

1 Introduction
Depuis quelques dizaines d’années, les techniques séparatives à membranes suscitent d’importants efforts de recherche et de développement. En effet, outre l’intérêt technologique que présentent les procédés à membranes, ils se révèlent plus économiques que les autres techniques séparatives conventionnelles (distillation, recristallisation, extraction par solvant...), car ils ne nécessitent ni changement d’état, ni élévation de température. Ces procédés sont de plus en plus utilisés et ce, dans des secteurs d’activités aussi nombreux que variés comme les industries chimiques, agroalimentaires, pharmaceutiques, textiles, la production d’eau potable, la protection de l’environnement...
Il existe actuellement une grande variété de membranes commerciales, à la fois de par la nature du matériau utilisé (organique ou inorganique) et de par leur structure (dense ou poreuse, symétrique ou asymétrique, souvent composite).
De façon générale, une membrane peut être décrite comme une barrière sélective permettant le passage préférentiel d’une espèce par rapport aux autres, sous l’effet d’une force de transfert. La micro-, l’ultra- et la nanofiltration (MF, UF et NF) désignent des procédés de séparation en phase liquide, pour lesquels la force de transfert est un gradient de pression. Ces procédés utilisent des membranes poreuses dont les diamètres de pores diminuent progressivement quand on passe de la MF (0,1–10 μm) à l’UF (2–100 nm), puis à la NF (< 2 nm).
La sélectivité (ou pouvoir de séparation) d’une membrane poreuse vis-à-vis d’un mélange de solutés chargés (ions, molécules) résulte à la fois d’un « effet tamis », c’est-à-dire d’une discrimination en fonction de la taille de l’espèce à retenir, et des effets de charges électriques découlant de la nature chimique du matériau membranaire. En effet, la plupart des membranes, organiques et inorganiques, acquièrent une charge de surface lorsqu’elles sont mises en contact avec un milieu aqueux. L’origine de cette charge peut provenir du caractère acide ou basique des groupements de surface (membranes organiques possédant par exemple des groupes amide, carboxyle ou sulfone) ou des propriétés amphotères des sites de surface (fonctions hydroxyle à la surface des oxydes métalliques constituant les membranes céramiques). L’adsorption d’espèces chargées (ions, polyelectrolytes, surfactants ioniques) à la surface du matériau peut également être responsable de la charge de surface, ou du moins y contribuer.
Il est donc indispensable, pour comprendre et maîtriser la sélectivité d’une membrane poreuse, de déterminer, non seulement ses caractéristiques structurales (diamètre de pore moyen, distribution de la taille des pores, tortuosité), mais aussi ses caractéristiques électriques de surface, telles que la charge ou le potentiel de surface de la paroi des pores.
Dans cet article sont présentées quatre méthodes expérimentales – potentiel d’écoulement, électro-osmose, potentiel de membrane et conductivité de pores – permettant la caractérisation des propriétés électriques des parois de pores d’une membrane. Des exemples illustrant l’influence du pH, de la concentration de l’électrolyte ou de sa nature sur les quatre grandeurs expérimentales sont ensuite présentés.
2 Interface membrane–solution
Avant d’aborder les différentes méthodes, rappelons que la présence de sites chargés à la surface d’un solide immergé dans une solution influence la distribution spatiale des espèces chargées en solution au voisinage de la surface. Les forces électrostatiques dues à la charge de surface attirent les espèces de charge opposée et repoussent celles de même charge. Le potentiel électrostatique exercé par la charge de surface varie progressivement au sein d’une zone appelée « double couche électrique ». Le modèle le plus couramment utilisé pour décrire la structure de la double couche électrique (« modèle Gouy–Chapman–Stern–Grahame ») 〚1〛, suggère que cette réorganisation conduise à la formation de deux zones distinctes, la couche compacte (ou couche de Stern) et la couche diffuse. L’épaisseur de cette dernière varie notamment avec la force ionique de la solution : elle diminue lorsque la force ionique augmente. L’épaisseur de la couche diffuse est généralement considérée comme étant égale à la longueur de Debye (κ–1). Cette grandeur constitue un paramètre utile pour estimer la portée des interactions électrostatiques. On utilise fréquemment le rapport entre le rayon de pore moyen d’une membrane et la longueur de Debye pour estimer la portion occupée par la couche diffuse dans le pore (comparativement à celle occupée par le canal électriquement neutre, c’est-à-dire par la solution de cœur). Ce rapport, appelé rayon électrocinétique, indique si les doubles couches électriques se formant à l’intérieur des pores peuvent se développer complètement ou si, au contraire, elles se chevauchent (phénomène de recouvrement de double couche 〚2〛).
Lorsque la phase liquide est mise en mouvement à travers les pores, la partie compacte de la double couche électrique reste solidaire de la surface du solide. La couche diffuse glisse donc par rapport à la couche compacte. Le plan de glissement entre les deux couches est appelé plan de cisaillement. Les méthodes permettant le déplacement tangentiel d’une phase (liquide ou solide) par rapport à l’autre (solide ou liquide) constituent les méthodes électrocinétiques. Le potentiel électrocinétique ou potentiel zêta (ζ) correspond à la valeur du potentiel électrostatique au niveau du plan de cisaillement. La position de ce plan n’est pas connue exactement, mais il est généralement admis que celui-ci est pratiquement confondu avec le plan externe de Helmholtz (PHE), qui sépare la couche compacte de la couche diffuse 〚3, 4〛. Le potentiel zêta est donc considéré comme une bonne approximation du potentiel Ψd, potentiel situé dans le PHE 〚5, 6〛.
Le potentiel zêta (ou potentiel Ψd) ne peut être mesuré directement, mais doit être déterminé à partir de mesures de grandeurs électrocinétiques telles que le coefficient de potentiel d’écoulement ou le débit électro-osmotique, de grandeurs électriques telles que la conductivité de pores ou de grandeurs électrochimiques telles que le potentiel de membrane. Ces méthodes se différentient par la nature de la force motrice utilisée et du signal mesuré (Tableau 1).
Méthodes de caractérisation des propriétés électriques des parois de pores d’une membrane.
| Nature de la force motrice | Nature du signal mesuré | ||
| Grandeurs électrocinétiques | coefficient de potentiel d’écoulement (SP) | mécanique (gradient de pression) | électrique (ddp) |
| débit électro-osmotique (V) | électrique (ddp) | hydraulique (flux volumique) | |
| Grandeurs électrochimiques | potentiel de membrane (Δϕm) | chimique (gradient de concentration) | électrique (ddp) |
| Grandeurs électriques | conductivité de pores (λpore) | électrique (courant) | électrique (ddp) |
3 Potentiel d’écoulement
La mesure du potentiel d’écoulement est la méthode la plus utilisée pour déterminer le potentiel électrocinétique de la paroi des pores d’une membrane 〚7–20〛. Cette technique présente l’avantage de permettre le suivi de l’évolution des propriétés électrocinétiques d’une membrane au cours du processus de filtration. Le potentiel d’écoulement peut ainsi être utilisé pour étudier l’influence des phénomènes de colmatage sur les propriétés de surface d’une membrane 〚21, 22〛, pour contrôler l’efficacité d’un cycle de nettoyage d’une surface membranaire 〚19〛, ou bien encore pour suivre le vieillissement d’un matériau membranaire au cours du temps 〚23〛.
3.1 Définition et description du phénomène
Pour introduire la notion de potentiel d’écoulement, considérons une membrane poreuse, chargée au contact d’un liquide contenant des espèces chargées. Un excès de charge s’installe dans la double couche électrique se développant à l’interface solide–liquide. Lorsqu’un gradient de pression est exercé à travers les pores de la membrane, les charges présentes dans la partie diffuse de la double couche électrique sont entraînées avec le liquide vers le compartiment basse pression, créant ainsi un courant d’écoulement Is (Fig. 1a). Le gradient de potentiel électrique résultant du déséquilibre de charges produit entre les extrémités des pores par le gradient de pression génère un courant de conduction Ic, opposé au flux convectif des charges 〚2〛. Un état stationnaire se traduisant par l’égalité des courants d’écoulement et de conduction est très vite atteint (I = Is + Ic = 0). La différence de potentiel électrique aux extrémités des pores représente le potentiel d’écoulement (Δϕ) (Fig. 1b) 〚24〛. La valeur du potentiel d’écoulement étant fonction de la différence de pression appliquée (ΔP), une autre grandeur lui est généralement préférée pour décrire le phénomène. Il s’agit du rapport Δϕ/ΔP, appelé coefficient de potentiel d’écoulement et noté SP :

Origine du potentiel d’écoulement.
3.2 Mesure du potentiel d’écoulement
Sur le plan expérimental, la différence de potentiel électrique (Δϕ) est mesurée à l’aide de deux électrodes réversibles (Ag/AgCl), placées de part et d’autre de la membrane et connectées à un millivoltmètre à haute impédance d’entrée, de telle sorte qu’aucun courant ne circule dans le système (I = 0). Il n’y a effectivement qu’à cette condition que la polarisation peut avoir lieu. La procédure usuelle de mesure du potentiel d’écoulement peut se résumer ainsi (Fig. 2) : la solution est tout d’abord mise en mouvement à travers la membrane sous une différence de pression transmembranaire ΔP0, de façon à équilibrer la membrane avec la solution d’étude (le rétentat et le perméat – ou filtrat – étant continuellement recyclés vers le réservoir d’alimentation pour maintenir constante la concentration de la solution d’alimentation). Une fois l’équilibre atteint, des échelons de pression d’amplitude croissante sont alors appliqués et la différence de potentiel électrique résultante enregistrée. Le coefficient de potentiel d’écoulement est déterminé en considérant les augmentations brusques de la différence de potentiel électrique transmembranaire (Δϕ) consécutives aux incréments de pression (ΔP). Le tracé de Δϕ en fonction de ΔP est une droite, dont la pente représente le coefficient de potentiel d’écoulement.

Représentation schématique d’une séquence de mesure du potentiel d’écoulement.
3.3 Influence du pH
Afin de déterminer la variation du potentiel zêta d’une membrane avec le pH de la solution (due à l’ionisation des sites de surface et à l’adsorption d’espèces chargées), une expérience consiste à mesurer le coefficient de potentiel d’écoulement en fonction du pH, à force ionique constante. Dans le cas des membranes céramiques, par exemple, le caractère amphotère des groupements hydroxyles de surface induit des valeurs positives ou négatives du coefficient de potentiel d’écoulement, selon la valeur du pH (Fig. 3). En effet, la variation du pH de la solution entraîne un déplacement des équilibres acido-basiques de surface, modifiant ainsi la charge de surface (charge due aux sites de surface). Il en résulte une modification de la quantité de contre-ions dans la couche diffuse (pour assurer l’électroneutralité) et donc une variation du coefficient de potentiel d’écoulement. Le pH particulier pour lequel le coefficient de potentiel d’écoulement s’annule définit le point isoélectrique (pie) du système membrane–solution, c’est-à-dire le pH pour lequel la charge nette (charge globale de la phase stationnaire « surface + couche compacte ») est nulle. À pH < pie, la charge nette est positive ; la contre-charge présente dans la couche diffuse est alors négative (excès d’anions par rapport aux cations). Lorsqu’un gradient de pression est imposé, les ions de cette couche sont déplacés vers le compartiment basse pression, d’où un coefficient de potentiel d’écoulement positif. À l’inverse, à pH > pie, la charge nette étant négative, la couche diffuse contient alors un excès de cations (par rapport aux anions), d’où un coefficient de potentiel d’écoulement négatif. On notera donc que le signe du coefficient de potentiel d’écoulement reflète le signe de la charge nette de la membrane et que le tracé de la courbe SP–pH permet la détermination du pie, grandeur aussi indispensable au choix d’une membrane, pour une application donnée, que le seuil de coupure.
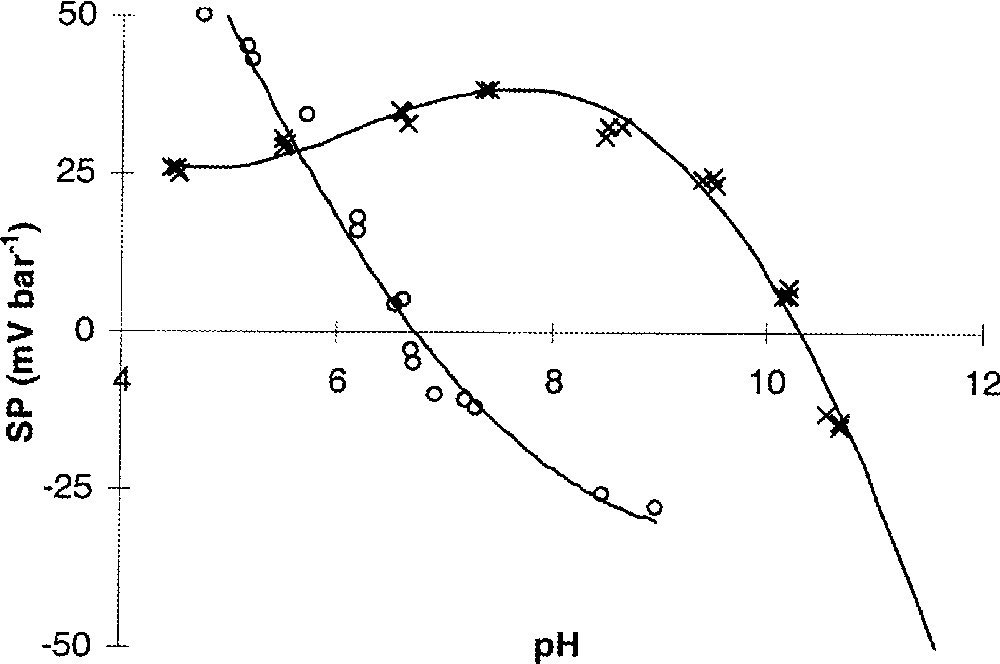
Coefficient de potentiel d’écoulement (SP) en fonction du pH ; KCl 0,006 mol l–1. ○ : Membrane UF Al2O3/TiO2/ZrO2 (seuil de coupure : 2 kD), Tami Industrie. × : Membrane UF Al2O3 non commercialisée.
La courbe SP–pH peut parfois présenter un maximum (exemple de la membrane Al2O3 sur la Fig. 3). Plusieurs phénomènes peuvent expliquer ce comportement. Considérons une membrane chargée positivement. La chute du coefficient de potentiel d’écoulement lorsque le pH diminue est souvent observée à bas pH. Dans ce cas, la diminution de SP résulte de l’augmentation de la force ionique due à l’ajout d’une solution acide pour abaisser le pH. L’augmentation de la force ionique s’accompagne d’une compression de la couche diffuse. Moins de contre-ions sont alors présents dans la couche diffuse et, par conséquent, moins de contre-ions seront déplacés sous l’action d’un gradient de pression. De surcroît, de par leur mobilité élevée, les ions hydronium augmentent aussi de façon notable la conductivité de la solution de cœur, ce qui a pour effet de diminuer la polarisation de la membrane (ce point sera discuté ultérieurement). Dans l’exemple donné sur la Fig. 3, ces deux causes (augmentation de la force ionique et de la conductivité de cœur) ne peuvent être invoquées, puisque la décroissance du coefficient de potentiel d’écoulement survient à des valeurs de pH où la quantité de protons n’est pas suffisante pour augmenter significativement la force ionique et la conductivité de la solution de cœur. Pour comprendre l’allure de ces courbes, il est nécessaire de considérer la relation entre le coefficient de potentiel d’écoulement et une grandeur fondamentale caractéristique de l’interface membrane–solution, telle que le potentiel zêta.
3.4 Coefficient de potentiel d’écoulement et potentiel zêta
Le coefficient de potentiel d’écoulement est le plus souvent relié au potentiel zêta par le biais de la relation d’Helmholtz–Smoluchowski 〚25〛 :
La relation d’Helmholtz–Smoluchowski montre bien que le coefficient de potentiel d’écoulement diminue lorsque la conductivité de la solution (λ0) augmente. Toutefois, cette relation souffre des restrictions suivantes : faible potentiel zêta et grands rayons électrocinétiques 〚26〛. Comme la valeur absolue du potentiel zêta n’est a priori pas connue, elle ne permet donc, d’une manière générale, que la détermination d’une valeur apparente du potentiel zêta.
Plusieurs auteurs ont proposé des corrections à apporter à l’équation d’Helmholtz–Smoluchowski afin d’accéder à la valeur réelle du potentiel zêta. Rice et Whitehead 〚27〛 ont établi de manière analytique un facteur de correction à appliquer à l’équation d’Helmholtz–Smoluchowski. Leur théorie est limitée aux faibles valeurs de potentiel zêta (ζ < 25 mV pour un électrolyte 1:1), du fait de l’emploi de l’approximation de Debye–Hückel, qui permet la linéarisation de l’équation de Poisson–Boltzmann. Levine et al. 〚28〛 ont appliqué la théorie de Rice et Whitehead aux fortes valeurs de potentiel zêta en développant des approximations analytiques pour la résolution de l’équation de Poisson–Boltzmann. Leur approche est cependant limitée à l’étude d’électrolytes symétriques monovalents, dont les deux espèces ioniques possèdent des mobilités identiques. Plus récemment, Bowen et Cao 〚29〛 ont étendu l’approche de Levine et al. 〚28〛 à des électrolytes symétriques, possédant des mobilités ioniques différentes, et à des valeurs quelconques du potentiel zêta, en résolvant numériquement l’équation non linéaire de Poisson–Boltzmann. Ces auteurs ont montré que l’emploi de l’équation d’Helmholtz–Smoluchowski peut conduire à une sous-estimation importante de la valeur réelle du potentiel zêta (principalement pour des faibles rayons électrocinétiques et/ou des surfaces fortement chargées) ainsi qu’à une interprétation erronée de l’évolution des propriétés électrocinétiques d’une membrane en fonction de son environnement physico-chimique.
Le coefficient de potentiel d’écoulement peut aussi être relié au potentiel zêta (sans aucune restriction sur le rayon électrocinétique ni sur le potentiel zêta), en s’appuyant sur la thermodynamique des processus irréversibles 〚30〛 pour décrire le transport d’un électrolyte à l’intérieur d’un capillaire chargé et en considérant un modèle de charge d’espace 〚31–33〛. Ce dernier repose sur les équations de Nernst–Planck, Navier–Stokes et Poisson–Boltzmann, pour décrire respectivement le transport des ions et du solvant ainsi que la variation radiale du potentiel électrostatique.
La Fig. 4 présente la variation du coefficient du potentiel d’écoulement en fonction du potentiel zêta obtenue par un modèle de charge d’espace 〚30, 34〛. On remarque que la relation d’Helmholtz–Smoluchowski n’est valable que pour des pores suffisamment grands et peu chargés, que le SP dépend de la taille des pores (ce que ne montre pas la relation d’Helmholtz–Smoluchowski) et que la courbe passe par un maximum. La diminution du coefficient du potentiel d’écoulement avec l’augmentation du potentiel zêta est due à la conduction de surface du matériau. En effet, pendant le processus du potentiel d’écoulement, le champ électrique créé par le gradient de pression engendre un flux retour des contre-ions dans les pores (par rétro-migration), entraînant ainsi une dépolarisation partielle de la membrane, le flux retour des contre-ions dans les pores étant d’autant plus important que la conductance de surface est élevée. La conductance de surface désigne un excès de conduction dans les pores, dû à la présence de charges excédentaires dans la double couche électrique 〚4〛. La conductance de surface croît de façon exponentielle avec le potentiel zêta 〚30〛.

Coefficient de potentiel d’écoulement SP (calculé à partir d’un modèle de charge d’espace 〚30, 34〛) en fonction du potentiel zêta ζ pour différents rayons de pore a ; KCl 0,001 mol l–1 (κ–1 ∼ 10 nm).
La représentation de l’évolution du coefficient de potentiel d’écoulement en fonction de la conductance de surface fait également apparaître un maximum (Fig. 5), ce qui prouve que la conductance de surface est bien à l’origine de la diminution du potentiel d’écoulement observée aux fortes valeurs du potentiel zêta (Fig. 4).

Coefficient de potentiel d’écoulement SP (calculé à partir d’un modèle de charge d’espace 〚30〛) en fonction de la conductance de surface Gs pour différents rayons de pore a ; KCl 0,001 mol l–1 (κ–1 ∼ 10 nm).
En résumé, l’augmentation du potentiel zêta conduit à une augmentation du nombre de contre-ions dans la double couche électrique, qui, à son tour, est à l’origine d’une augmentation de la conductivité du pore. Tant que la conductance de la région située près de la paroi du pore reste suffisamment faible, le coefficient de potentiel d’écoulement croît avec le potentiel zêta, ce qui est dû à un plus grand nombre de charges déplacées par le gradient de pression. Le phénomène d’écoulement des charges l’emporte sur le phénomène d’électromigration. À partir d’une certaine valeur du potentiel zêta, c’est le phénomène d’électromigration qui prédomine, du fait d’une conductance de surface très élevée. Dans ce cas, le flux retour des contre-ions devient si important que la polarisation axiale du pore diminue, d’où la diminution du coefficient de potentiel d’écoulement.
On notera que l’allure des courbes SP–ζ (Fig. 4) implique que deux valeurs de potentiel zêta peuvent être associées à une même valeur de coefficient de potentiel d’écoulement. Expérimentalement, cette ambiguïté peut être levée si l’on effectue plusieurs mesures de potentiel d’écoulement en fonction du pH. En effet, à force ionique constante, seul le pH influence la valeur du coefficient du potentiel d’écoulement. Dans ce cas, le pH le plus proche du point isoélectrique correspond au potentiel zêta le plus faible. Le potentiel d’écoulement correspondant à ce pH sera alors associé au potentiel zêta le plus petit.
La dépendance du coefficient de potentiel d’écoulement en fonction du rayon de pore (Fig. 4) est également due à la conduction de surface, dont la contribution au phénomène global de conduction à l’intérieur des pores est d’autant plus élevée que le rayon électrocinétique est petit.
On notera, par ailleurs, que la courbe SP–ζ est d’autant plus aplatie que le rayon de pore diminue (Fig. 4). En conséquence, la méthode de mesure du potentiel d’écoulement s’avère être plus adaptée à la caractérisation de membranes présentant des pores de grandes dimensions plutôt que des pores de faibles dimensions (incertitude sur la valeur du potentiel zêta plus faible pour les grands pores) 〚34〛.
La Fig. 6 montre clairement que l’utilisation de la relation d’Helmholtz–Smoluchowski, qui ne prend pas en compte le phénomène de conductance de surface, peut dans certains cas conduire à une sous-estimation importante de la valeur du potentiel zêta.

Potentiel zêta (ζ) en fonction du pH ; KCl 0,006 mol l–1 ; membrane Al2O3 (identique à celle de la Fig. 3). ○ : ζ calculé à partir d’un modèle de charge d’espace 〚30〛. × : ζ calculé à partir de l’équation (2) (relation d’Helmholtz–Smoluchowski).
4 Électro-osmose
L’examen de la littérature montre que le potentiel zêta d’une membrane peut également être déterminé à partir de mesures de débit électro-osmotique 〚16, 35–44〛. Toutefois, la technique d’électro-osmose est peu utilisée, en raison notamment des difficultés expérimentales de mise en œuvre des mesures 〚45〛. Par ailleurs, les dispositifs expérimentaux développés jusqu’à présent sont limités à l’étude de membranes planes.
4.1 Définition et description du phénomène
Le principe de l’électro-osmose repose sur la mise en mouvement d’une phase liquide adjacente à un solide chargé sous l’action d’une force électrique (différence de potentiel électrique ou courant), appliquée parallèlement à la surface du solide chargé.
L’application d’un champ électrique à travers une membrane poreuse chargée contenant une solution électrolytique provoque la migration des espèces ioniques situées au-delà du plan de cisaillement. Les ions, selon le signe de leur charge, se déplacent en direction des électrodes de polarité opposée, entraînant le solvant par viscosité. Si cations et anions possèdent des mobilités voisines, le flux global dans le canal électriquement neutre est nul. Le flux de solvant mesuré, résultant alors des seuls ions présents dans la partie diffuse de la double couche électrique, est appelé « débit électro-osmotique ».
4.2 Mesure du débit électro-osmotique
Sur le plan expérimental, la détermination du flux électro-osmotique se fait donc en réalisant l’électrolyse d’une solution électrolytique, l’anode et la cathode étant positionnées de part et d’autre de la membrane. Il est préférable de conduire l’électrolyse à intensité de courant constante plutôt qu’à tension constante. En effet, au cours d’une électrolyse à tension constante, l’intensité du courant baisse considérablement dans les premières minutes de l’électrolyse ; il n’est pas possible de la maintenir constante en fonction du temps 〚46〛.
Le débit électro-osmotique peut être déterminé, par exemple, en pesant le liquide traversant la membrane au cours du temps (Fig. 7) 〚16, 44〛. La masse de liquide recueilli croît linéairement avec le temps d’électrolyse (Fig. 8), la pente de la droite correspondant au débit électro-osmotique. Celui-ci diminue lorsque la concentration électrolytique augmente. En effet, l’augmentation de la concentration s’accompagne d’une compression de la couche diffuse. Davantage de contre-ions s’accumulent dans le PHE (ou pénètrent plus ou moins dans la couche compacte), neutralisant ainsi une part plus importante de la charge de surface. Il en résulte une diminution de l’excès de contre-ions (par rapport aux co-ions) dans les pores. Puisque co-ions et contre-ions migrent dans des directions opposées, le débit électro-osmotique résultant est plus faible.

Dispositif expérimental de mesure du débit électro-osmotique, selon 〚16, 44〛.

Masse de liquide déplacé par électro-osmose en fonction du temps ; NaCl ; membrane Al2O3/SiO2 (MF) 〚16, 44〛.
Il est à noter que les manipulations d’électro-osmose nécessitent des volumes de solution importants (∼10 l) ainsi que des temps de mesure relativement courts, afin que les variations de concentration et de pH dues à l’électrolyse soient négligeables.
4.3 Débit électro-osmotique et potentiel zêta
Dans le cas de l’électro-osmose, la relation d’Helmholtz–Smoluchowski s’écrit :
Comme pour le potentiel d’écoulement, la relation d’Helmholtz–Smoluchowski ne permet de déterminer la valeur exacte du potentiel zêta que si les pores sont suffisamment grands devant l’épaisseur de la double couche électrique et relativement peu chargés ; sinon, l’utilisation d’un modèle plus réaliste, tel qu’un modèle de charge d’espace, s’avère nécessaire.
4.4 Influence de la nature de l’électrolyte
Les mesures de débit électro-osmotique (comme les mesures de potentiel d’écoulement) permettent de mettre en évidence l’adsorption spécifique d’ions à la surface du matériau. Pour ces ions (dits non indifférents), l’interaction avec la surface n’est pas de nature purement électrostatique. Les forces supplémentaires mises en jeu peuvent être de nature physique ou chimique. L’adsorption spécifique de cations déplace le pie vers les pH plus élevés, alors que l’adsorption spécifique d’anions déplace celui-ci vers les pH plus bas (Fig. 9). Le déplacement est d’autant plus important que la concentration des ions non indifférents est élevée. Le pie ne peut donc être considéré comme une propriété intrinsèque d’un matériau membranaire amphotère. Il est caractéristique d’un couple membrane–solution.
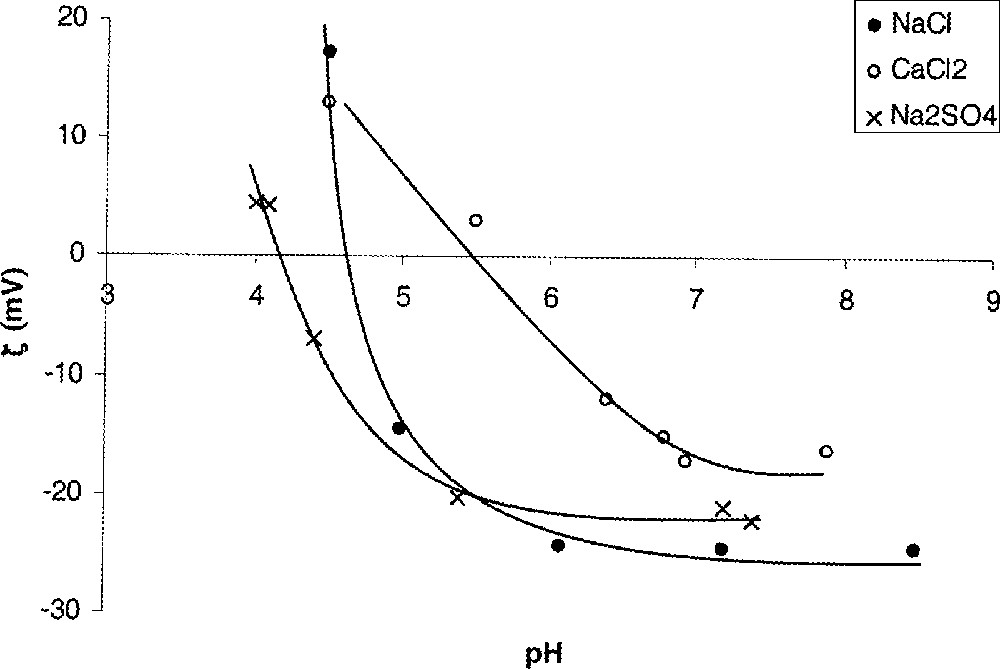
Potentiel zêta (ζ) – déterminé à partir de mesures de débit électro-osmotique – en fonction du pH pour différents électrolytes ; membrane MF Al2O3/SiO2 (identique à celle de la Fig. 8).
5 Conductivité de pores
La conductivité d’une solution à l’intérieur des pores d’une membrane chargée est généralement plus élevée que la conductivité de la solution située à l’extérieur des pores (conductivité de cœur), ce qui est dû à l’excès de charges présentes dans la double couche électrique. Il est donc possible, à partir des mesures de conductivité de pores, d’accéder au potentiel zêta par le biais d’un modèle tel qu’un modèle de charge d’espace 〚47, 48〛.
5.1 Mesure de résistance électrique et détermination de la conductivité de pores
La détermination expérimentale de la conductivité de pores requiert des mesures de résistance électrique de membrane. La méthode galvanostatique à quatre électrodes est la plus adaptée pour ce type de mesures 〚34, 47〛. Elle utilise une cellule de mesure à quatre électrodes (deux électrodes de courant et deux électrodes de tension) sans contact électrodes–membrane. L’absence de contact direct entre la membrane et les électrodes évite que les mesures soient éventuellement perturbées par l’existence d’une impédance d’interface (entre les électrodes et la membrane), qui est par ailleurs fonction de la force de serrage exercée sur les électrodes.
L’utilisation d’une cellule sans contact électrodes–membrane nécessite deux mesures pour déterminer la résistance de la membrane, puisque la résistance mesurée Rcell est alors la somme de deux résistances : la résistance de la membrane Rm et la résistance des deux colonnes d’électrolytes Rsol, qui se trouvent entre la membrane et les électrodes de tension (Fig. 10) :
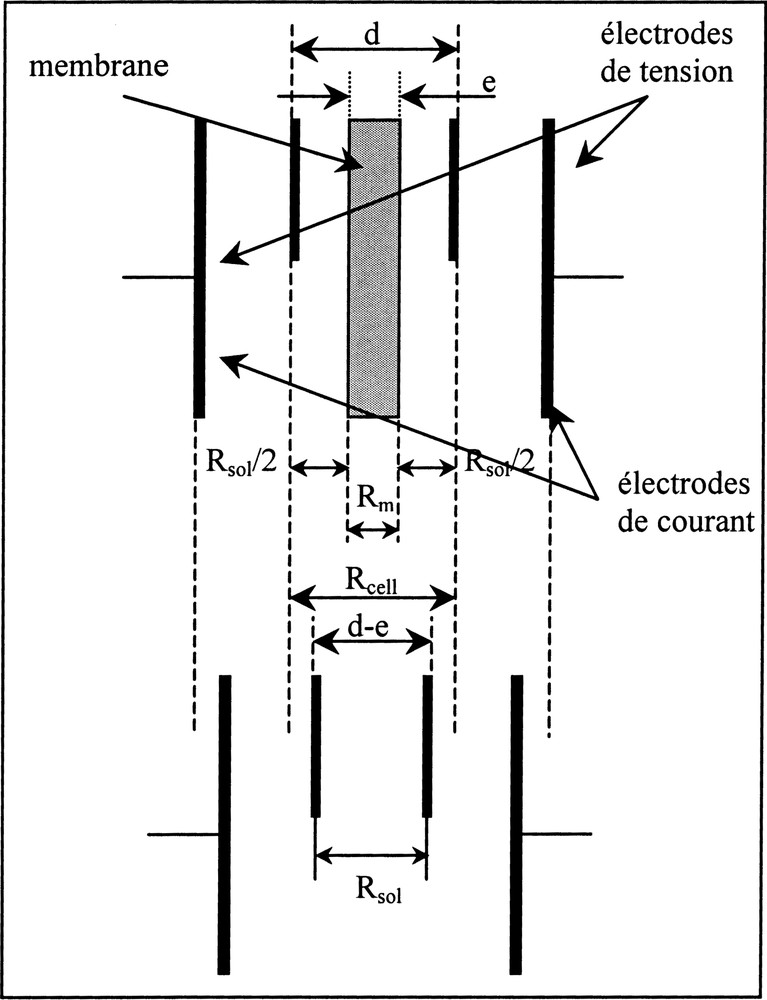
Principe de mesure de la résistance électrique d’une membrane (méthode galvanostatique à quatre électrodes).
La distance entre les électrodes de tension doit être la plus courte possible, de façon à minimiser la valeur de Rsol par rapport à celle de Rm.
Deux mesures de résistance électrique sont donc réalisées successivement : l’une sur le système membrane–solution (Rcell) et l’autre sur la solution seule (Rsol) (Fig. 10).
La résistance de la membrane est reliée à la conductivité de pores par la relation :
Le rapport l/Ap est une constante, qui ne dépend que des caractéristiques structurales du matériau poreux.
Pour une concentration électrolytique donnée, la détermination de la conductivité de pores (λpore) se fait en mesurant la résistance de la membrane à cette concentration, puis en milieu très concentré (en général, 1 mol l–1), pour lequel on a :
En milieu très concentré, l’influence de la surface devient négligeable et on considère que la conductivité de l’électrolyte est la même à l’intérieur et à l’extérieur des pores 〚2〛. Ainsi, en identifiant les équations (5) et (6), on obtient :
5.2 Influence du pH et de la concentration
L’étude de la variation du rapport λpore/λ0 en fonction du pH permet la détermination du pie du système membrane–solution (Fig. 11). En effet, au pie, la courbe passe par un minimum correspondant à un rapport λpore/λ0 égal à 1. Lorsque la membrane n’est pas chargée (potentiels ζ ou Ψd = 0 mV), la zone située près de la surface contient autant de cations que d’anions et par conséquent, la conductivité de pores (λpore) est identique à celle de la solution externe (λ0). Lorsqu’on s’éloigne du pie, la charge nette de surface augmente. La double couche électrique renferme alors un excès de contre-ions, qui rend la solution dans les pores plus conductrice que la solution se trouvant à l’extérieur des pores (λ0).

Rapport de la conductivité de pores sur la conductivité de la solution de cœur (λpore/λ0) en fonction du pH ; KCl 0,006 mol l–1 ; membrane UF Al2O3 (a = 75 nm) 〚48〛.
À la différence du coefficient du potentiel d’écoulement et du débit électro-osmotique, on notera qu’une seule mesure de conductivité de pores ne permet pas de conclure quant au signe de la charge membranaire (Fig. 12). Une étude de la conductivité en fonction du pH s’avère alors nécessaire pour lever cette incertitude. Pour une membrane chargée positivement, l’augmentation du pH s’accompagnera d’une diminution de la conductivité de pores, alors que pour une membrane chargée négativement, une augmentation de la conductivité de pores sera au contraire observée.

Rapport de la conductivité de pores sur la conductivité de la solution de cœur (λpore/λ0), calculé à partir d’un modèle de charge d’espace 〚30, 34〛, en fonction du pH ; KCl 0,006 mol l–1 ; a = 75 nm.
On conçoit aisément que ce type de mesures reste cependant limité aux faibles rayons électrocinétiques. En effet, pour un rayon de pore donné, l’augmentation de la concentration de l’électrolyte s’accompagne, d’une part, d’une diminution de l’épaisseur de la couche diffuse et donc, d’une augmentation du volume du canal électriquement neutre à l’intérieur du pore et, d’autre part, d’un accroissement de la conductivité de la solution (λ0) dans le canal électriquement neutre. En conséquence, la contribution de la surface à la conductivité du pore diminue par rapport à celle du canal électriquement neutre. La conductivité de pores λpore se rapproche alors de celle de la solution λ0 (Fig. 13).
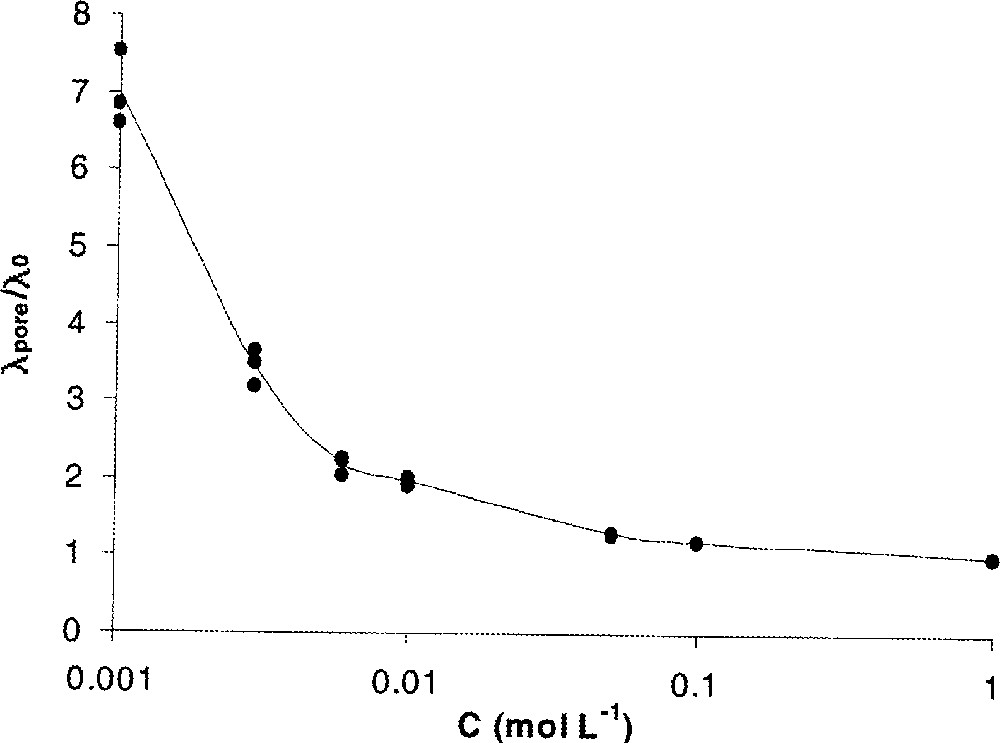
Rapport de la conductivité de pores sur la conductivité de la solution de cœur (λpore/λ0) en fonction de la concentration ; KCl ; pH = 5,6 ; membrane UF Al2O3 (a = 75 nm) 〚48〛.
Comme pour les mesures électrocinétiques (coefficient de potentiel d’écoulement et débit électro-osmotique), le potentiel zêta peut également être déduit des mesures de conductivité de pores via un modèle de charge d’espace.
6 Potentiel de membrane
L’étude du transport d’un soluté à travers une membrane poreuse sous l’action d’un gradient de concentration permet aussi d’obtenir des informations sur l’état de charge des parois de pores d’une membrane 〚18, 49〛. Toutefois, cette méthode ne peut être appliquée à n’importe quelle membrane. En effet, si les rayons des pores sont trop grands (membranes de MF ou de haute UF), la diffusion des espèces chargées à travers les pores de la membrane ne sera pas perturbée par la charge nette de surface. Il est cependant possible, dans certains cas, de réduire le rayon électrocinétique du pore, en travaillant à faible force ionique, de manière à ce que la portée des interactions électrostatiques soit plus grande. Le transport des espèces chargées est alors régi par un phénomène mixte d’exclusion–diffusion 〚50〛.
6.1 Description du phénomène d’exclusion–diffusion
Lorsque deux solutions d’un même électrolyte à des concentrations différentes (CI et CII) sont séparées par une membrane poreuse chargée, une différence de potentiel électrique, appelée potentiel de membrane, s’établit entre les deux solutions. Ce potentiel résulte de la différence de mobilité des cations et anions (phénomène de diffusion) ainsi que de l’influence de la charge électrique de la paroi des pores sur le transport des ions (phénomène d’exclusion).
Si la membrane n’est pas chargée, à l’instant initial, le transport des ions résulte seulement du phénomène de diffusion des ions, depuis la solution la plus concentrée vers la solution la moins concentrée. Comme les mobilités des ions sont généralement différentes, l’un des deux types d’ions diffuse plus rapidement à travers les pores et les ions les plus lents restent en arrière. La séparation de charges ainsi créée induit un champ électrique à travers la membrane (établissement d’un potentiel de diffusion), qui s’oppose au déplacement des espèces les plus mobiles et, au contraire, accélère les plus lents jusqu’à ce que leurs flux soient égaux. Ainsi, après un régime transitoire très bref, il s’établit un régime stationnaire de diffusion, pendant lequel le flux ionique est électriquement neutre.
Le potentiel de diffusion (Δϕdiff), résultant de la différence de mobilité des cations et des anions, peut être exprimé par l’équation de Planck–Henderson, qui, pour un système binaire, s’écrit (avec CII > CI) :
Si la membrane est chargée, les forces électrostatiques dues à la charge de surface attirent les contre-ions et repoussent les co-ions. La double couche électrique renferme donc un excès de contre-ions et un déficit de co-ions. Sous l’action d’une différence de concentration appliquée de part et d’autre de la membrane, les contre-ions tendent à diffuser vers la solution la plus diluée, alors que les co-ions, exclus (partiellement ou totalement) du pore, communiquent à la solution la plus concentrée le signe de leur charge.
Lorsque les pores de la membrane sont petits et fortement chargés, le phénomène d’exclusion devient prépondérant ; en revanche, lorsque les pores sont grands ou peu chargés, c’est le phénomène de diffusion qui prédomine.
6.2 Mesure du potentiel de membrane
La membrane est tout d’abord équilibrée en faisant circuler sous pression une solution de concentration moyenne Cm (moyenne arithmétique ou logarithmique des concentrations CI et CII), à un pH identique à celui des solutions I et II. La membrane est ensuite insérée entre les deux compartiments d’une cellule de mesure contenant les solutions I et II. Dans chaque compartiment, la solution doit être continuellement renouvelée, afin de maintenir constante sa concentration au cours de l’expérience. De même, une agitation efficace des solutions au voisinage de la membrane s’avère nécessaire afin de limiter la création d’un gradient de concentration à l’interface membrane–solution (phénomène de polarisation de concentration). Deux électrodes Ag/AgCl placées de part et d’autre de la membrane permettent la mesure du potentiel de cellule (Δϕcell), lequel peut être décomposé de la manière suivante :
Dans l’équation (9), le terme (ϕ′ – ϕII) + (ϕI – ϕ) représente la différence de potentiel due aux concentrations différentes de part et d’autre de la membrane. Cette différence de potentiel, appelée potentiel de concentration (Δϕc), est égale, pour des solutions diluées d’un électrolyte symétrique (z+ = –z– = z), à :
Le potentiel de membrane (Δϕm) est défini comme la différence du potentiel interne de la solution la plus concentrée et du potentiel interne de la solution la moins concentrée 〚51〛, soit :
L’équation (9) peut donc être réécrite sous la forme :
Le potentiel de membrane est donc obtenu expérimentalement, en soustrayant le potentiel de concentration au potentiel de cellule.
Dans le cas d’une membrane amphotère séparant deux solutions diluées d’un même électrolyte, le potentiel de membrane peut être exprimé sous la forme suivante 〚52, 53〛 :
En comparant la relation (13) à la relation (8), le potentiel de membrane apparaît donc comme un potentiel de diffusion modifié, traduisant l’influence de la charge de surface sur le transport des ions à l’intérieur des pores de la membrane. Ainsi, pour des pores non chargés, le potentiel de membrane sera égal au potentiel de diffusion, tandis que, dans le cas limite de l’exclusion totale du co-ion, il prendra une valeur égale au potentiel de Nernst (ΔϕN), soit :
6.3 Influence du pH et de la concentration
Comme le montre la Fig. 14, une seule mesure de potentiel de membrane suffit à déterminer le signe de la charge membranaire : selon que la valeur mesurée est supérieure ou inférieure à la valeur correspondant au potentiel de diffusion, on déduira que la membrane est chargée positivement ou négativement. Le pH pour lequel le potentiel de membrane est égal au potentiel de diffusion correspond au pie. Lorsque la force ionique augmente, la portée des interactions électrostatiques diminue (phénomène de compression de double couche), du fait d’un écrantage plus important de la charge de surface par les contre-ions. Il en résulte que le potentiel de membrane se rapproche de la valeur du potentiel de diffusion.

Potentiel de membrane (Δϕm) en fonction du pH pour différentes concentrations ; CII/CI = 2 ; NaCl ; membrane UF Al2O3/TiO2/ZrO2.
L’utilisation d’un modèle tel qu’un modèle de charge d’espace permet, là encore, de pouvoir calculer le potentiel zêta (ou Ψd) à partir de mesures de potentiel de membrane 〚55〛.
Les principales caractéristiques des quatre méthodes présentées dans cet article sont regroupées dans le Tableau 2.
Caractéristiques des différentes méthodes permettant la détermination du potentiel zêta d’une membrane.
| Méthodes | Types de membranes | Géométrie des membranes | Informations obtenues à partir d’une seule mesure | Avantages/inconvénients |
| Potentiel d’écoulement | MF et UF | Plane ou tubulaire | • Signe de ζ. | • Mesures pouvant être effectuées sur le pilote de filtration lui-même. |
| • Deux valeurs de ζ sont possibles. | • Simplicité expérimentale. | |||
| Electro-osmose | MF et UF | Plane | • Signe de ζ. | • Mesures ne pouvant être effectuées sur le pilote de filtration. |
| • Deux valeurs de ζ sont possibles. | • Difficultés expérimentales 〚45〛. | |||
| Conductivité de pores | UF et NF | Plane | • Ni signe, ni valeur de ζ. | • Mesures ne pouvant être effectuées sur le pilote de filtration. |
| • Peu sensible aux faibles valeurs de ζ 〚34〛. | ||||
| Potentiel de membrane | UF et NF | Plane | • Signe et valeur de ζ. | • Mesures ne pouvant être effectuées sur le pilote de filtration. |
| • Peu sensible aux fortes valeurs de ζ 〚34〛. | ||||
| • Nécessité de maîtriser le phénomène de polarisation de concentration aux interfaces membrane–solution. |
7 Conclusion
Le potentiel zêta ou la charge de surface d’une membrane sont des grandeurs particulièrement recherchées par les spécialistes des procédés à membranes. La connaissance de ces grandeurs est en effet indispensable à une meilleure compréhension, à la prévision et à l’optimisation de la sélectivité d’une membrane, puisque les performances de celle-ci ne sont en général pas régies par les seules caractéristiques structurales du matériau filtrant. Par ailleurs, ces grandeurs constituent aussi des paramètres de contrôle de l’efficacité d’un traitement de régénération de surface membranaire. Dans cet article ont été présentées quatre méthodes expérimentales – potentiel d’écoulement, électro-osmose, potentiel de membrane et conductivité de pores – permettant d’obtenir des informations sur l’état de charge des parois des pores d’une membrane. Il a été montré que ces quatre méthodes renseignaient sur le signe du potentiel zêta (ou de la charge nette) et, par conséquent, permettaient la détermination du point isoélectrique. L’utilisation d’un modèle (tel qu’un modèle de charge d’espace) reliant les grandeurs expérimentales au potentiel zêta s’avère toutefois nécessaire pour la détermination de ce dernier. Dans le cas des grandeurs électrocinétiques (coefficient de potentiel d’écoulement et débit électro-osmotique), l’utilisation de la relation d’Helmholtz–Smoluchowski ne permet en effet de déterminer qu’une valeur apparente du potentiel zêta.