

1 Introduction
Les bicycloalcanols optiquement actifs peuvent être des précurseurs intéressants de composés énantiomériquement enrichis ; aussi, dans ce travail, nous nous proposons en premier lieu de préparer et de dédoubler par voie enzymatique des bicyclo[n,1,0]hex(hept)an-2-ols (n = 3,4) différemment substitués, montrant ainsi l'influence de la taille du cycle et des substituants sur l'énantiosélectivité. En second lieu et en vue de synthétiser des composés spiraniques chiraux optiquement actifs, nous avons tenté de mettre au point une méthode de synthèse sélective du 6-(but-3-ényl)bicyclo[4,1,0]heptan-2-ol. En effet, celui-ci doit permettre d'accéder à des composés spiraniques chiraux par cyclisation d'un radical hexényl, issu de l'ouverture d'un cyclopropyl, radical formé, soit à partir du composé cétonique correspondant et l'iodure de samarium [1], soit par le traitement avec Bu3SnH d'un composé phénylsélénié issu de la substitution de l'hydroxyle [2]. Au cours de ces cyclisations radicalaires, la configuration du carbone asymétrique qui est quaternaire doit rester inchangée et la préparation de bicyclo[4,1,0]heptan-2-ols optiquement actifs permettrait de synthétiser de tels composés spiraniques énantiomériquement enrichis.
2 Résultats et discussion
Dans un précédent travail, il a été montré qu'il était possible de préparer des bicyclo[n, 1,0]hex(hept)an-2-ol substitués en position 6 par des groupements à longueurs de chaînes variables et énantiomériquement enrichis en effectuant des réactions d'estérification ou de transestérification catalysées par la lipase de Mucor ou par la lipase Pseudomonas cepacia, les temps de réactions de plusieurs jours ou plusieurs semaines nécessaires pour atteindre 50 % de conversion dans les transformations ne permettant pas d'utiliser ce type de dédoublement dans une séquence réactionnelle.
Le premier objectif de ce travail a donc été de mettre au point les conditions expérimentales du dédoublement par voie enzymatique des bicyclo[n,1,0]hex(hept)an-2-ol, et plus précisément de réduire au maximum le temps de réaction des transformations pour atteindre 50 % de conversion, tout en obtenant les excès énantiomériques les plus élevés possibles. Ce double objectif a été atteint d'une manière satisfaisante pour la série des bicycloheptanols (Tableau 3) en utilisant la lipase de Candida antarctica (novozyme) ; en ce qui concerne les bicyclohexanols, les résultats ne sont pas tout autant appréciables, ce qui nous a conduits à restreindre les exemples dans cette série (Tableau 4). Au vu de ces résultats, il semble donc que la tendance générale, va dans le sens de la progression de l'énantiosélectivité par rapport à la longueur des chaînes des substituants (E > 60) et au contraire à diminuer en fonction de la taille du cycle (E < 20). Ces premiers travaux permettront ultérieurement de préparer le 6-(but-3-ényl)bicyclo[4,1,0]heptan-2-ol optiquement actif, généralement préparé par cyclopropanation d'alcool allylique à partir d'un mélange racémique [2–5], car on notera que, si la réaction de cyclopropanation des cyclopent(hex)-2-ène-1-ol substitués en position 1 ou 6 par des chaînes alkylées est relativement aisée, il n'en est pas de même pour le 3-(but-3-ényl)cyclohex-2-énol, où il y a compétition entre les deux insaturations et où, en plus, la séparation des substrats et des produits est délicate à réaliser (Fig. 1). Aussi et afin d'améliorer la sélectivité en intervenant sur les conditions réactionnelles, des essais de cyclopropanation ont été réalisés et reportés dans les Tableaux 1 et 2, où l'on notera que la présence d'oxygène était nécessaire pour améliorer le rendement en produit monocyclopropanique 7
Résultats des résolutions enzymatiques sur les bicyclo[4,1,0]heptan-2-ols 4a–4f
| no | R | Substrats | Temps | Alcool résiduel 4'* | Ester 5'* | C | E | ||||
| (h) | Rdt (%) | a[α]D 20 | ees (%) | Rdt (%) | a[α]D 20 | eep (%) | |||||
| 1 | H | 4a | 1,45 | 41 | –76 (1,8) | 99,3 | 51 | +62 (1,6) | 87 | 0,53 | 80 |
| 2 | CH3 | 4b | 2 | 48 | –71 (0,9) | 96 | 44 | +65 (0,8) | 88 | 0,52 | 61 |
| 3 | C2H5 | 4c | 1,30 | 42 | –63 (1,3) | 99 | 47,5 | +57 (0,7) | 92 | 0,52 | 126 |
| 4 | C4H9 | 4d | 1,15 | 41 | –54 (1,3) | 99,7 | 43 | +50 (0,9) | 96 | 0,51 | 317 |
| 5 | C6H13 | 4e | 1,25 | 45 | –43 (0,5) | 99,8 | 45 | +55 (0,9) | 96 | 0,50 | 189 |
| 6 | C11H23 | 4f | 1,20 | 50 | –27 (2,7) | 88 | 45 | +28 (2,3) | 99,6 | 0,47 | > 800 |
Resultats des resolutions enzymatiques sur les bicyclo[3,1,0]hexan-2-ols 4'd et 4'e
| N° | R | Substrats | Temps | Alcool résiduel 4* | Ester 5* | C | E | ||||
| (h) | Rdt (%) | a[α]D 20 | ees (%) | Rdt (%) | a[α]D 20 | eep (%) | |||||
| 1 | C4H9 | 4’d | 2 | 52 | –39 (0,9) | 71 | 44 | +49,7(0,8) | 80 | 0,47 | 19 |
| 2 | C6H13 | 4’e | 1,30 | 42 | –63 (1,3) | 69 | 47,5 | +57(0,7) | 75 | 0,47 | 12 |
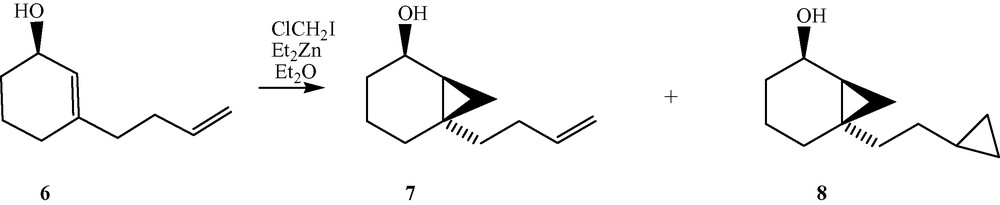
Cyclopropanation du 3-(but-3-ényl)cyclohex-2-énol.
Essais de cyclopropanation du 3-(but-3-ényl)cyclohex-2-énol 6 avec Et2Zn–ICH2X
| Conditions réactionnelles | ||||||||
| Exp. | Et2Zn | CH2IX | Solvant | Temp. | Temps de réaction | Proportions | ||
| (Eq) | (Eq) | (°C) | (h) | 6 | 7 | 8 | ||
| 1 | 2 | X = Cl ; 3,3 | DCE | 20 | 2,5 | 0 | 50 | 50 |
| 2 | 2 | X = Cl ; 2,1 | DCE | 20 | 1 | 1 | 83 | 16 |
| 3 | 2 | X = Cl ; 2,1 | DCE | 20 | 6 | 3 | 73 | 24 |
| 4. | 2 | X = Cl ; 2,1 | DCE | 20 | 1 | 11 | 79 | 10 |
| 5. | 2 | X = Cl ; 2,1 | Et2O | 35 | 2,5 | 55 | 45 | 0 |
| 6 | 2 | X = I ; 1,2 | Et2O | 35 | 5 | 25 | 75 | 0 |
| 7 | 2 | X = I ; 2,1 | Et2O | 35 | 7 | 21 | 62 | 14 |
Essais de cyclopropanation du 3-(but-3-ényl)cyclohex-2-énol 6 avec Zn/Ag et CH2I2
| Conditions réactionnelles | ||||||||
| Exp. | Zn–Ag | CH2I2 | Solvant | Température | Temps de réaction | Proportions | ||
| (Eq) | (Eq) | (°C) | (h) | 6 | 7 | 8 | ||
| 1 | 4,8 | X=Cl ; 2,4 | Et2O | reflux sans O2 | 2 | 63 | 37 | 0 |
| 2 | 4,8 | X=Cl ; 2,4 | Et2O | reflux avec O2 | 2 | 46 | 54 | 0 |
| 3 | 4,8 | X=Cl ; 2,4 | Et2O | reflux avec O2 | 3 + 3 | dégradation |
Notons enfin, pour terminer, que la réaction de cyclopropanation des alcools allyliques à 5 ou 6 atomes de carbone est diastéréosélective ; en effet le cyclopropane se trouve en position cis par rapport au groupe hydroxyle [3,6].
Les endobicycloheptanols 4a–f ont été préparés (Fig. 2) sous atmosphère inerte [7] à partir de cyclohex-2-ène-1-ol 3a–f par addition de ClCH2I à Et2Zn [8]. Les cyclohexénols 3a–f ont été obtenus par réduction avec NaBH4, en présence CeCl37 H2O [9] de leur correspondant cyclohex-2-ène-1-one 2a–f. La réaction de la 3-éthoxycyclohex-2-ène-1-one 1 [10] avec un réactif de Grignard [11] ou avec un composé halogéné en présence de lithium [12], suivie par un traitement aqueux, donne les cyclohexénones substituées 2a–f. Les endobicyclohexanols 4'd et 4'e ont été obtenus de la même manière.

Préparation des bicycloalcanols 4 et 4.
Les réactions de transestérification (Fig. 3) de l'hexanoate d'isopropényle qui ont été conduites dans le méthyl tertiobutyléther à 35 °C en présence de la lipase de C. antarctica ont permis d'atteindre des énantiosélectivités et des vitesses de réaction élevées, les résultats sont consignés dans le Tableau 3.
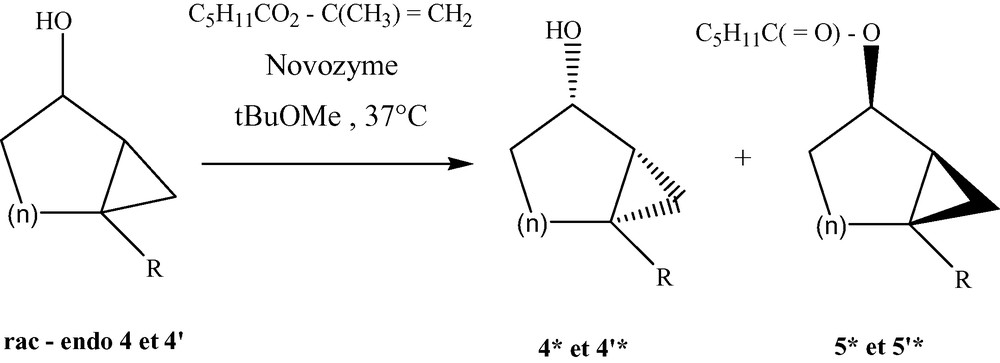
Résolution enzymatique des bicycloalcanols 4 et 4.
L'hexanoate d'isopropényle a été préparé en chauffant à reflux un mélange d'acide hexanoïque et d'acétate d'isopropényle en présence d'acide paratoluènesulfonique [13]. Généralement, après une conversion autour de 50% et séparation des produits par chromatographie sur une colonne de gel de silice, les excès énantiomériques des bicycloalcanols 4*a–f et 4'*d–e sont déterminés par chromatographie gazeuse sur une colonne capillaire (Cydex B) et ceux des bicycloalkyl hexanoates 5*a–f et 5'*d–e ont été mesurés à partir de leurs correspondants bicycloalcan-2-ols isolés après traitement des esters avec LiAlH4. Les résultats affichés dans les Tableaux 3 et 4 ont été vérifiés par la formule du facteur d'énantiosélectivité E = ln (1 – C) (1 – ees)/ln (1 – C) (1 + ees) et du taux de conversion C : C = ees/(ees + eep) [14]. Il reste à noter que la détermination de la stéréochimie a été établie par comparaison avec le signe de rotation spécifique et le temps de rétention des énantiomères enrichis en (1R, 2S, 6S) et (1R, 2R, 6R) [13,14], respectivement synthétisés par cyclopropanation des (S)-3-éthyl cyclohex-2-énol et (R)-3-butylcyclohex-2-énol préparés [15] à partir des (S)- et (R)-3-iodo cyclohex-2-énol selon la méthode décrite par Mori et al. [16] (Fig. 4).
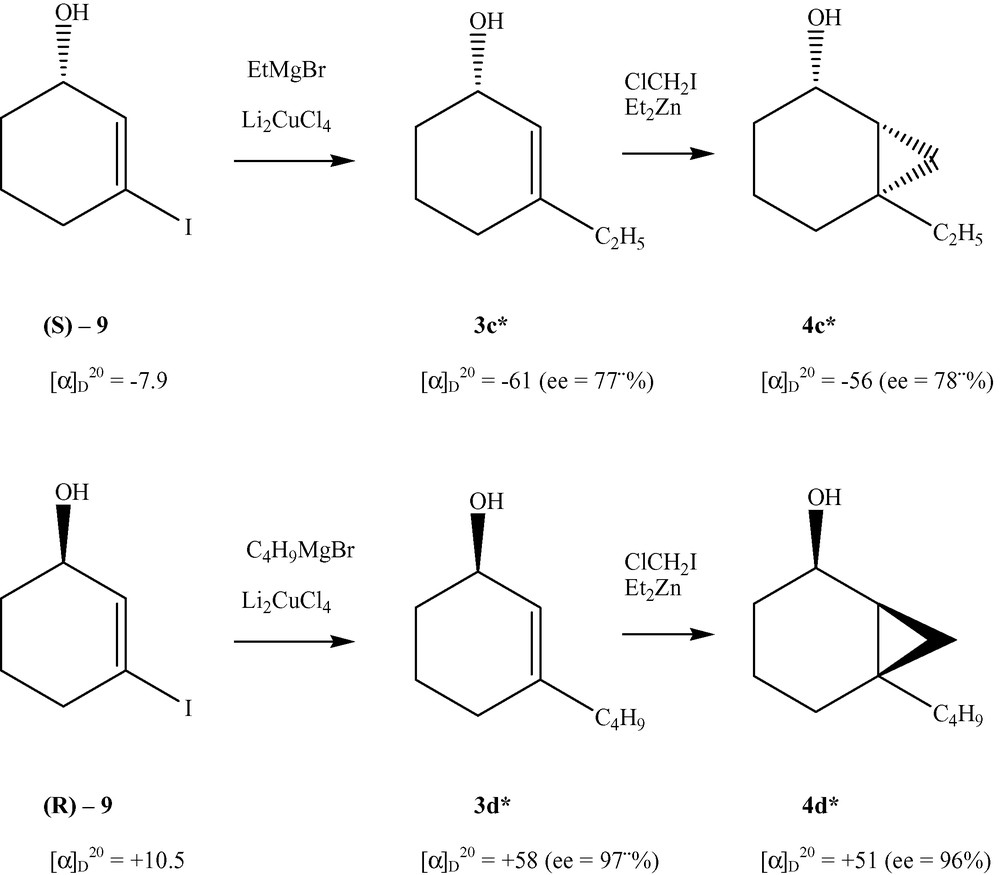
Corrélation stéréochimique.
3 Conclusion
Le dédoublement des bicyclohex(hept)an-2-ols a pu être réalisé en quelques heures en effectuant la transestérification de l'hexanoate d'isopropényle par ces alcools en présence de la lipase de C. antarctica (novozyme).
L'énantiosélectivité de ces réactions est excellente, du moins dans la série des bicycloheptanols, pour permettre d'isoler le substrat ou l'ester formé avec de très bons excès énantiomériques. Cette méthode est donc intéressante pour l'accès à des composés optiquement actifs dérivés de bicycloalcanols.
Il reste à améliorer la mauvaise chimiosélectivité observée lors de la cyclopropanation du 3-(but-3-ényl)cyclohex-2-énol.
4 Partie expérimentale
4.1 Méthodes générales
Les spectres de RMN 1H et 13C ont été réalisés sur des spectromètres Bruker AC200 (200 MHz) et Bruker AC250 (250 MHz). Les déplacements chimiques δ sont indiqués en ppm par rapport au TMS. Les abréviations utilisées sont les suivantes : s, singulet ; d, doublet ; dd, doublet de doublet ; t, triplet ; tt, triplet de triplet ; q, quartet ; m, multiplet et b pour les signaux élargis. Les spectres infrarouges ont été enregistrés sur un spectromètre PerkinElmer 682. Les spectres de masse ont été déterminés sur une GC-MS Nermag R10-10 (colonne capillaire : CPCIL5, 25 m) à ionisation par impact électronique de 70 eV. Les chromatographies sur colonne ont été faites avec du gel de silice 70–230 mesh. La CCM a été réalisée sur gel de silice 0,25 mm (Merck 60F254). Une colonne Cydex B de 25 m × 0.25 mm ID a été utilisée pour les mesures des excès énantiomériques (gaz vecteur : hélium). Les cyclohexane-1,3-dione, cyclohex-2-én-1-one et le 3-méthyl cyclohex-2-én-1-one proviennent de Acros Organics.
4.2 Préparation du lbicyclo[4,1,0]heptan-2-ol 4a
À une solution de 196 mg de cyclohex-2-ène-1-ol 3a dans 6 ml de 1,2-dichloroéthane maintenu à 0 °C, on ajoute sous argon 4 ml d'une solution 1 M de Et2Zn (4 mmol, 2 equiv) dans l'hexane. Après 15 min sous agitation à 0 °C, on additionne goutte à goutte 600 μl de ClCH2I (8 mmol, 4 equiv) et on laisse revenir le mélange à température ambiante. Après 15 min on remplace l'arrivée d'argon par un tube à chlorure de sodium et on agite pendant 1 h. Le mélange réactionnel est ensuite versé sur une solution saturée de chlorure d'ammonium et on extrait à l'éther. Les phases organiques sont lavées à l'eau et séchées sur du sulfate de sodium. On évapore sous vide le solvant et on purifie le produit brut par chromatographie sur gel de silice en éluant avec pentane–diéthyléther : 80:20 pour obtenir à 67% de rendement le bicyclo[4,1,0]heptan-2-ol 4a. Les bicycloalcanols 4b–4f et 4'd, 4'e ont été obtenus selon le même protocole.
4.2.1 6-méthylbicyclo[4,1,0]heptan-2-ol 4b
Rendement : 60%.
RMN 1H (250 MHz, CDCl3) δ 4,22 (dd, J = 6,3, 12,3 Hz, 1H), 1,5 1–1,12 (m, 8H), 1,09 (s, 3H), 0.51 (t, J = 4,9, 4,9 Hz, 1H), 0,31 (dd, J = 4,9, 8,9 Hz, 1H) ; RMN 13C (59,3 MHz, CDCl3) δ 67,7, 31,8, 29,6, 29,1, 28,3, 25,4, 22,2, 14,0 [9].
4.2.2 6-éthylbicyclo[4,1,0]heptan-2-ol 4c
Rendement : 98%.
RMN 1H (250 MHz, CDCl3) δ 4,19 (dd, J = 6,2, 11,8 Hz, 1H), 1,7 1–1,29 (m, 6H), 1,29–0.94 (m, 4H), 0,90 (bt, J = 7,84 Hz, 3H), 0,49 (t, J = 4,8, 4,8 Hz, 1H), 0,35 (dd, J = 4,8, 8,7 Hz, 1H) ; RMN 13C (62,9 MHz, CDCl3) δ 66,8, 33,7, 29,6, 27,2, 25,1, 24,2, 20,3, 13,4, 10,4 ; IR (film) 3380, 3080, 3010, 2960, 2840, 1460 cm−1 ; EI-MS (m/z) 140 (M+, 1,3), 125 (2,6), 123 (5,5), 122 (22), 111 (39), 107 (29), 94 (26), 93 (100).
4.2.3 6-butylylbicyclo[4,1,0]heptan-2-ol 4d
Rendement : 88%.
RMN 1H (250 MHz, CDCl3) δ 4,20 (dd, J = 6,2, 12,0 Hz, 1H), 1,73–0,96 (m, 14H), 0,89 (bt, J = 7,6 Hz, 3H), 0,48 (t, J = 4,7, 4,7 Hz, 1H), 0,34 (dd, J = 4,7, 8,7 Hz, 1H) ; RMN 13C (62,9 MHz, CDCl3) δ 66,8, 40,9, 29,6, 28,7, 27,6, 25,3, 23,1, 22,7, 20,2, 14,0, 13,7 ; IR (film) 3400, 3060, 3000, 2960, 2840, 1460 cm−1 ; EI–MS (m/z) 168 (M+, 0,9), 151 (3,3), 150 (135), 125 (9,1), 111 (46), 94 (23), 93 (99),82 (58),79 (100).
4.2.4 6-hexylbicyclo[4,1,0]heptan-2-ol 4e
Rendement : 60 %.
RMN 1H (250 MHz, CDCl3) δ 4,20 (dd, J = 6,2, 11,82 Hz, 1H), 1,71–0,95 (m, 18H), 0.89 (bt, J = 7.5 Hz, 3H), 0,49 (t, J = 4.6, 4,6 Hz, 1H), 0,34 (dd, J = 4.6, 8,5 Hz, 1H); 13C RMN (62,9 MHz, CDCl3) δ 66,8, 41,3, 31,8, 29,8, 29,7, 29,6, 29,3, 27,7, 26,5, 25,5, 23,3, 14,0, 13,7; IR (film) 3360, 3000, 2960, 2840, 1460 cm−1 ; EI–MS (m/z) 224 (M+, 1,5), 207 (9,33), 206 (21), 111 (25), 97 (19), 93 (64), 82 (36).
4.2.5 6-undécylbicyclo[4,1,0]heptan-2-ol 4f
Rendement : 60 %
RMN 1H (250 MHz, CDCl3) δ 4,18 (dd, J = 6,2, 12,0 Hz, 1H), 1,71–0,95 (m, 28H), 0,89 (bt, J = 7,5 Hz, 3H), 0,45 (dd, J = 4,7, 4,7 Hz, 1H), 0,34 (dd, J = 4,7, 8,6 Hz, 1H) ; RMN 13C (62,9 MHz, CDCl3) δ 67,1, 41,3, 31,9, 29,9, 29,8, 29,7, 29,65, 29,6, 29,5, 29,3, 27,8, 26,6, 25,6, 23,4,22,7, 20,3, 14,0, 13,6 ; IR (film) 3370, 3070, 3005, 2960, 2840, 1460 cm−1 ; EI–MS (m/z) 247 (4,2), 92 (263), 91 (41), 8 (7,9), 79 (100).
4.2.6 5-butylbicyclo[3,1,0]hexan-2-ol 4'd
Rendement : 51 %.
RMN 1H (200 MHz, CDCl3) δ 4,52 (m, 1H), 1,96 (m, 1H), 1,72 (m, 1H), 1,63 (m, 1H), 1,50 (m, 2H), 1,28 (m, 7H), 1,10(m, 1H), 0,92 (t, J = 4,7 Hz, 3H), 0,34 (m, 1H) ; RMN 13C (62,9 MHz, CDCl3) δ 72,8, 40,3, 32,9, 32,0, 28,5, 27,8, 23,7, 20,2, 14,0, 12,7 ; IR (film) : 3380, 3060, 3010, 2960, 2840, 1460 cm−1 ; EI–MS (m/z) 154 (M+, 2,10), 125 (17), 97 (55), 85 (27), 79 (76), 70 (100), 55 (53).
4.2.7 5-hexybicyclo[3,1,0]hexan-2-ol 4'e
Rendement : 50 %
RMN 1H (250 MHz, CDCl3) δ 4,59–4,44 (m, 1H), 2,01–1,84 (m, 1H), 1,84–1,68 (m, 1H), 1,55–1,45 (m, 2H), 1,45–1,19 (m, 11H), 1,19–1,00 (m, 1H), 0,87 (t, J = 6,84 Hz, 3H), 0,35–0,26 (m, 1H) ; RMN 13C (62,9 MHz, CDCl3) δ 72,8, 40,6, 32,9, 32,5, 32,0, 30,6, 28,5, 25,6, 23,1, 20,2, 14,0, 12,7 ; IR (film) : 3360, 3020, 3000, 2960, 2840, 1460 cm−1 ; EI–MS (m/z) 128 (M+, 3,82), 165 (8), 164 (21), 97 (23), 80 (17),79 (100), 67 (11), 66 (21).
4.3 Cyclopropanation du 3-(but-3-ényl)cyclohex-2-en-1-ol 6
La cyclopropanation de ce composé, d'après les essais rapportés dans les Tableaux 1 et 2, montre un mélange de deux produits 7 et 8 révélés par RMN 1H.
Composé 7 : 5,9–5,70 (ddt, J = 19, 11,7 Hz, 1H), 5,08–4,95 (d, J = 19 Hz, 1H), 4,95–4,9 (d, J = 11 Hz, 1H), 4,28–4,12 (m, 1H), 0,53–0,45 (m, 2H), 1,45–0,3 (m, 2H).
Composé 8 : Les signaux des protons cyclopropaniques résultant de la seconde cyclopropanation se trouvent à 0,90 ppm et entre 0 et –0,05 ppm.
4.4 Résolution enzymatique du bicyclo[4,1,0]heptan-2-ols 4a
(100 mg, 0,78 mmol) de 3-méthylbicyclo[4,1,0]heptan-2-ol 4a, sont dissous dans 1 ml de méthyltertiobutyléther dans un ballon à agitation magnétique. À cette solution, on ajoute 50 mg de Novozym® et 61,9 mg (0,39 mmol) d'hexanoate d'isopropényle sous agitation à 35 °C. La réaction est suivie par CCM ; à environ 50% de conversion, la réaction est interrompue et, par filtration, on récupère l'enzyme, qu'on lave plusieurs fois avec du tert-BuOMe. Après évaporation du solvant sous pression réduite, les produits de la réaction sont séparés sur une colonne de gel de silice (pentane–diéthyléther : 70:30) pour donner l'alcool résiduel (rendement : 41%) et l'ester (rendement : 51%). L'hexanoate de bicycloheptyl 5*a est ensuite traité à 0 °C dans du diéthyléther avec l'hydrure de lithium aluminium. Après agitation pendant 1 h on ajoute, à température ambiante, un peu de sulfate de sodium afin d'obtenir une solution plus claire ; celle-ci est alors filtrée et concentrée sous pression réduite pour donner le bicyclo[4,1,0]heptan-2-ol (rendement ~90%) optiquement actif, séparé auparavant de l'héxan-1-ol par chromatographie sur colonne de gel de silice (pentane–diéthyléther : de 80:20 à 50:50).Les transestérifications avec les bicycloalcanols 4b-f et 4'd-e ont été obtenues de la même manière.
4.5 Corrélation chimique
4.5.1 Préparation de 3-éthylcyclohex-2-ène-1-ol 3c*
À une solution de 84 mg de Li2CuCl4 (0,38 mmol, 0,15 equiv) dans 4 ml de THF, maintenue à –20 °C sous argon, on ajoute 10,5 ml d'une solution 1 M de EtMgBr dans du THF (10,5 mmol, 4 equiv) et 586 mg de (–)-3-iodocyclohex-2-en-1-ol 9 [8] dans 4 ml de THF. Après agitation pendant 14 h à –20 °C, le mélange réactionnel est versé dans une solution de chlorure d'ammonium et est extrait avec du diéthyléther. Le solvant est évaporé sous vide et le produit brut purifié par chromatographie sur colonne à gel de silice (pentane–diéthyléther : 90:10) pour donner le (–)-3-éthylcyclohex-2-ène-1-ol 3c* (rendement 66 %) ; ee = 77%, déterminé par GLC sur une colonne Cydex B (P = 0,8 bar, 7 °C), les temps de rétention notés sont : (S)-3c* : 27 min, (R)-3c* : 30 min.
4.5.2 Préparation du 3-butylcyclohex-2-ène-1-ol 3d*
Ce composé a été obtenu à partir de (+)-3-iodocyclohex-2-ène-1-ol 9 [8] et de BuMgBr, selon le même protocole ci-dessus, (72%) de (+)-3-butyl cyclohex-2-ène-1-ol 3d* (rendement 72%) ; ee = 99%, déterminé par GLC sur une colonne Cydex B (P = 0,8 bar, 85 °C), les temps de rétention sont : (S)-3d* : 39 min, (R)-3d* : 40 min.