

1 Introduction
Les réseaux bimétalliques coordinés par des ponts oxalates, de formule générale {〚MIIMIII(C2O4)3〛–C+〛}n (ox = (C2O4)2–, C+ = monocation) constituent une famille importante d’aimants à base moléculaire 〚1–7〛. Dans ces composés, le sous-réseau anionique 〚MIIMIII(C2O4)3〛–, résultat d’un enchaînement d’espèces monomères 〚M(ox)3/2〛n– de symétrie D3, présente deux structures différentes selon les configurations relatives de ces sous-unités hexacoordinées (Δ ou Λ) :
- • bidimensionnelle et organisée en nid d’abeille si cet enchaînement est hétérochiral (Δ-Λ) 〚6–7〛 (Fig. 1a) ;
- • tridimensionnelle et hélicoïdale s’il est homochiral (Δ-Δ ou Λ-Λ) 〚1–5〛 (Fig. 1b).
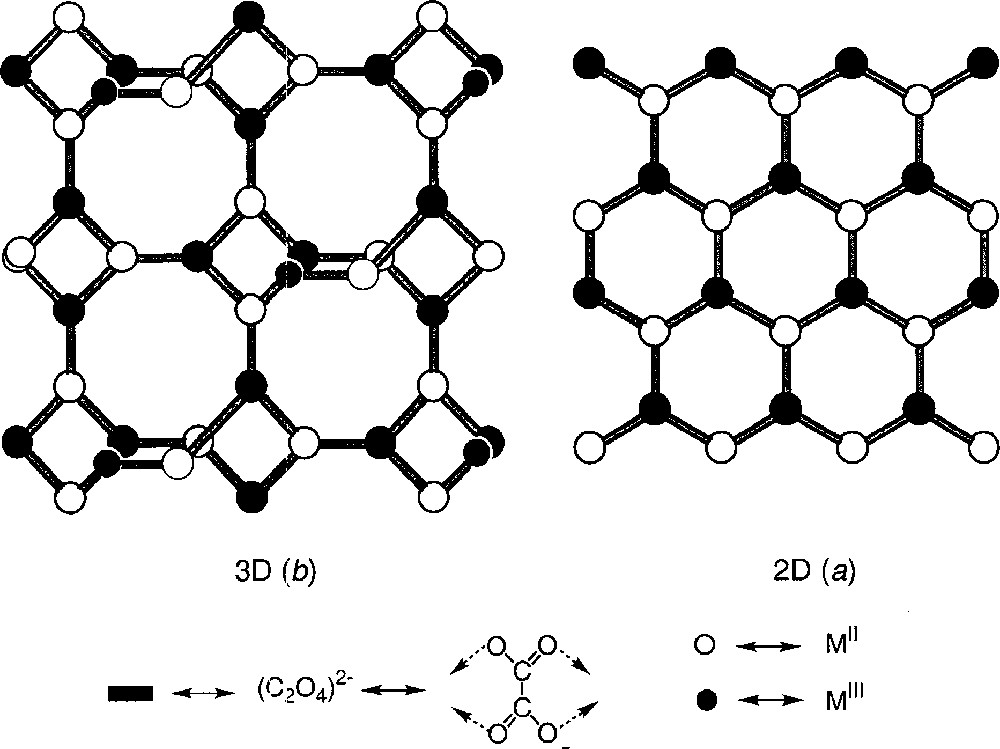
Représentation schématique des polymères 2D (a) et 3D (b).
L’orientation vers l’une ou l’autre de ces deux structures dépend de la nature du cation C+, qui joue dans la synthèse de ces polymères un rôle fondamental de template. En effet, l’utilisation d’un monocation achiral (N(Bu)4+…) aboutit à un réseau bidimensionnel, tandis que celle d’un cation de symétrie D3, comme 〚Ru(bpy)3〛2+, mène à la formation d’un composé trimensionnel. En partant de réactifs racémiques, le polymère bidimensionnel est constitué d’une alternance de plans anioniques montrant des configurations opposées les unes aux autres : par exemple, à trois plans MnΛ-CrΔ succèdent trois plans MnΔ-CrΛ, rendant ainsi le polymère globalement racémique 〚6–7〛 (Fig. 2). En série tridimensionnelle, l’utilisation de réactifs non dédoublés aboutit également à la formation d’un produit racémique 〚1–5〛 (Fig. 2).
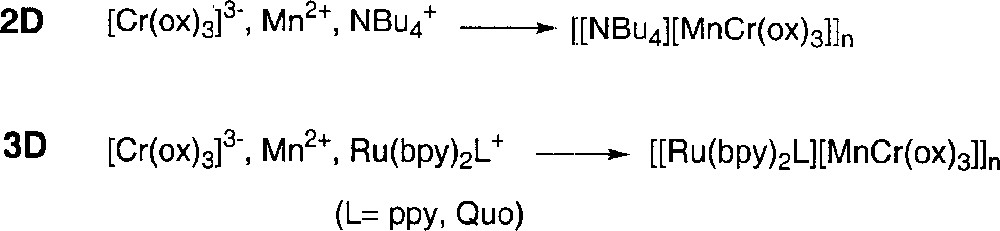
Synthèse des polymères 2D et 3D.
Nous avons montré récemment 〚8–10〛 que la synthèse de ces polymères sous forme optiquement active est possible à partir de 〚MIII(ox)3〛3– (MIII = CrIII, CoIII) dédoublé. Par exemple, pour la structure bidimensionnelle, tous les plans présenteront alors un enchaînement MnΛ–CrΔ si l’on part de 〚CrΔ(ox)3〛3–. Pour le réseau tridimensionnel, nous avons montré que chacun des énantiomères Δ et Λ de la brique monomère 〚Cr(ox)3〛3– est capable de produire un auto-assemblage conduisant à des polymères optiquement actifs où tous les métaux hexacoordinés possèdent alors la configuration de l’anion de départ 〚8–9〛.
Il est important de noter que la synthèse de ces composés est hautement diastéréosélective et qu’avec la seule configuration absolue de l’anion monomère dédoublé, on induit la configuration absolue des autres éléments chiraux (métaux hexacoordinés) dans le polymère final. Il reste cependant à prouver 〚8–9〛 que cette induction de chiralité peut s’effectuer également par le monocation sous forme optiquement active en utilisant une brique anionique 〚Cr(ox)3〛3– racémique. Il faut pour cela synthétiser des monocations de type 〚Ru(bpy)2L〛+ (L = ppy, Quo) et les dédoubler en leurs énantiomères Δ et Λ, ce qui n’avait jamais été réalisé auparavant.
Dans cette note, nous présenterons donc le dédoublement des espèces 〚Ru(bpy)2ppy〛+ 1 et 〚Ru(bpy)2Quo〛+ 2 en leurs énantiomères respectifs 〚ΔRu(bpy)2ppy〛+ (1Δ), 〚ΛRu(bpy)2ppy〛+ (1Λ) et 〚ΔRu(bpy)2Quo〛+ (2Δ), 〚ΛRu(bpy)2Quo〛+ (2Λ). Dans une deuxième partie, nous montrerons le dosage par RMN 1H grâce à l’utilisation de l’anion 〚ΔTrisphat〛 {Trisphat = tris(tetrachlorobenzenediolato)phosphate(V)} 〚11–13〛 des excès énantiomériques de ces produits enrichis.
2 Résultat et discussion
2.1 Obtention des énantiomères des cations 〚Ru(bpy)2ppy〛+ 1 et 〚Ru(bpy)2Quo〛+ 2
La séparation des énantiomères de 〚Ru(bpy)2(Cl)2〛 〚14〛 est possible par chromatographie sur colonne chirale. Les complexes de type 〚Ru(bpy)2L〛PF6 (1Δ), (1Λ), (2Δ) et (2Λ) peuvent être ensuite obtenus 〚15〛 par remplacement des ligands chlores, avec rétention de configuration. Cette méthode, qui nécessite un lourd investissement en chromatographie liquide haute pression, n’étant pas très accessible, nous avons proposé une voie chimique mettant en jeu la formation des réseaux bimétalliques à ponts oxalates décrits précédemment.
La formation des polymères tridimensionnels 〚8–10〛 est, nous l’avons vu, hautement diastéréosélective. C’est cette propriété que nous avons utilisée pour dédoubler les monocations (1) et (2).
L’utilisation de K3〚ΔCo(ox)3〛 (〚α〛D = +1 667) (rotation maximale décrite dans la littérature 〚16〛 : 〚α〛D = +1 928), de MnCl2 et de deux équivalents de 〚Ru(bpy)2L〛PF6 racémique (1) ou (2) dans un mélange DMSO/H2O mène à la précipitation du polymère {〚ΔRu(bpy)2(L)〛〚MnΔCo(ox)3〛}n (rendement = 90%). Ainsi, la solution mère est maintenant enrichie en énantiomères opposés de 〚Ru(bpy)2L〛PF6, c’est-à-dire, respectivement, (1Λ) et (2Λ), qui sont récupérés ensuite du filtrat en les précipitant par ajout d’éther (Fig. 3). Partant de la forme Λ de K3〚Co(ox)3〛 (〚α〛D = –1 892) (rotation maximale décrite dans la littérature 〚16〛 : 〚α〛D = –1 940), la même stratégie conduit à la précipitation du polymère {〚ΛRu(bpy)2L〛〚MnΛCo(ox)3〛}n et à l’obtention des 〚Ru(bpy)2L〛PF6 enrichis en énantiomère Δ (1Δ) ou (2Δ).
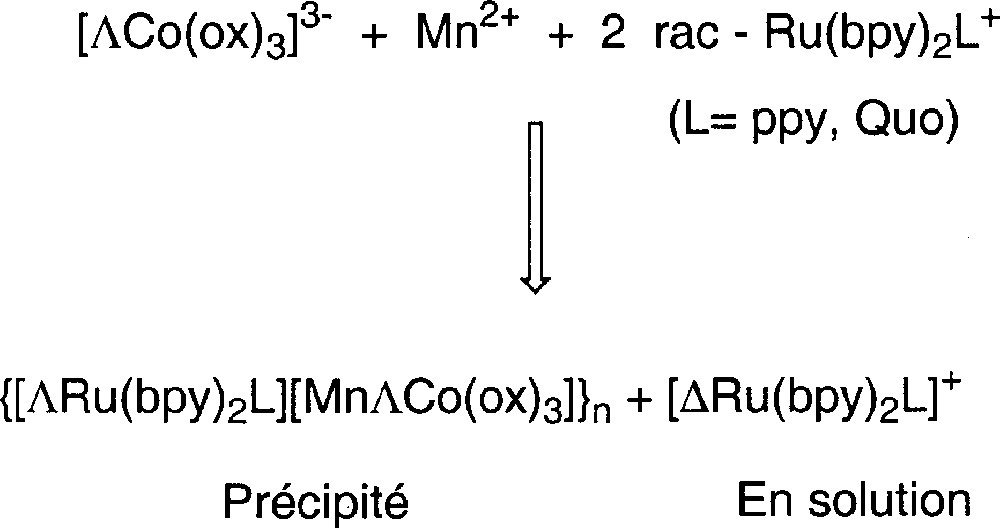
Préparation du polymère optiquement actif {〚ΛRu(bpy)2L〛〚MnΛCo(ox)3〛}n (L = ppy, Quo) avec dédoublement partiel du complexe 〚Ru(bpy)2L〛+ en son énantiomère Δ.
Les cations 〚Ru(bpy)2L〛+ étant obtenus sous leur forme optiquement active, il faut ensuite mesurer leur excès énantiomérique. Le dichroïsme circulaire ne le permet pas car, même si cette méthode permet souvent de déterminer les configurations absolues, on ne peut connaître l’excès énantiomérique s’y rapportant. De même, le pouvoir rotatoire absolu (e.e. = 100%) de ces espèces étant inconnu, il est impossible également de faire une comparaison quelconque avec les échantillons afin d’évaluer leur excès énantiomérique. De plus, ces produits sont très colorés, ce qui rend difficile une mesure précise de leur pouvoir rotatoire. Nous avons donc utilisé un agent chiral de déplacement en RMN 1H, le 〚ΔTrisphat〛– 〚11–13〛, de symétrie D3, qui interagit fortement avec les cations étudiés en formant des paires d’ions distéréomériques. Ainsi, cet anion induit le doublement de certains pics en RMN 1H et permet la mesure de l’intensité relative des pics des deux formes diastéréomères et donc de doser l’excès diastéréomérique. Cet excès diastéréomérique des espèces 〚ΛRu(bpy)2L〛〚ΔTrisphat〛 et 〚ΔRu(bpy)2L〛〚ΔTrisphat〛 est approximativement égal à l’excès énantiomérique dans les fractions enrichies (1Δ), (1Λ), (2Δ) et (2Λ), car l’excès énantiomérique du 〚ΔTrisphat〛– est de 98%.
2.2 Détermination de l’excès énantiomérique de (1Δ), (1Λ), (2Δ) et (2Λ) par RMN 1H grâce à l’anion 〚ΔTrisphat〛–
Les spectres RMN 1H des cations 〚Ru(bpy)2L〛+ (1) et (2) ont été enregistrés sous leur forme racémique en l’absence (Figs 4a et 5a) et en présence de 〚ΔTrisphat〛– (Figs 4b et 5b) et sous leurs formes enrichies (1Δ), (1Λ), (2Δ) et (2Λ) en présence de 〚ΔTrisphat〛– en solution dans l’acétone (L = ppy) ou dans CH2Cl2 (L = Quo) (Figs 4c–4d et 5c–5d).

Spectres RMN 1H de 1 en solution dans l’acétone d6 (c = 0,014 mol·l–1). a, b. Forme racémique, respectivement en l’absence et en présence de 〚ΔTrisphat〛–. c, d. Formes enrichies 1Δ et 1Λ en présence de 〚ΔTrisphat〛–. À gauche, région des protons aromatiques et pyridiniques. À droite, agrandissement du signal du proton H-10 ; tous les agrandissements sont centrés sur le milieu du multiplet.
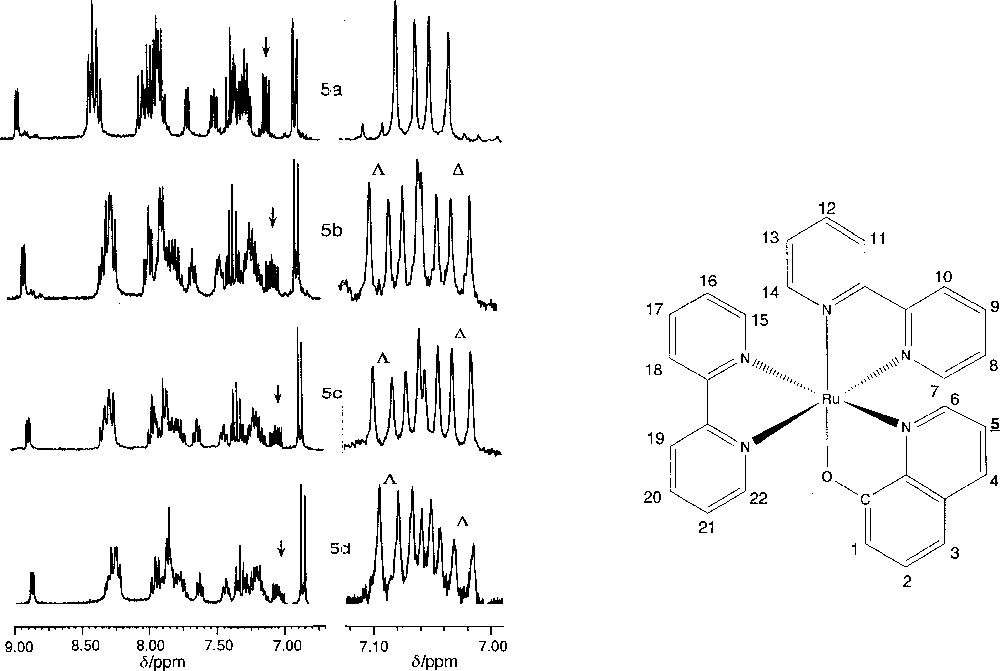
Spectres RMN 1H de 2 en solution dans CD2Cl2 (c = 0,014 mol·l–1). a, b. Forme racémique, respectivement en l’absence et en présence de 〚ΔTrisphat〛–. c, d. Formes enrichies 2Δ et 2Λ en présence de 〚ΔTrisphat〛–. À gauche, région des protons aromatiques et pyridiniques. À droite, agrandissement du signal du proton H-5 ; tous les agrandissements sont centrés sur le milieu du multiplet.
Ces études de RMN 1H permettent de déterminer l’excès énantiomérique des composés enrichis (1Δ), (1Λ), (2Δ) et (2Λ) par formation de paires diastéréomériques avec l’anion chiral 〚ΔTrisphat〛–. Les spectres des composés racémiques (Figs 4a et 5a) se compliquent après ajout de 〚ΔTrisphat〛– (Figs 4b et 5b), car les paires diastéréomériques montrent des déplacements chimiques différents. Les spectres des composés (1Δ), (1Λ), (2Δ) et (2Λ) montrent clairement un enrichissement en un des deux diastéréomères (Figs 4c–4d et 5c–5d). Pour mesurer l’excès diastéréomérique s’y rapportant, nous avons choisi de faire le dosage sur les multiplets centrés à δ = 7,57 ppm pour 1 (proton H-10 〚17〛) et à δ = 7,05 ppm pour 2 (proton H-5 〚18〛), qui sont les plus sensibles à la présence de l’agent chiral. L’évolution de ces multiplets dans les différents spectres est illustrée sur les Figs 4a–4d et 5a–5d. Nous avons pu attribuer chaque pic à chacun des deux diastéréomères, en comparant les spectres des formes enrichies (1Δ), (1Λ), (2Δ) et (2Λ) en présence de 〚ΔTrisphat〛–. Ainsi, les doublets le plus et le moins déblindés du multiplet à 7,57 ppm de 〚Ru(bpy)2ppy〛+ correspondent respectivement aux diastéréomères 〚ΛRu(bpy)2(ppy)〛〚ΔTrisphat〛 et 〚ΔRu(bpy)2(ppy)〛〚ΔTrisphat〛. Pour le cation 〚Ru(bpy)2Quo〛+, le singulet le plus déblindé du multiplet à 7,00 ppm est attribuable à la forme 〚ΛRu(bpy)2(Quo)〛〚ΔTrisphat〛, tandis que le moins déblindé correspond à 〚ΔRu(bpy)2(Quo)〛〚ΔTrisphat〛. Ces signaux sont suffisamment bien séparés pour permettre, par intégration, une mesure précise des proportions entre les deux diastéréomères et donc de leur excès diastéréomérique. Dans le spectre des cations racémiques 〚Ru(bpy)2L〛+ en présence de 〚ΔTrisphat〛–, les signaux des deux diastéréomères apparaissent de même intensité (e.d. = 0%), tandis que, dans les mêmes conditions, les proportions dans celui des formes enrichies (1Δ), (1Λ), (2Δ) et (2Λ) sont respectivement de 72:28 et 25:75 pour (1Δ) et (1Λ) et de 60:40 et 40:60 pour (2Δ) et (2Λ). L’excès diastéréomérique des paires diastéréomériques et donc l’excès énantiomérique est ainsi respectivement de 44 et 50% dans (1Δ) et (1Λ) ; il est de 20% dans (2Δ) et (2Λ).
3 Conclusion
Nous avons présenté dans cet article une méthode originale de dédoublement des cations 〚Ru(bpy)2ppy〛+ et 〚Ru(bpy)2Quo〛+ par formation diastéréosélective de polymères tridimensionnels du type {〚Ru(bpy)2(L)〛〚MnCo(ox)3〛}n. Ces cations optiquement actifs pourront par la suite être utilisés comme templates dans la construction de réseaux optiquement actifs du même type, pour y induire les configurations absolues des cations MII et MIII dans le sous-réseau anionique. MII et MIII seront alors choisis pour générer dans ces structures des propriétés d’aimants moléculaires. Ces polymères alors pourront posséder des propriétés de dichroïsme naturel en même temps qu’un ordre magnétique à longue distance.
Nous avons également montré que l’anion chiral 〚ΔTrisphat〛– permet la différenciation systématique en RMN 1H des deux énantiomères des cations du type 〚Ru(bpy)2L〛+ et, ainsi, de déterminer la pureté énantiomérique des produits enrichis.
4 Section expérimentale
4.1 Réactifs
Les composés suivants ont été préparés selon les procédés de la littérature : K3〚Co(ox)3〛·3 H2O 〚19〛, racémique 〚Ru(bpy)2ppy〛PF6 〚14,17〛, racémique 〚Ru(bpy)2Quo〛PF6 〚14,20〛, 〚l-cinchonidinium〛〚ΔTrisphat〛 〚11,12〛, 〚NBu4〛〚ΔTrisphat〛 〚13〛. La résolution des énantiomères Δ et Λ K3〚Co(ox)3〛·3 H2O a été effectuée selon la procédure décrite dans la littérature 〚16〛. Les autres réactifs ont été achetés et utilisés tels quels.
4.2 Procédures générales
Les pouvoirs rotatoires des composés de départ ont été effectués en solution à 20 °C, dans un tube de 1 dm, en utilisant la raie D du sodium sur un polarimètre Ameria AA5. Les excès énantiomériques ont été calculés par comparaison des pouvoirs rotatoires maximum trouvés dans la littérature 〚11–15〛. Les spectres de RMN 1H (300 MHz) ont été obtenus à 300 K dans des tubes de 5 mm de diamètre externe sur un spectromètre Bruker AC 300 équipé d’une sonde QNP (solutions d’acétone d6 pour (1) et de CD2Cl2 pour (2) c = 0,014 mol·l–1). Les déplacements chimiques sont donnés d’après la convention IUPAC par rapport au tétraméthylsilane.
4.3 Préparation de (1Δ), (1Λ), (2Δ) et (2Λ)
4.3.1 Préparation de (1Δ) et (1Λ) 〚10〛
Dans un tube à hémolyse, une solution aqueuse (0,2 ml) de K3〚ΛCo(ox)3〛·3 H2O {〚α〛D = –1 892 (c = 0,02 dans H2O) (rotation maximale d’après 〚16〛 : 〚α〛D = –1 940)} (39,4 mg, 0,08 mmol) et de MnCl2·4 H2O (15,7 mg = 0,08 mmol) est ajoutée rapidement à une solution (0,4 mL) de 〚Ru(bpy)2ppy〛PF6 (1) racémique (100,2 mg = 0,14 mmol) dans le DMSO. De l’acétone et de l’eau sont successivement ajoutés pendant 5 min, en grattant continuellement les parois du tube avec un pipette Pasteur (les additions sont faites dans cet ordre : acétone–H2O–acétone–H2O–acétone) (1,2 ml à chaque fois). Le précipité est filtré, lavé à l’eau (5 ml) et à l’acétone jusqu’à ce que le filtrat devienne incolore, puis séché à l’air. On obtient ainsi le solide vert {〚ΛRu(bpy)2(ppy)〛〚MnΛCo(ox)3〛}n (68,6 mg = 0,072 4 mmol) (rendement : 91%). Le filtrat violet est concentré à l’évaporateur rotatif pour éliminer l’acétone et, après précipitation par addition d’éther (200 ml), 〚ΔRu(bpy)2ppy〛PF6 (1Δ) enrichi (43,7 mg = 0,061 2 mmol) est filtré et séché à l’air. La même procédure a été employée pour l’autre énantiomère 1Λ, en utilisant le complexe K3〚ΔCo(ox)3〛·3 H2O {〚α〛D = +1 667 (c = 0,02 dans H2O) (rotation maximale d’après 〚16〛 : 〚α〛D = +1 928)}.
C31H24N5F6PRu RMN 1H (300,13 MHz, C3D6O, 300 K, TMS) : δ = 6,48 (d, 1H), 6,85 (m, 2H), 7,02 (t, 1H), 7,36 (m, 3H), 7,57 (t, 1H), 7,70 (d, 1H), 7,76 (t, 1H), 7,91 (m, 6H), 8,03 (d, 1H), 8,12 (m, 3H), 8,57 (pseudo-t, 2H), 8,65 (d, 1H), 8,73 (d,1H).
4.3.2 Préparation de (2Δ) et (2Λ)
Dans un tube à hémolyse, une solution aqueuse (0,2 ml) de K3〚ΛCo(ox)3〛·3 H2O {〚α〛D = –1 892 (c = 0,02 dans H2O) (rotation maximale trouvée d’après 〚16〛 : 〚α〛D = –1 940)} (39,4 mg, 0,08 mmol) et de MnCl2·4 H2O (15,7 mg = 0,08 mmol) est ajoutée à une solution de 〚Ru(bpy)2Quo〛PF6 (2) racémique (100,2 mg = 0,14 mmol) ; après dissolution partielle de 2 dans 0,4 mL de DMSO, on complète la dissolution du complexe par un ajout d’acétone). Le mélange est agité pendant 45 min, puis on ajoute de l’éther (10 mL). Le précipité rouge–brun est filtré, lavé à l’acétone et à l’éther, puis séché à l’air. On obtient ainsi le solide rouge {〚ΛRu(bpy)2(Quo)〛〚MnΛCo(ox)3〛}n (63,5 mg = 0,067 9 mmol) (rendement : 84%). Le filtrat rouge est concentré à l’évaporateur rotatif pour éliminer l’acétone et l’éther puis, après précipitation par addition d’éther (200 ml), 〚ΔRu(bpy)2Quo〛PF6 (2Δ) enrichi (50,1 mg = 0,071 3 mmol) est filtré et séché à l’air. La même procédure a été employée pour l’autre énantiomère 2Λ, en utilisant le complexe K3〚ΔCo(ox)3〛·3 H2O {〚α〛D = +1 667 (c = 0,02 dans H2O) (rotation maximale trouvée d’après 〚16〛 : 〚α〛D = +1 928)}.
C29H22N5OF6PRu RMN 1H (300,13 MHz, CD2Cl2, 300 K, TMS) : δ = 6,88 (d, 2H), 7,09 (q, 1H), 7,29 (m, 4H), 7,46 (t, 1H), 7,66 (d, 1H), 7,92 (m, 7H), 8,41 (m, 5H), 8,88 (d, 1H).
4.3.3 Préparation des sels de 〚ΔTrisphat〛 (1Δ), (1Λ), (2Δ) et (2Λ)
Nous donnerons à titre d’exemple la préparation de 〚ΔTrisphat〛 (1Δ) : 128 mg de (1Δ) PF6– sont dissous dans un minimum d’acétone avec 193 mg de 〚ΔTrisphat〛Cinconidium. La solution est agitée 30 min, puis, après évaporation du solvant, le résidu est chromatographié sur colonne d’alumine neutre (éluent : chlorure de méthylène). 0,156 g (76%) de 〚ΔTrisphat〛 (1Δ) sont ainsi récupérés.
Remerciements
Nous remercions B. Malézieux, C. Train et R. Andrès pour leur aide et leurs conseils, ainsi que O. Convert pour les études RMN des complexes 〚Ru(bpy)2L〛PF6. Nous remercions également le CNRS et l’université Pierre-et-Marie-Curie pour leur support financier. Ce travail représente une partie du travail de thèse de Muriel Brissard.