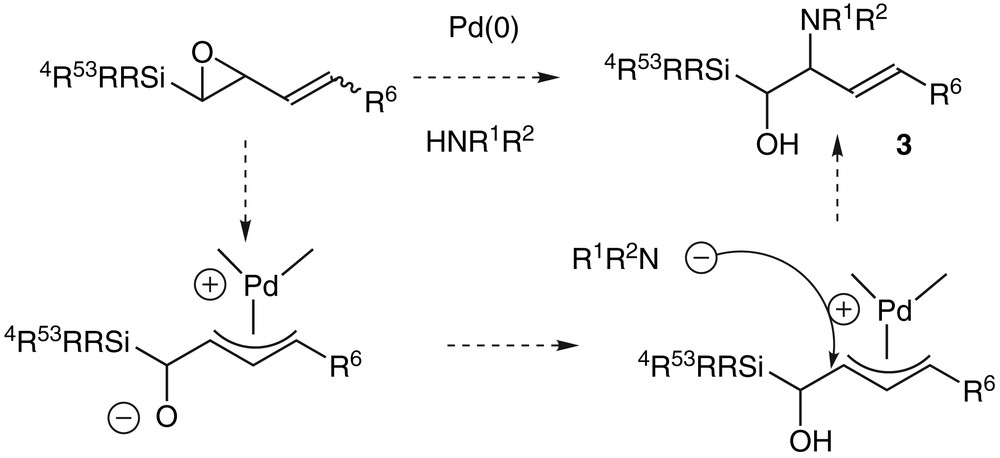
We were not able to isolate any amino-alcohol 3 using phtalimide, which can give its acidic proton to the oxirane in the ring-opening reaction [20]. We obtained some aldehyde 5 instead, in a yield of 10%, which was increased when the reaction was conducted only with palladium(0) without any nitrogen reagent. Under these palladium-catalysed conditions, we therefore reported the first 1,2-shift of a silicon atom that occurred under smooth conditions (room temperature and neutral medium) [21] (Scheme 3).
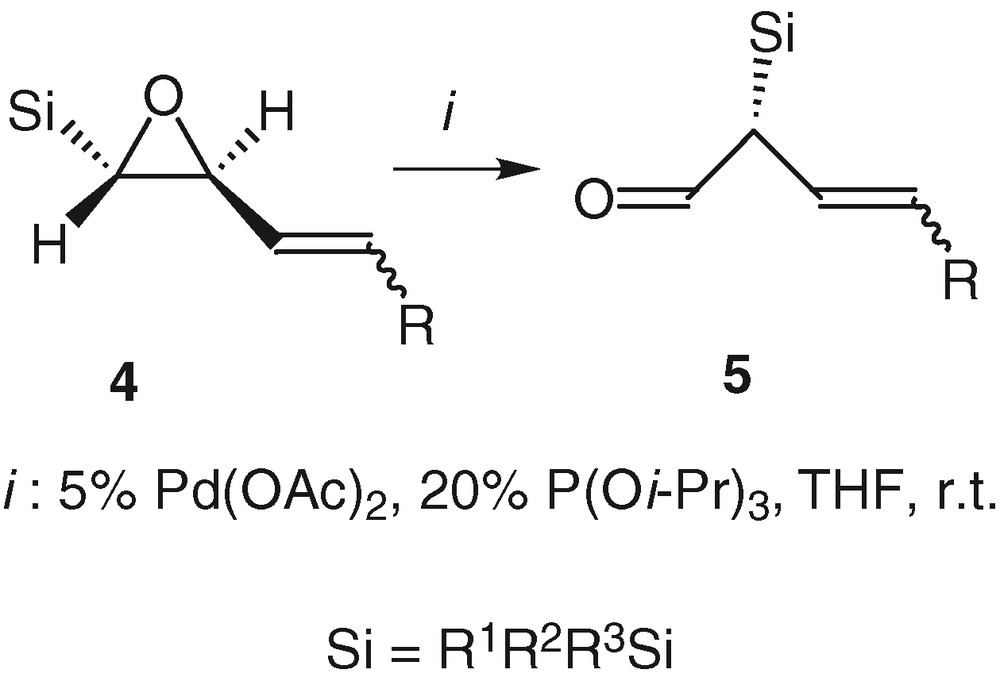
2.1 Silylated vinyl-trans-oxiranes: general mechanistic study
This rearrangement reaction can follow two pathways after the formation of the π-allyl palladium complex 6. One way goes through the formation of a pentavalent silicon intermediate 7 and therefore yields the product of Brook rearrangement 8, which is not really interesting. The second way is the migration of silicon, which has proven to occur anti to the palladium [22] and which gives the aldehyde 5 with a total chirality transfer and the retention of the configuration of the double bond [23]. The substituents of the silicon atom and the ligand of the palladium are directly responsible for the orientation towards the unsaturated enol ether 8 or the aldehyde 5 [24]. In order to favour the enantioselective rearrangement of compounds 4 into the aldehydes 5, we reported the importance of having bulky groups on the silicon atom, namely dimethyltert-butyl or triisopropyl and of reducing the metal with π-acceptor ligands like triphenyl- or triisopropylphosphite [24] (Scheme 4).
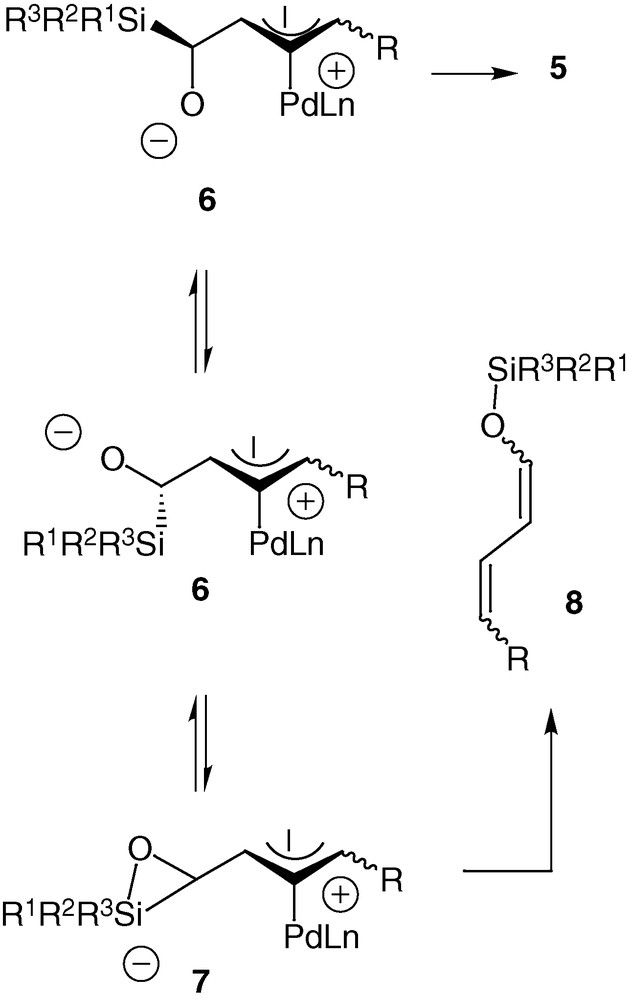
Our interest in this reaction is based upon the smooth experimental conditions, the short reaction times and the high degree of functionalization of the formed aldehydes. Indeed α-silylated aldehydes are difficult to isolate and to handle [25,26]. With the compounds 5, we benefit from stable ambident molecules that may react on electrophiles by the double bond, with nucleophiles by the aldehyde moiety, and which may be oxidized in Tamao or Fleming transformations (Scheme 5).
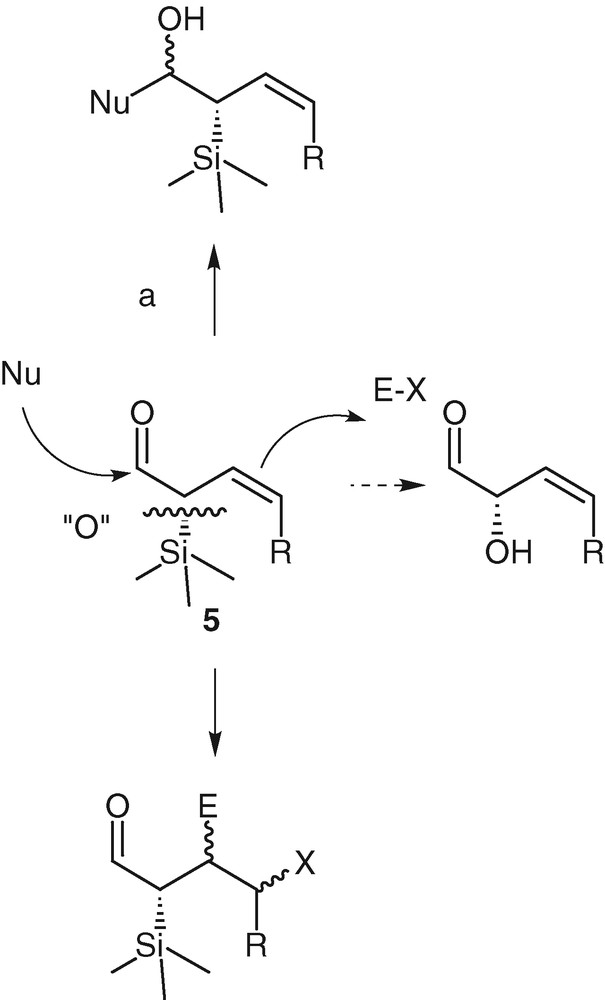
Alkylation of the aldehydes 5 (reaction a of Scheme 5) has been thoroughly studied and gave interesting results in terms of diastereoselectivity, which will be developed in section 2.3 [27,28].
2.2 Scope and limits of the rearrangement reaction
In this part we show the influence of the structure of the starting silylated vinyloxirane on the rate of the rearrangement. The substituent on the double bond may accelerate the rearrangement, the configuration cis or trans of the epoxide plays as well a role in the rate and therefore in the choice of experimental conditions of the reaction. All these structural factors will be studied and rationalized in order to better understand the mechanism of this transposition reaction.
The first results in this field are shown in Table 1 with the trans-α,β-epoxy-δ-substituted-γ,δ-vinylsilanes 4a–e that rearrange quickly into α-silylated-δ-substituted-γ,δ-unsaturated-aldehydes 5.
Quick rearrangement of trans-α,β-epoxy-δ-substituted-γ,δ-vinylsilanes 4a–e into α-silylated-δ-substituted-γ,δ-unsaturated-aldehydes 5.
The reaction conditions and the time of the reaction indicate that this rearrangement occurs easily and under smooth conditions. Compounds 4d–e rearrange very rapidly on silicagel and need special basic conditions for their purification to avoid spontaneous migration of the silicon atom [29].
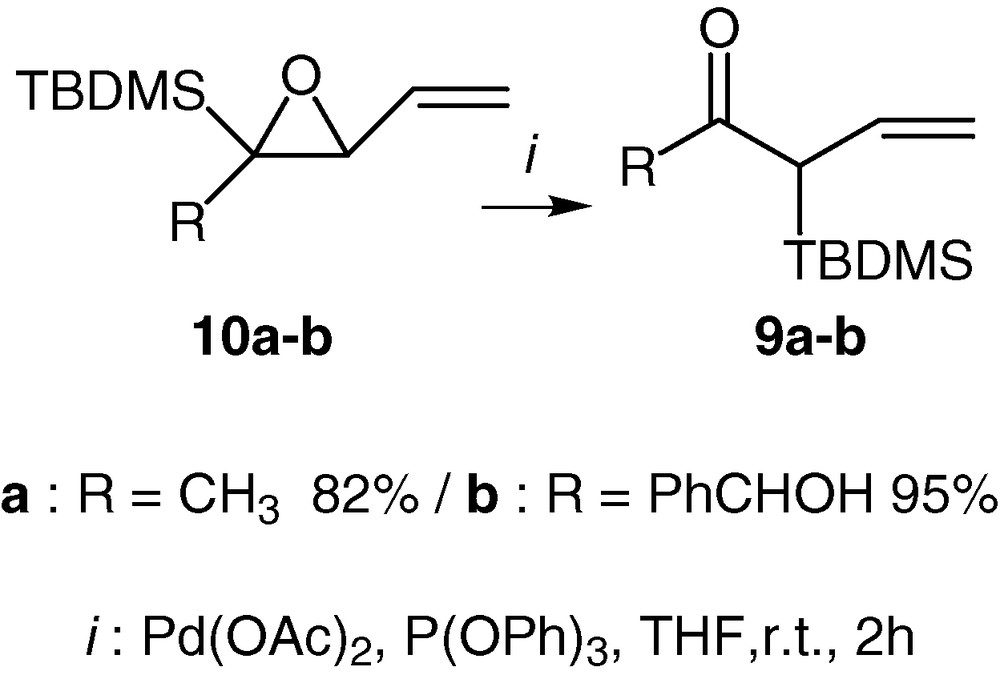
This reaction can provide α-silylated-β,γ-unsaturated-ketones when the silylated vinyloxiranes are substituted in the position α to the silicon atom. In this case the ketones 9a and 9b are formed in good yields and under similar smooth conditions [30,31] (Scheme 6).
When the silylated vinyloxiranes display a quaternary allylic carbon in the β-position to the silicon atom, the experimental conditions of the rearrangement may be more drastic, as shown in Table 2 [29].
Experimental conditions of the rearrangement of compounds 11a–f into compounds 12–14.
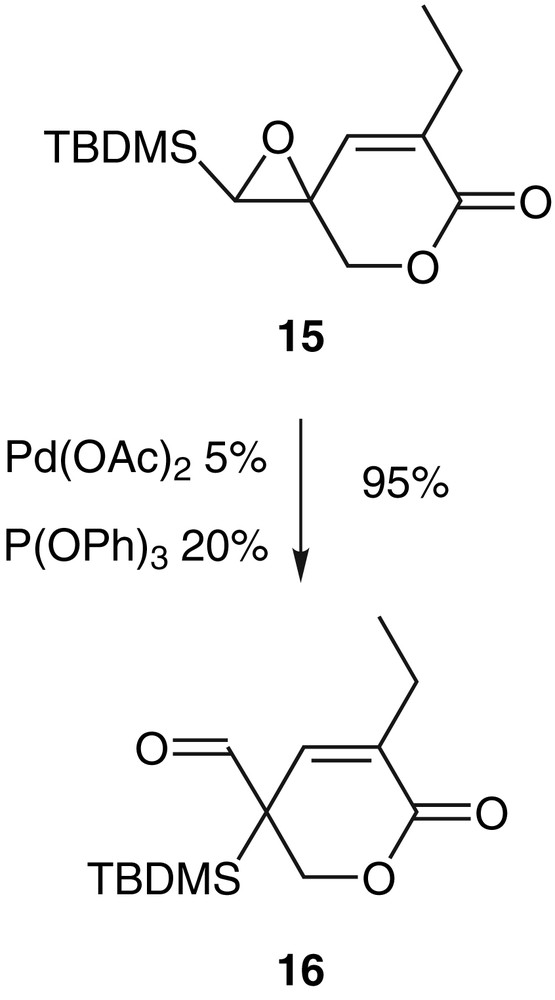
The degradation or the lack of reactivity in the case of vinylepoxysilanes 11c, 11e or 11f indicate the importance of the conformation of the oxirane in the rate of the rearrangement. Therefore, the double bond has been frozen into a cycle like in the epoxylactone 15 which reacted very easily in THF at room temperature with palladium(0) and gave the aldehyde 16 with a very good yield (Scheme 7) [29].
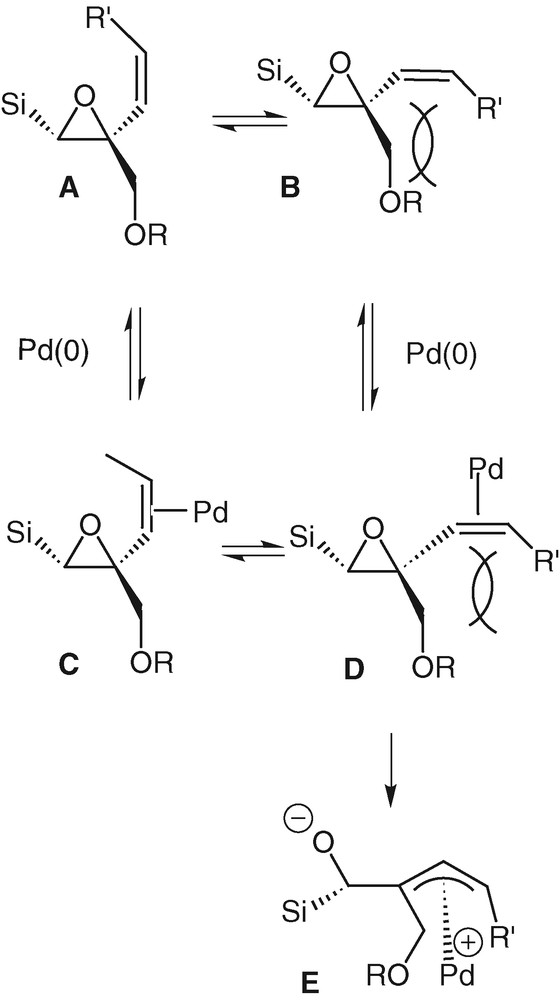
A limiting factor of the rearrangement reaction is probably the steric hindrance brought by the substituents on the quaternary carbon atom. In the case of the trisubstituted olefins 11e and 11f, this steric factor prevents the double bond from lining up with the epoxide, the palladium can still bind to the vinyl group but there is not the planarness required for the formation of a π-allyl complex. This factor is less important in the case of unsubstituted olefins. In the case of the cyclic compound 15, the blocked conformation of the double bond imposes this planarness and allows easily the occurrence of the rearrangement in a good yield. Formation of palladium π-allyl complex E can only derive from the palladium-associated-form D that exists only if the steric hindrance between the double bond and the ether function is sufficiently low (Scheme 8).
Indeed this 1,2-silicon migration proves to be a versatile tool in the field of synthetic chemistry with some limits due to the steric hindrance of some β-disubstituted-α,β-epoxy-γ,δ-vinylsilanes.
2.3 Synthetic applications
This palladium-catalysed rearrangement of silylated vinyloxiranes was associated in a one-pot procedure with the alkylation of the aldehydes 5 that has been thoroughly studied and gave interesting results in terms of diastereoselectivity [27]. When the starting oxirane has a Z double bond substituted by an electron-withdrawing group as a methyl ester, alkylation gives an intermediary alcoolate that can cyclise into a δ-lactone [32] (Scheme 9).
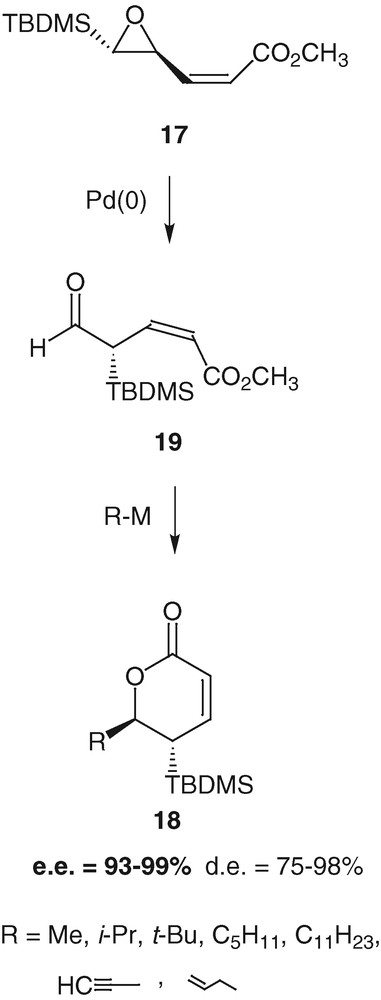
These highly functionalized lactones are efficient Michael acceptors that undergo conjugated additions and further electrophilic trapping with a high diastereoselectivity. When the electrophile is prochiral like the benzaldehyde, there is even an extracyclic stereogenic carbon atom whose configuration is controlled in the process of the trapping [33,34] (Scheme 10).
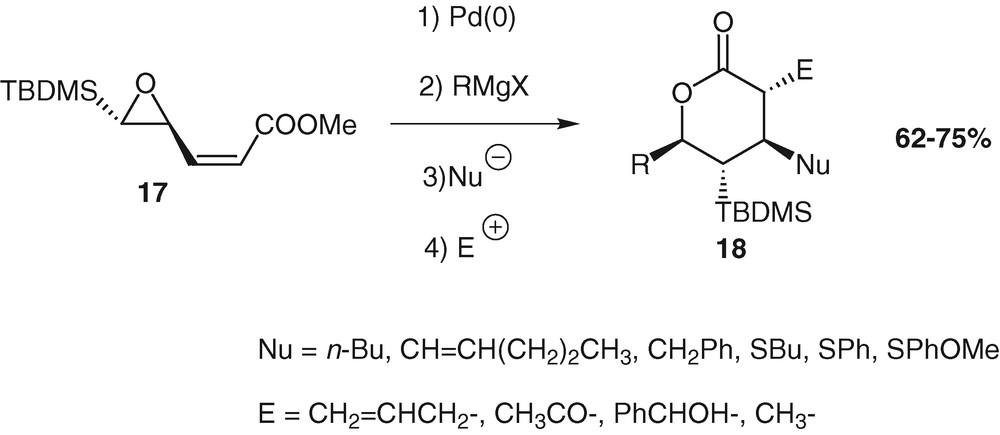
Tetrasubstituted δ-lactones 18 can be prepared from compound 17 in four steps accomplished in a one-pot procedure with formation of four contiguous stereogenic carbon atoms in good yield [33].
These results confirm the efficiency of the palladium-catalysed rearrangement of α,β-epoxy-γ,δ-vinylsilanes in the field of organic synthesis in terms of stereoselectivity and high functionalisation.
3 Silyl-substituted 1,4-butadienol derivatives and palladium-catalysed alkylations
3.1 α′,β′-vinyl-α,β-epoxysilanes
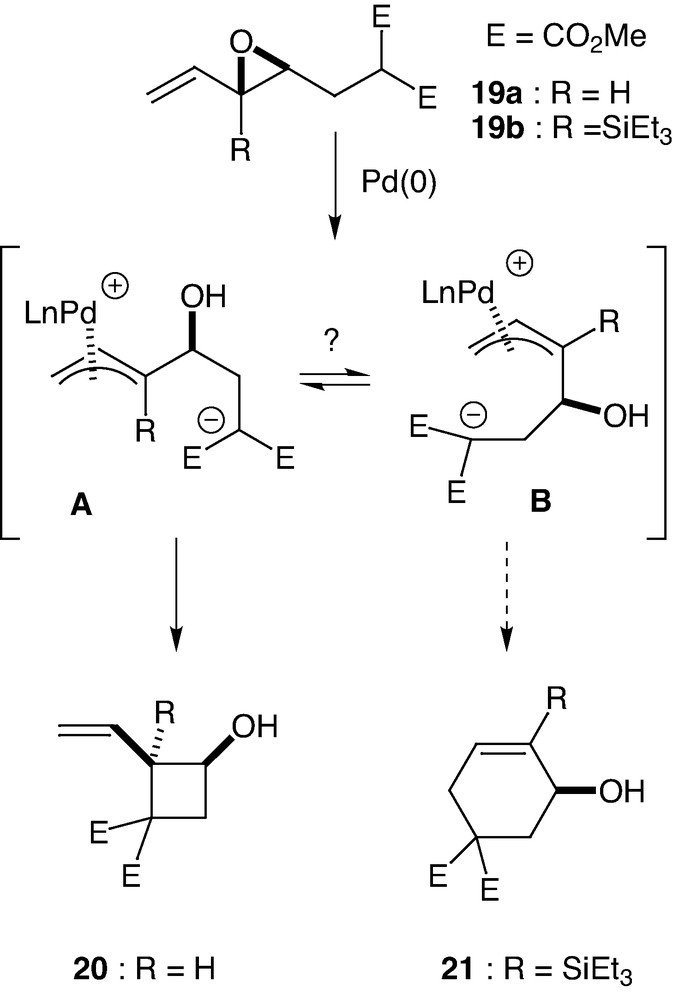
During our study on the behaviour of a malonate derivative 19a bearing a vinylepoxide moiety, we observed a stereoselective 4-exo-trig cyclisation leading to the corresponding cyclobutanol 20 in 70% yield (Scheme 11). In order to check the possibility to reverse the chemoselectivity of the reaction, we decided to prepare a silylated substrate 19b. Indeed, based on the previous work of Corriu [35], Trost [18], Tsuji [36], Sato [37] and others [9], silicon atom in one of the terminal positions of a π-allyl palladium complex plays a curious role. Thus, attacks by nucleophiles occur at the remote position with respect to the silicon even if a bulkier substituent is present [9,38]. Starting from vinylsilane 19b, the attack of a palladium(0) species should afford the zwiterionic π-allylpalladium intermediates A and/or B (Scheme 11). The presence of a silicon group should favour the formation of the cyclohexenol 21, since steric and electronic factors act in the same direction.
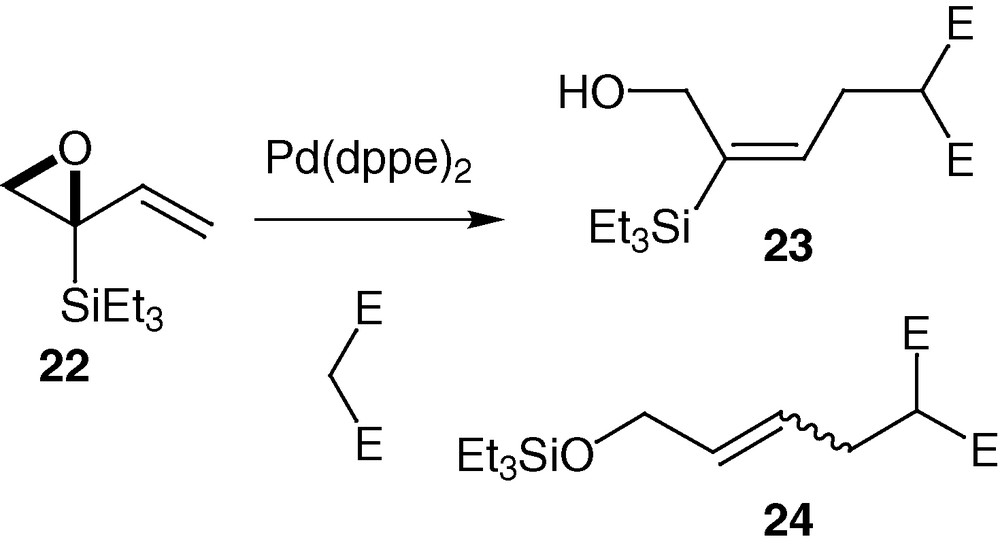
On the other hand, having already observed silyl migration with vinyl epoxides 5 (vide supra), we first investigated the reactivity of the 2-triethylsilyl vinylepoxide 22 in the presence of palladium catalyst and dimethylmalonate as nucleophile (Scheme 12) [39]. Two products are isolated after 4 h. The expected one 23 in a low yield (11%) as well as a second one 24, in 16% yield, resulting from a 1,2 C-to-O silyl migration, followed by the addition of the dimethyl malonate anion (Scheme 12); C-desilylation during palladium-catalysed alkylation has been reported [40].
3.2 Chemo- and stereoselective alkylations of 2-silylbut-2-ene diol derivatives
With the precedent experiment we have shown that 2-triethylsilyl vinylepoxide underwent a migration of the silicon group from carbon to oxygen during palladium-catalysed reactions. However, in order to validate our strategy where a silicon group would control the chemo- and stereoselectivity of palladium-catalysed reactions, we prepared a C4 analogue of our desired silylated vinylepoxide (Scheme 13). In this context, we chose a general synthesis that could give access to different silyl substituted C4 synthons from commercially available cheap materials. Our approach started with the 2-butyne-1,4-diol diacetate 25 (easily prepared by diacetylation of 2-butyne-1,4-diol). Stereoselective syn or anti hydrosilylation could be achieved in high yields by choosing either platinum or ruthenium complexes as catalyst [41,42]. The obtained vinylsilanes 26 present two potential leaving groups that are differentiated only by their position relative to the silicon group. We have shown that in the presence of catalytic amounts of palladium (0) complexes, alkylations, with stabilized carbanions as nucleophiles, occurred in good to excellent yields (70–100%). Gratifying, the silicon group totally differentiated the two potential leaving groups: only the β-acetate relative to the silicon was displaced. In addition, very high stereoselectivities were observed. The stereoselectivity was dependent on the temperature of the reaction but in all the substrates studied, the configuration of the double bond was mainly retained starting either from Z or E 26 (Scheme 12). As an example, using dimethyl malonate as nucleophile, the E/Z ratio of 27 is varying from 25:75 at r.t. to 10:90 at 0 °C, starting from Z-26, whereas it is varying from 90/10 to 98/2 starting from E-26.
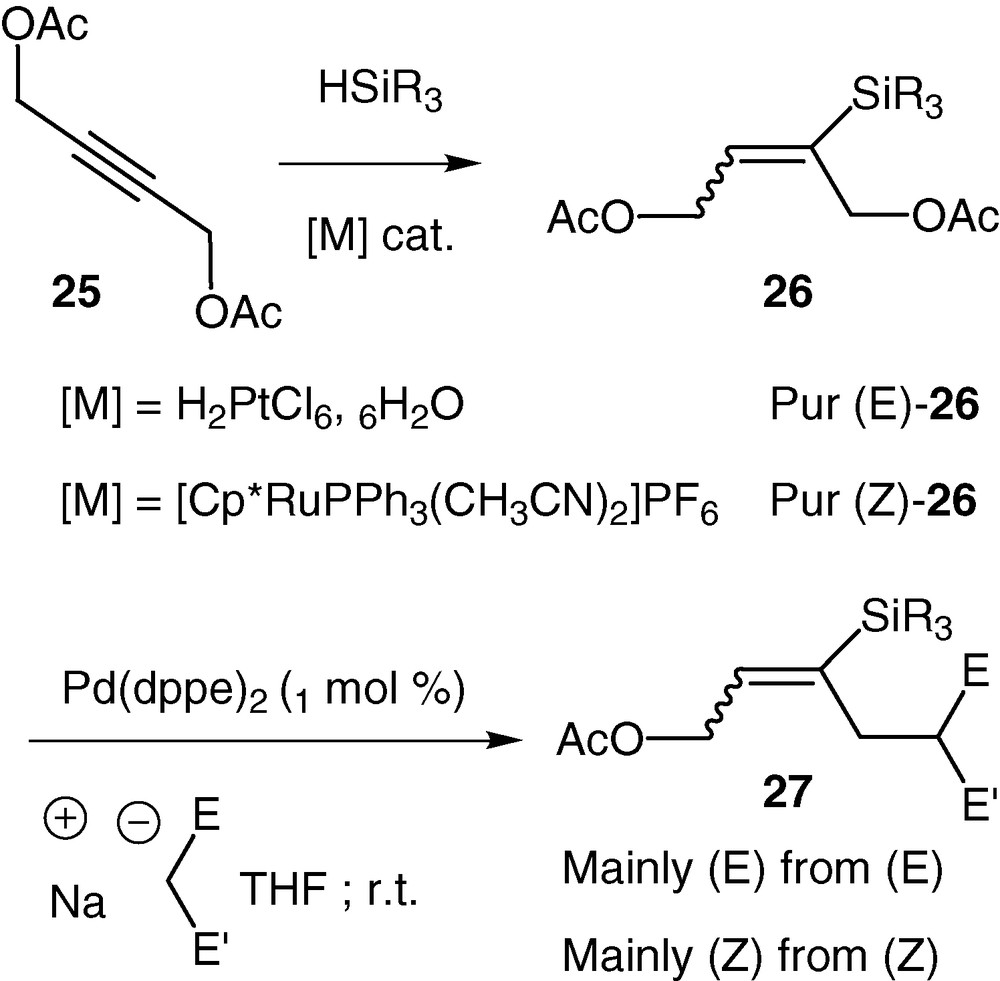
The allylation of dimethyl malonate was performed with the same efficiency with variously substituted (E)-vinylsilanes 26 and some representative examples are reported in Table 3.
Representative examples of the he allylation of dimethyl malonate with variously substituted (E)-vinylsilanes 26
| Entry | Substrate | SiR3 | Vinylsilane (%) | E/Z ratio |
| 1 | 26a | SiEt3 | 95 | 98/2 |
| 2 | 26b | Si(i-Pr)3 | 75 | 97/3 |
| 3 | 26c | SiMe2t-Bu | 98 | 100/0 |
| 4 | 26d | SiMe2Ph | 98 | 100/0 |
| 5 | 26e | SiMe2(4-MeO–C6H4) | 82 | 95/5 |
| 6 | 26f | SiMe2(4-CF3–C6H4) | 64 | 98/2 |
The preparation and reactivity of two analogues in which the silyl group has been replaced by a tert-butyl group were also examined. Their lower reactivity toward the nucleophile as well as the lost of stereoselectivity can be ascribed to the ability of the silicon group to stabilize a β−carbocation. Our interest in the use of these bis–allylic substrates was originally stimulated when we discovered the behaviour of a dissymmetric precursor 28 bearing a carbonate in place of the acetate in the γ position relative to the silicon (Scheme 14). With either dimethyl malonate or methyl acetoacetate sodium salts, acetate acts as the leaving group, in spite of the recognized better ability of carbonates as leaving groups in this type of chemistry [43,44].
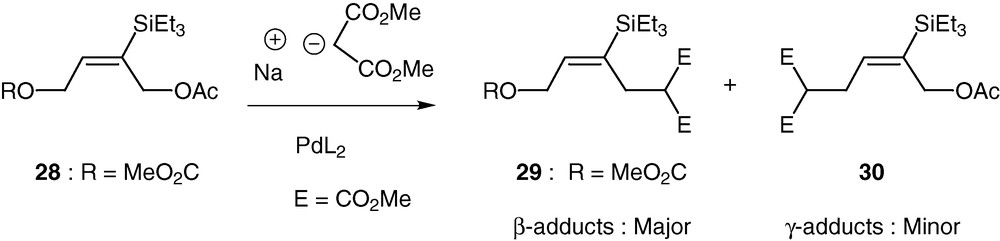
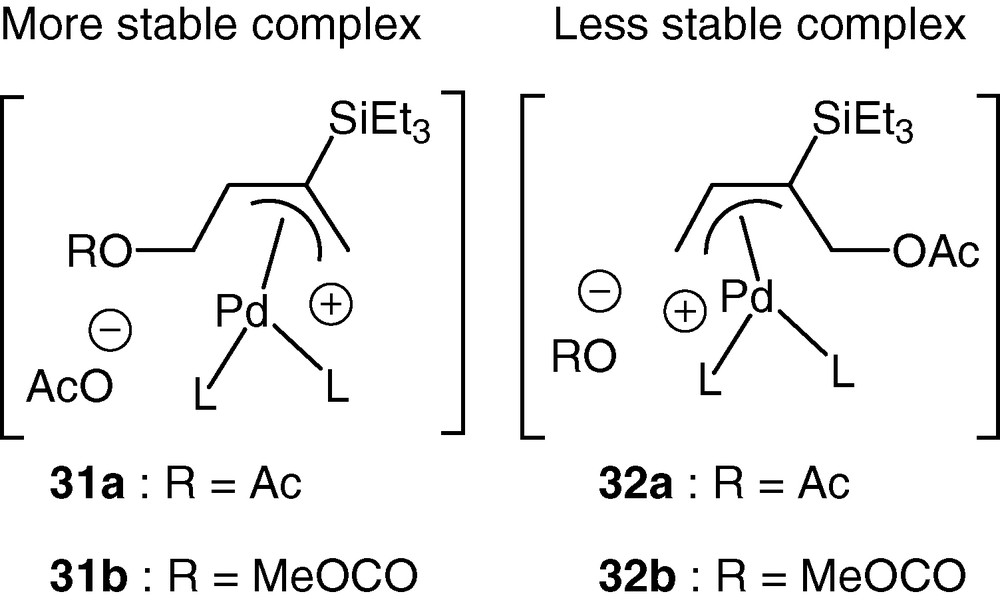
As a representative example, dimethyl malonate gave respectively the β and γ adducts in a ratio of 80:20. Some theoretical calculations have been done and, as expected, have shown that the presence of silicon in the central carbon atom of π-allyl palladium cations causes stronger Pd–allyl interaction, therefore justifying that cations 31a and 31b are more stable than their isomers 32a and 32b possessing a silicon at the terminal atom (Scheme 15) [45].
3.3 Competitive 5-endo-trig vs 3-exo-trig palladium-catalysed cyclisations. Preparation of cyclopentenes
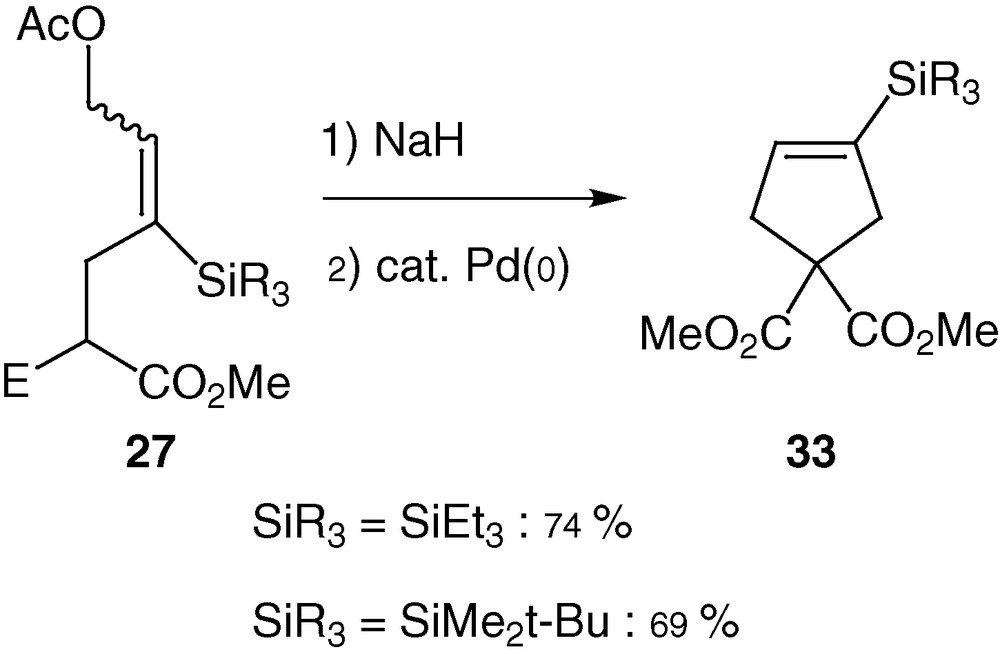
The described silylated allylic diacetates still present a leaving group after reaction with nucleophiles. Moreover, after reaction with malonates, the products 27 are themselves nucleophiles due to the acidic hydrogen of the malonate function. Treatment with sodium hydride followed by addition of palladium catalyst gave after one night at reflux the expected cyclopentenes 33 in good yields as the unique product (Scheme 16). No trace of the corresponding cyclopropane could be observed [46]. Varying the silyl substituents from alkyl to aryl was dramatic for the course of the reaction. Alkyl groups are tolerated and allowed the formation of the expected cyclopentene, whereas aryl one totally inhibits the cyclisation [47]. We thus have confirmed that silyl group at a proper position can drive palladium-catalysed cyclisation to the 5-endo-trig process. Balance has to be found between the substituents of the silicon as well as the size and the basicity of the internal nucleophile. Indeed, when the keto-ester function is the nucleophile, competitive hetero-5-exo-trig cyclisation occurs and dihydrofuran derivatives were isolated in 69% yield.
3.4 Competitive 5-exo-trig vs 7-endo-trig cyclisations. Preparation of pyrrolidones
In 1998, G. Poli et al. prepared 3,4-disubstituted pyrrolidones in high yields by a palladium-catalysed cyclisation based on the interaction between a resonance-stabilized carbanion and an allylic acetate function, tethered by a nitrogen atom [48]. Having successfully prepared cyclopentenes by a 5-endo-trig palladium-catalysed reaction, we started a project to study the effect of silicon on these kinds of molecules. In particular, we were curious to verify if such modifications could still permit cyclisations, and, in positive case, to determine a possible 5-exo versus 7-endo preference. The precursors have been prepared from the 1,4-butyne diol by first monoacetylation followed by a regioselective platinum-catalysed hydrosilylation giving 34 in 80% yield. A palladium-catalysed reaction with benzylamine afforded in 52% yield the expected aminoalcohol 35 bearing the amine function at the less substituted carbon (Scheme 17). Some additional steps delivered the acyclic precursor 36 bearing the triethylsilyl group.
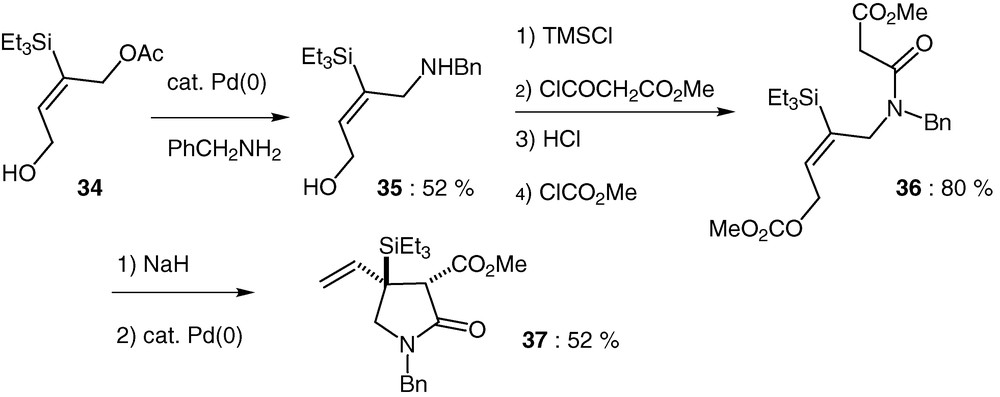
Treatment of the sodio enolate of compound 36 with 5 mol% Pd(OAc)2 and 10 mol% of dppe in DMF at 100 °C for 30 min gave the pyrrolidone 37 as the unique product in 52% yield [49]. Exclusive formation of pyrrolidone structures from the above amide precursor indicates that a 7-endo type approach is in this case forbidden. Such a phenomenon, which contrasts with what was observed with the related β-ketoesters [50], is certainly associated with the amide bond. Indeed, the nature of such a function is expected to force the C–C(O)–N–C dihedral angle value of the reacting cyclisation precursors toward a planar arrangement (0° or 180°), thereby disfavouring the 7-endo cyclisation [51].
4 Conclusion
This paper has summarized the results acquired by us in the field of silylated oxygenated molecules. We pointed out that substituents on the silicon atom of the γ,δ-vinyl-α,β-epoxysilanes could direct the reaction towards a Brook rearrangement or an enantioselective 1,2-carbon-to-carbon silyl migration. We described the silicon effects in the chemoselectivity of Pd-catalysed alkylations on different substrates like α,β-vinyl-α,β-epoxysilanes or silylated butene diols. Finally, we stressed the point of synthetic applications with the preparation of highly functionalized δ-lactones and easily accessible cyclopentenes.