

1 Introduction
Les apatites, de formule chimique Me10(XO4)6Y2, où Me représente un cation divalent (Ca2+, Sr2+, Ba2+, etc.), XO4 un anion trivalent (PO43−, VO43−, etc.) ou tétravalent (SiO44−, GeO44−) et Y un anion monovalent (Cl−, F−, OH−, etc.) ou divalent (O2−, CO32−), cristallisent généralement dans le système hexagonal (groupe d’espace P63/m) [1]. Dans cette structure, les groupements XO4, agencés en un empilement quasi compact, forment deux types de tunnels, parallèles à l’axe c, dans lesquels sont localisés les cations Me2+. Les tunnels du premier type, de plus petite taille, sont occupés par quatre Me2+ (sites 4f, Me(1)) qui sont entourés par neuf atomes d’oxygène. Les tunnels du deuxième type sont bordés par des atomes d’oxygène et les six Me2+ restants (sites 6h, Me(2)). Ces cations se trouvant aux sommets de deux triangles équilatéraux, alternés à la cote ¼ et ¾, sont entourés par six atomes d’oxygène et un atome Y. Ces apatites font l’objet de plusieurs études, aussi bien expérimentales que théoriques [2–6], en raison de leurs nombreuses applications et de la faculté de leur structure à générer de nouveaux matériaux, grâce aux diverses substitutions qu’elle peut accepter. Ces substitutions sont aussi bien anioniques que cationiques, conduisant à la formation de solutions solides totales ou partielles [5–7].
Les britholites sont des dérivés apatitiques, obtenues par substitution couplée du cation divalent par une terre rare trivalente et du groupement trivalent PO43− par le groupement tétravalent SiO44− [8,9]. Des observations réalisées sur des réacteurs nucléaires naturels tels que ceux d’Oklo, au Gabon [10], ont montré que ces composés sont capables de piéger des radionucléides comme le césium et les actinides mineurs. Aussi, le strontium 90 est issu de la fission de l’uranium et du plutonium dans les réacteurs nucléaires [11–16]. Il est particulièrement nocif pour la vie des animaux et des êtres humains, en particulier quand il est incorporé dans les cellules osseuses et celles des dents, par substitution du calcium. Par ailleurs, les apatites sont caractérisées par une forte mobilité des ions localisés dans les tunnels du deuxième type. Ils sont les principaux porteurs de charges de ces matériaux [17,18]. Leur mobilité joue certainement un rôle dans leur lixiviation ou dissolution [14,15]. Elle est aussi responsable de leur conductivité électrique [2,19–21].
Ces apatites sont généralement obtenues par réaction à l’état solide à haute température [5,22], méthode très énergivore. C’est pourquoi il nous a paru intéressant de procéder à leur préparation par mécanosynthèse [23]. Cette méthode, qui implique seulement une réaction à l’état solide à température ambiante, semble être attrayante. En effet, outre sa simplicité, sa rapidité et sa faible consommation d’énergie, elle peut se prêter à la production industrielle.
Après avoir préparé et caractérisé les composés de formule générale Sr7−xCaxLa3(PO4)3(SiO4)3F2, avec x = 0, 1 et 2, nous procéderons à une étude structurale du composé correspondant à x = 0, préparé par broyage et traitement thermique, afin de déterminer l’influence de la méthode de synthèse sur la distribution des cations entre les deux sites cristallographiques. En effet, des études ont montré que le mode opératoire adopté pour leur préparation peut avoir une influence sur cette répartition. Enfin, nous clôturons ce travail par une étude de ses propriétés électriques.
2 Protocole expérimental
2.1 Préparation des poudres
Les réactifs utilisés sont SrCO3 (> 96 % Riedel de Haen), La2O3 (> 99,5 % Prolabo), SiO2 (> 99,5 % Alfa), SrF2 (> 99,5 % Prolabo), CaCO3 (> 99 % Fluka), et Sr2P2O7. Ce dernier composé a été obtenu à partir de SrCO3 et (NH4)2HPO4. Après avoir été homogénéisés dans un mortier en agate et mis en forme par pressage uniaxial à froid, ils ont été calcinés à 900 °C pendant 10 h.
En raison de sa sensibilité à l’humidité et au gaz carbonique de l’air [24], l’oxyde de lanthane a été calciné à 1000 °C pendant 12 h, juste avant la pesée.
Les échantillons, de formule générale Sr7−xCaxLa3(PO4)3(SiO4)3F2, avec x = 0, 1 et 2, ont été préparés selon l’équation suivante :
| (3–x) SrCO3 + x CaCO3 + 3/2 La2O3 + 3/2 Sr2P2O7 + 3 SiO2 + SrF2 → Sr7−xCaxLa3(PO4)3(SiO4)3F2 + 3 CO2 | (1) |
Deux voies de synthèse ont été mises en œuvre, la mécanosynthèse, en utilisant un broyeur planétaire de type Retsch PM 200, et la réaction à l’état solide à haute température.
Dans la première méthode, les réactifs pris dans des proportions stœchiométriques ont été introduits dans une jarre de 45 cm3 contenant cinq billes de 12 mm de diamètre. La jarre et les billes sont en acier. Le rapport masse des billes sur masse de la poudre est 34/1. Les vitesses de rotation du plateau et de la jarre étaient respectivement égales à 500 et 1000 tr/min. Ces conditions opératoires correspondent à une énergie cinétique de choc de 0,151 J/choc, une fréquence de choc de 100 Hz et une puissance injectée de choc de 15,1 W/g. La durée de broyage était égale à 25 h. Cette durée a été déterminée suite à différentes expérimentations [25].
L’échantillon Sr7La3(PO4)3(SiO4)3F2 a été également préparé par réaction à l’état solide à haute température. Après avoir été broyés et homogénéisés dans un mortier en agate, les réactifs ont été mis en forme par pressage uniaxial sous une pression de 100 MPa. Ils ont été ensuite calcinés à 900 °C puis à 1300 °C sous une atmosphère dynamique d’argon. La durée de chaque palier isotherme était de 12 h. La vitesse de la montée en température pour tous les traitements thermiques était de 10 °C/min.
Dans la suite de l’article, les échantillons Sr7La3(PO4)3(SiO4)3F2, Sr6CaLa3(PO4)3(SiO4)3F2 et Sr5Ca2La3(PO4)3(SiO4)3F2 seront notés, respectivement, Sr7La3F2, Sr6CaLa3F2 et Sr5Ca2La3F2.
2.2 Caractérisation des poudres
Le strontium et le lanthane ont été dosés au moyen d’un spectromètre d’émission atomique Shimadzu ICPQ/V-1014S, alors que le silicium l’a été à l’aide d’un spectromètre de type PerkinElmer 3110. La teneur en ions phosphate a été déterminée par colorimétrie selon la méthode de GEE et DEITZ à 460 nm à l’aide d’un spectromètre de type Jenway 6400. Le dosage des ions fluorures a été effectué par ionométrie en utilisant une électrode spécifique au fluor INGOLD de type PF4-L.
L’analyse par diffraction des rayons X (DRX) des échantillons a été effectuée au moyen d’un diffractomètre PANalytical X’Pert Pro, en utilisant la radiation Kα du cuivre (λ = 1,5406 Å). Les diffractogrammes ont été acquis dans l’intervalle 0–75°C en 2θ par une acquisition pas à pas à la vitesse de 0,02 °/s. Le logiciel X’Pert HighScore Plus de la firme PANalytical a été mis à profit pour l’identification des phases cristallines en comparant les diagrammes DRX obtenus aux fichiers de la base de données JCDD PDF-2.
Les spectres infrarouge ont été enregistrés dans le domaine 400–4000 cm−1 à l’aide d’un spectromètre de type PerkinElmer 1283 à transformée de Fourier, en utilisant la technique du KBr.
L’analyse thermogravimétrique a été réalisée entre la température ambiante et 1400 °C, au moyen d’un appareil de type Setaram 92. La montée en température a été effectuée avec une vitesse de 10 °C/min.
Les spectres RMN–MAS du solide de 31P et 29Si ont été acquis à l’aide d’un spectromètre de type Bruker MSL 300 aux fréquences respectives de 121,5 et 59,6 MHz. Les références utilisées sont l’acide phosphorique (85 %) pour 31P et le tétraméthylsiliane pour 29Si.
Les mesures de l’impédance complexe ont été effectuées à différentes températures sur une pastille frittée à 1300 °C pendant 12 h. La pastille dont les faces ont été préalablement enduites d’une laque d’argent a été placée entre deux électrodes de platine, reliées à l’analyseur d’impédance (Hewlett-Packard 4192 A) par des fils également en platine. La gamme de fréquences utilisée est comprise entre 10 Hz et 13 MHz.
3 Résultats et discussion
3.1 Préparation des poudres par mécanosynthèse
3.1.1 Diffraction des rayons X
Les diagrammes DRX des échantillons sont présentés sur la Fig. 1. Leur identification par référence à la fiche JCPDS no 17-0609, correspondant à la fluorapatite strontique de symétrie hexagonale et de groupe d’espace P63/m, indique que tous les échantillons sont formés par une phase apatitique pure. Aucune phase secondaire n’a été détectée. Comme le montre le Tableau 1, les paramètres de maille, déterminés par la méthode des moindres carrés, diminuent avec la substitution. Cette diminution qui est à relier à la taille des ions en substitution – le cation Ca2+ (coord. 9 : r.i. = 1,18 Å) étant plus petit que Sr2+ (coord. 9 : r.i. = 1,31 Å) [26] – constitue une preuve de l’insertion du calcium dans la structure apatitique. Aussi, l’insertion des ions carbonate (cf. section 3.1.2), de plus petite taille que celles des groupements PO43− et SiO44−, a sûrement une influence sur les paramètres de maille.

Diagrammes DRX des échantillons préparés par mécanosynthèse : a : Sr7La3F2 ; b : Sr6CaLa3F2 ; c : Sr5Ca2La3F2.
Paramètres cristallographiques des échantillons préparés.
| Échantillon | a (Å) | c (Å) | V (Å3) |
| Sr7La3F2 | 9,739 | 7,258 | 595,986 |
| Sr6CaLa3F2 | 9,720 | 7,255 | 593,834 |
| Sr5Ca2La3F2 | 9,696 | 7,249 | 590,132 |
3.1.2 Spectroscopie d’absorption infrarouge
Les spectres d’absorption infrarouge sont illustrés sur la Fig. 2. Les bandes d’absorption dont les valeurs sont consignées dans le Tableau 2 ont été attribuées en se référant à la littérature [5,8,27–29].
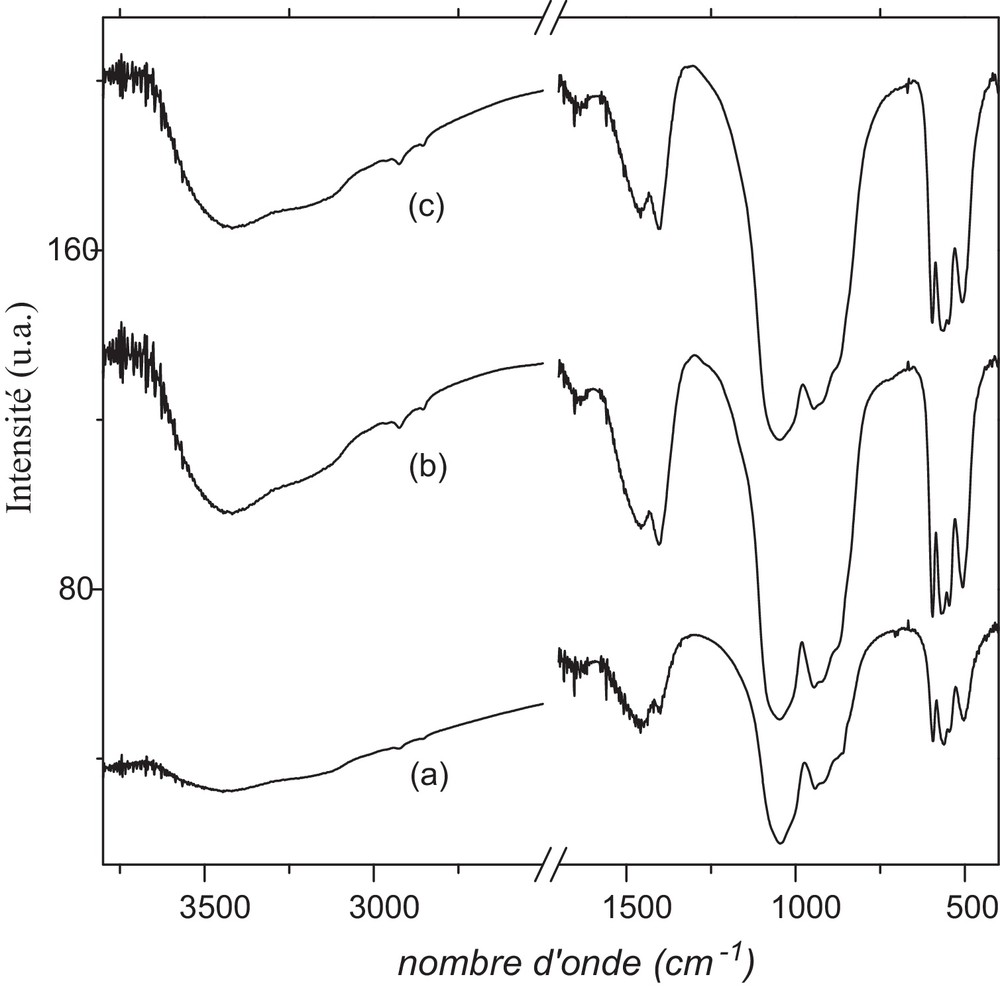
Spectres d’absorption infrarouge des échantillons préparés par mécanosynthèse : a : Sr7La3F2 ; b : Sr6CaLa3F2 ; c : Sr5Ca2La3F2.
Attribution des bandes d’absorption infrarouge des apatites préparées.
| υ (cm−1) | ||||||||
| PO43− | SiO44− | |||||||
| Échantillon | υ1 | υ2 | υ3 | υ4 | υ1 | υ2 | υ3 | υ4 |
| Sr7La3F2 | 926 – | 490 530 | 1030 1084 | 550 590 | 830 – | 425 440 | 940 901 | 498 – |
| Sr6Ca1La3F2 | 920 – | 495 535 | 1030 1088 | 545 580 | 850 – | 430 460 | 960 −907 | 505 – |
| Sr5Ca2La3F2 | 918 – | 498/ 540 | 1030 1090 | 540 575 | 850 – | 440 470 | 990 912 | 508 – |
Pour le composé non substitué, les bandes de vibration vers 926, 490–530, 1030–1084 et 550–590 cm−1 correspondent respectivement aux modes ν1, ν2, ν3 et ν4 des groupements PO4 dans une structure apatitique. En plus de ces bandes, le spectre comportent aussi des bandes associées aux groupements SiO4, situées à 830 (ν1), 425–440 (ν2), 901–940 (ν3) et 498 (ν4) cm−1. Les bandes positionnées à 1453–1406 et 863 cm−1, attribuées au groupement CO32− [25,30,31], donnent à penser que des ions carbonate provenant des réactifs ont été incorporés dans la structure apatitique et, plus précisément, dans les sites de PO43−/SiO44−. Dans ces conditions, une quantité de SiO2 ou/et de Sr2P2O7 doit se retrouver dans la poudre broyée après réaction. Toutefois, ces composés n’ont pas été détectés. Ceci est probablement dû à leurs faibles quantités. Des apatites carbonatées de type B ont été donc obtenues. Les bandes vers 3450 et 1640 cm−1 sont dues aux molécules d’eau adsorbées sur la surface des particules. Pour les composés substitués, peu de changement sont observés sur les spectres (Fig. 2).
Comme il est observé sur ces derniers, toutes les bandes sont larges et mal résolues, en raison, d’une part, de la mauvaise cristallinité des poudres et, d’autre part, du désordre provoqué par les différentes substitutions ayant lieu dans la structure apatitique.
3.1.3 Analyse thermique
Les courbes thermogravimétriques des trois échantillons sont données sur la Fig. 3. La perte de masse totale (4,75 % environ) est pratiquement la même pour les trois échantillons. Cette perte est inférieure à celle prévue (7,49, 7,82 et 7,92 % respectivement pour les composés Sr7La3F2, Sr6CaLa3F2 et Sr5Ca2La3F2) d’après l’équation de la réaction (1). Ceci donne à penser qu’une quantité non négligeable de CO2 s’est dégagée au cours du broyage. Par ailleurs, les allures des courbes sont légèrement différentes. En effet, les différentes pertes ne sont pas de la même amplitude et n’interviennent pas dans les mêmes domaines de température. Ceci est en rapport avec la nature des réactifs carbonatés utilisés.
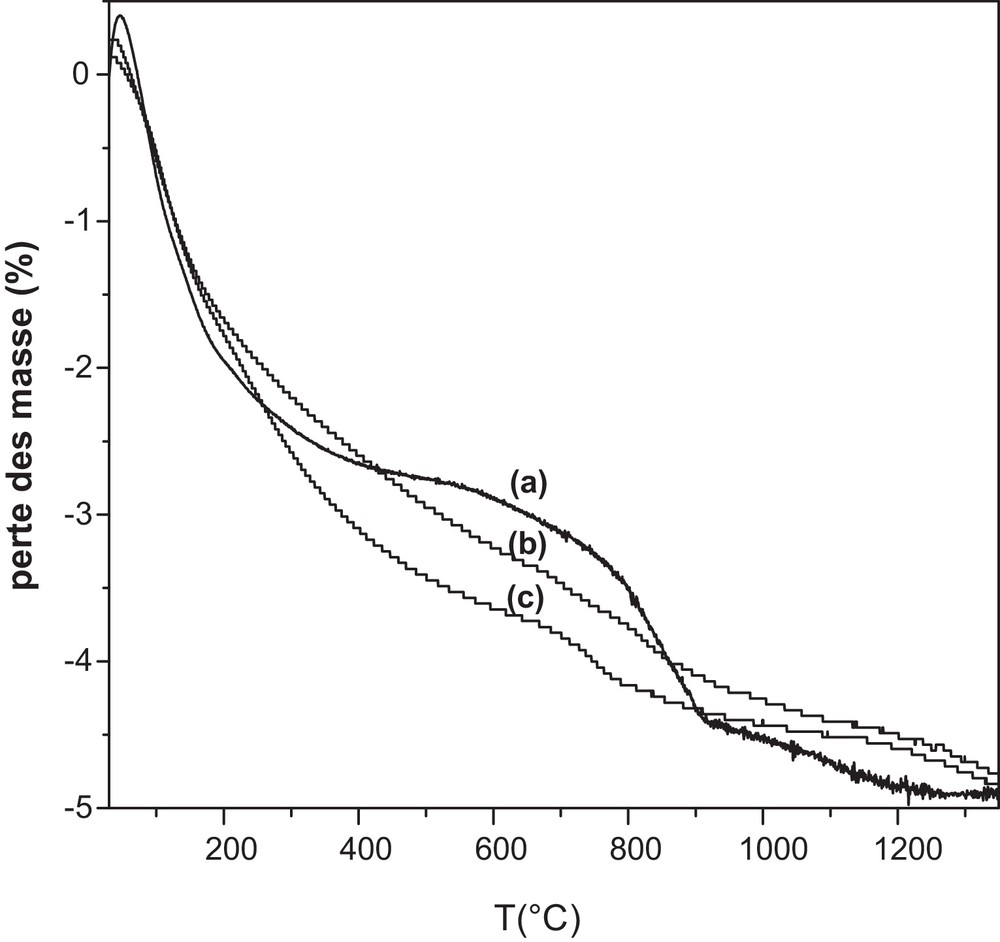
Analyses thermogravimétrique des échantillons préparés par mécanosynthèse : a : Sr7La3F2 ; b : Sr6CaLa3F2 ; c : Sr5Ca2La3F2.
Sur la courbe de l’échantillon non substitué au calcium, la première perte de masse de 2,80 %, entre 25 et 530 °C, est due au départ de l’eau moléculaire et CO2 adsorbés sur la surface des poudres [32]. Les deux pertes suivantes, de valeurs respectives 1,55 et 0,40 % et intervenant dans les domaines 530–920 et 920–1200 °C, sont attribuées au départ des carbonates insérés dans la structure apatitique. Quant aux courbes des échantillons substitués, elles présentent une première perte de masse, respectivement de 3,65 et 3,75 %, dans les domaines 25–850 et 25–730 °C. Elle est due au départ des mêmes espèces que pour l’échantillon non substitué. Toutefois, sa valeur plus importante et son domaine de température plus étalé du côté des hautes températures donnent à penser qu’une certaine quantité de La2O3, qui se serait hydratée, et/ou de SrCO3 et CaCO3, qui n’auraient pas participé à la réaction, auraient contribué à cette perte. Une deuxième perte de 1,10 % et de 1,00 %, se produisant respectivement dans les domaines 850–1400 et 730–1400 °C, est due au départ des carbonates insérés dans la structure apatitique. Aussi, le départ de CO2 dû à ces ions peut intervenir à des températures plus basses que 730 °C [33].
3.2 Étude structurale et électrique de Sr7La3(PO4)3(SiO4)3F2
3.2.1 Analyse chimique
Les résultats de l’analyse chimique, rapportés dans le Tableau 3, montrent que pour l’échantillon Sr7La3F2, obtenu par traitement thermique, la valeur du rapport (Sr + La/P + Si) est voisine de la valeur stœchiométrique (1,667). On constate également que la quantité de fluor déterminée est comparable à celle introduite initialement dans le mélange. En revanche, pour l’échantillon obtenu par mécanosynthèse, la valeur du rapport (Sr + La/P + Si) est plus faible que la valeur théorique. De même, la teneur en fluor dosée est inférieure à celle utilisée initialement.
Nombre d’atomes par maille de l’échantillon de formule Sr7La3F2.
| Sr | La | P | Si | F | Sr + La/P + Si | |
| Préparé par mécanosynthèse | 6,70 | 2,86 | 2,98 | 3,02 | 0,98 | 1,593 |
| Préparé par traitement thermique | 6,99 | 3,00 | 2,99 | 3,01 | 1,98 | 1,665 |
3.2.2 Affinement structural
Afin de déterminer l’effet de la méthode de synthèse sur la distribution des cations entre les deux sites cristallographiques, une étude structurale du composé Sr7La3F2, obtenu par broyage et traitement, a été réalisée par la méthode de Rietveld [34], en utilisant le programme Fullprof [35]. L’affinement structural a été effectué dans les groupes d’espace P63/m, P63 et
Données de l’affinement pour l’échantillon Sr7La3F2.
| Préparé par mécanosynthèse | Préparé par traitement thermique | |
| Masse molaire (g) | 1629,115 | 1629,115 |
| Système | hexagonal | hexagonal |
| Groupe d’espace | P63/m | P63/m |
| Z | 1 | 1 |
| Paramètres cristallins | ||
| *a (A°) | 9,718(2) | 9,729(2) |
| *c (A°) | 7,268(1) | 7,257(1) |
| volume V (Å3) | 594,554(9) | 594,917(9) |
| Densité calculée (g·cm3) | 4,5500 | 4,547 |
| Décalage de zéro point 2θ (°) | 0,00110(1) | 0,0557(1) |
| Nombre de paramètres affinés | 33 | 34 |
| Facteurs de confiance | ||
| Rp | 2,08 | 6,09 |
| Rwp | 2,69 | 8,36 |
| RB | 3,34 | 6,85 |
| RF | 3,21 | 4,90 |
Diagrammes DRX de l’échantillon Sr7La3F2 : observé, calculé et leur différence.
Comme l’analyse par spectroscopie infrarouge a montré que les apatites sont carbonatées et de type B, une convergence de l’affinement a été observée, en localisant l’ion CO32− dans le même site que le groupement PO43−. La quantité de carbonate déterminée est égale à 0,24 mol par maille. Elle est comparable à celle déterminée par l’analyse thermogravimétrique.
Par rapport aux paramètres de maille de l’échantillon préparé à haute température, le paramètre a de l’échantillon broyé est plus petit, alors que c est plus grand (Tableau 4). Une telle évolution est observée lorsque des ions carbonate se substituent aux ions phosphate (apatite type B) [30]. En effet, l’axe a est à la fois influencé par la taille des cations du site Me(2) et par celle des tétraèdres PO43−/SiO44−. Pour s’accommoder de la contraction de l’axe a, due à l’insertion des ions CO32− dans les sites PO43−/SiO44−, la liaison C–O étant plus courte (1,25 Å) que les liaisons P–O (1,51 Å) et Si–O (1,62 Å) [26], la maille doit s’expanser suivant l’axe c. Ainsi, cette évolution des paramètres de maille constitue une preuve supplémentaire de l’insertion des ions carbonate dans la structure apatitique.
En bloquant l’ion fluorure dans sa position idéale 2a (0 0 ¼), l’affinement a conduit, pour les deux échantillons, à des facteurs d’agitation thermique de cet ion élevés. En revanche, en le plaçant dans la position 4e (0 0 z), des valeurs convenables de ces facteurs ont été obtenues. De plus, un décalage appréciable de cet ion le long de l’axe z a été observé. Un tel déplacement, rapporté pour diverses fluorapatites substituées [36,37], est lié aux interactions entre cet ion et les cations occupant les sites Me(2) [38]. Les positions atomiques et les coefficients d’agitation thermique sont rassemblés dans le Tableau 5.
Positions atomiques, nombre d’atomes par maille et paramètres d’agitation thermique isotrope de l’échantillon Sr7La3F2.
| Apatite | Atomes | M | X | Y | Z | N | Beq [Å2] |
| Préparé par mécanosynthèse | Sr(I) | 4f | ⅓ | ⅔ | 0,2068) | 2,111 | 0,332(1) |
| La(I) | 4f | ⅓ | ⅔ | 0,206(8) | 1,660 | 0,332(1) | |
| Sr(II) | 6h | 0,239(9) | −0,009(6) | ¼ | 4,423 | 0,332(1) | |
| La(II) | 6h | 0,239(9) | −0,009(6) | ¼ | 1,184 | 0,332(1) | |
| P/Si/ | 6h | 0,402(4) | 0,358(6) | ¼ | 2,88 | 2,280(6) | |
| C | 6h | 0,402(4) | 0,358(6) | ¼ | 0,24 | 2,280(6) | |
| O1 | 6h | 0,384(8) | 0,484(6) | ¼ | 6 | 0,604(9) | |
| O2 | 6h | 0,598(8) | 0,447(6) | ¼ | 6 | 0,604(9) | |
| O3 | 12i | 0,362(37) | 0,267(3) | 0,064(3) | 12 | 0,604(9) | |
| F | 4e | 0 | 0 | 0,136(10) | 0,96 | 1,104(9) | |
| Préparé par traitement thermique | Sr(I) | 4f | ⅓ | ⅔ | 0,0001(8) | 3,208 | 0,187(9) |
| La(I) | 4f | ⅓ | ⅔ | 0,0001(8) | 0,792 | 0,187(9) | |
| Sr(II) | 6h | 0,238(7) | −0,011(8) | ¼ | 3,792 | 0,187(9) | |
| La(II) | 6h | 0,239(9) | −0,009(6) | ¼ | 2,208 | 0,187(9) | |
| P/Si | 6h | 0,393(8) | 0,365(6) | ¼ | 6 | 0,989(5) | |
| O1 | 6h | 0,330(6) | 0,487(8) | ¼ | 6 | 3,139(7) | |
| O2 | 6h | 0,591(9) | 0,4716(2) | ¼ | 6 | 3,139(7) | |
| O3 | 12i | 0,343(10) | 0,262(10) | 0,057(11) | 12 | 3,139(7) | |
| F | 4e | 0 | 0 | 0,186(9) | 2 | 1,769(7) |
L’analyse systématique de la distribution des cations entre les deux sites cristallographiques, consécutive à une substitution cationique dans les fluorapatites, hydroxyapatites et chlorapatites, montre que de nombreux facteurs interviennent dans cette distribution, parmi lesquels il y a lieu de citer la taille du substituant, sa polarisabilité et son électronégativité [39]. Les terres rares préfèrent généralement les sites Me(2) [37,38,40]. Cependant, il a été constaté que la nature de l’apport de la terre rare peut avoir une influence sur sa répartition entre les deux sites cationiques. Ainsi, dans la fluorapatite dopée au néodyme, cet élément est distribué entre les deux sites, s’il est introduit sous la forme NdF3. En revanche, il est uniquement localisé dans le site Me(2), lorsqu’il est introduit sous la forme Nd2O3 [41].
Aussi, il a été observé que la teneur en substituant utilisée peut avoir une influence sur sa localisation. Ainsi, l’étude conduite sur un monocristal de fluorapatite contenant 2,9 % en masse de Gd2O3, synthétisé à partir d’un bain fondu, a montré une préférence marquée de Gd pour le site Me(2) [42]. Il en est de même pour la fluorapatite riche en Gd, préparée par la méthode hydrothermale [42]. Lorsque la quantité insérée est très faible, Gd est préférentiellement localisé dans le site Me(1) [42].
L’affinement des taux d’occupation du lanthane des deux sites cationiques a été réalisé sans qu’aucune contrainte ne soit imposée. Le Tableau 5 montre que la distribution de cet élément entre les sites Me(1) et Me(2) est en étroite relation avec la méthode de préparation utilisée. Ceci est confirmé en comparant les rapports des facteurs d’occupation affinés des deux sites [La(1)/La(2)] (0,358 pour le composé calciné et 1,402 pour le composé broyé) à la valeur du rapport théorique statistique (0,666). Ainsi, le lanthane occupe préférentiellement le site Me(2) pour l’échantillon préparé par traitement thermique et le site Me(1) pour celui obtenu par mécanosynthèse.
Les distances interatomiques et les angles O–(P/Si)–O sont regroupés dans le Tableau 6. La valeur moyenne de la distance interatomique <(P/Si)–O> de l’échantillon calciné est égale à 1,636 Å, alors qu’elle est de 1,499 Å pour celui broyé. Cette valeur plus faible plaide aussi en faveur de l’insertion des ions CO32− dans les sites du groupement PO43−. Les valeurs moyennes des angles <O–(P/Si)–O> des deux composés broyés et calcinés sont aussi plus faibles que celle dans un tétraèdre régulier (Tableau 6). Les cations localisés dans les sites 4f sont coordonnés chacun à neuf atomes d’oxygène [3O(1), 3O(2), 3O(3)], six atomes se répartissent au-dessus et en dessous du plan contenant les cations Sr2+/La3+, alors que les trois autres, plus éloignés, se placent presque dans le même plan. Quant aux cations Sr2+/La3+, localisés dans les sites 6h, ils sont coordonnés par six atomes d’oxygène et un atome de fluor. La distance moyenne <Sr/La(1)–O> est respectivement égale à 1,650 et 1,646 Å pour les deux échantillons. Si ces valeurs sont voisines, il n’en est pas de même pour la liaison <Sr/La(2)–O> . Pour l’échantillon obtenu par réaction à haute température, cette dernière est égale à 2,57 Å. Elle est beaucoup plus faible que la valeur (2,72 Å), déterminée pour l’autre échantillon. La distance Sr/La(2)–F est respectivement égale à 2,52 et 2,42 Å pour les deux échantillons broyés et calcinés. Cette distance, plus grande que celle observée dans la fluorapatite non substituée, est consécutive au déplacement de l’ion fluorure le long de l’axe c, en dehors du centre des triangles formés par les cations du site S(2).
Distances interatomiques (Å) et angles de liaisons (°) de l’échantillon Sr7La3F2.
| Préparé par mécanosynthèse | Préparé par traitement thermique | |
| (P, Si)–(O1) | 1,315(10) | 1,579(2) |
| (P, Si)–(O2) | 1,660(7) | 1,676(2) |
| (P, Si)–(O3) (×2) | 1,511(3) | 1,646(1) |
| <(P, Si)–O> | 1,499 | 1,636 |
| (O1)–(P, Si)–(O2) | 99,01 (6) | 107,12 (8) |
| (O1)–(P, Si)–(O3) (×2) | 112,02 (4) | 110,77 (6) |
| (O2)–(P, Si) (O3) (×2) | 102,01 (3) | 105,78 (5) |
| (O3)–(P, Si)–(O3) | 125,70(8) | 116,01(9) |
| <O–(P, Si)–O> | 108,78 | 108,37 |
| (Sr1, La1)–O(1) (×3) | 2,65(7) | 2,50(2) |
| (Sr1, La1)–O(2) (×3) | 2,50(5) | 2,57(5) |
| (Sr1, La1)–O(3) (×3) | 2,79(4) | 2,89(3) |
| <(Sr1, La1)–O> | 2,65 | 2,65 |
| (Sr2, La2)–O(1) | 3,20 (9) | 2,79(6) |
| (Sr2, La2)–O(2) | 2,63(7) | 2,45(2) |
| (Sr2, La2)–O(3) (×2) | 2,76(3) | 2,71(1) |
| (Sr2, La2)–O(3) (×2) | 2,49(3) | 2,38(1) |
| <(Sr2, La2)–O> | 2,72 | 2,57 |
| (Sr2, La2)–F | 2,52(4) | 2,42(1) |
| DI ((P, Si)–O) | 0,061 | 0,017 |
| DI (O–(P, Si)–O) | 0,071 | 0,013 |
3.2.3 Spectroscopie RMN 31P et 29Si
L’analyse par spectroscopie RMN–MAS de 31P de l’échantillon Sr7LaF2 préparé par traitement thermique révèle la présence d’un seul signal isotrope vers 3,00 ppm (Fig. 5a), prouvant l’existence d’un seul type de phosphore dans un environnement apatitique. L’existence d’un seul pic montre que l’échantillon est exempt de phase secondaire phosphatée, corroborant les résultats obtenus par DRX. Le spectre de 29Si comporte, aussi, un seul pic (Fig. 5b). Ceci indique que le silicium occupe le même site que le phosphore.

Spectres RMN de 31P (a) et 29Si (b) de l’échantillon Sr7La3F2 préparé par traitement thermique.
3.2.4 Conductivité ionique
Les spectres d’impédance complexe obtenus dans le plan de Nyquist, acquis dans le domaine 400–700 °C, sont regroupés dans la Fig. 6. Ils sont constitués d’un seul demi-cercle centré sur l’axe représentant les parties réelles de l’impédance. À partir des résistances à haute fréquence mesurées sur les spectres d’impédance, il a été possible de déterminer la conductivité ionique pour les différentes températures en utilisant la relation suivante :
| (2) |

Diagrammes d’impédance complexe de l’échantillon Sr7La3F2.
À titre d’exemple, la conductivité mesurée à 700 °C est de 2,42·10−5 S·cm−1. Elle est du même ordre de grandeur que celle déterminée pour la fluorapatite strontique (3,35·10−5 S·cm−1) [2].
Les valeurs de la conductivité ionique obtenues ont été utilisées pour tracer les diagrammes d’Arrhenius selon la relation suivante :
| (3) |
La Fig. 7 représente la courbe d’Arrhenius de la conductivité ionique de l’échantillon. Cette figure montre que la conduction électrique est un processus thermiquement activé et que la courbe présente un changement de pente à 535 °C. Les énergies d’activation déduites de cette courbe sont égales à 0,49 eV pour les basses températures et à 1,52 eV pour les hautes températures. Cette dernière valeur est aussi peu différente de celle de la fluorapatite strontique (1,47 eV) [2].
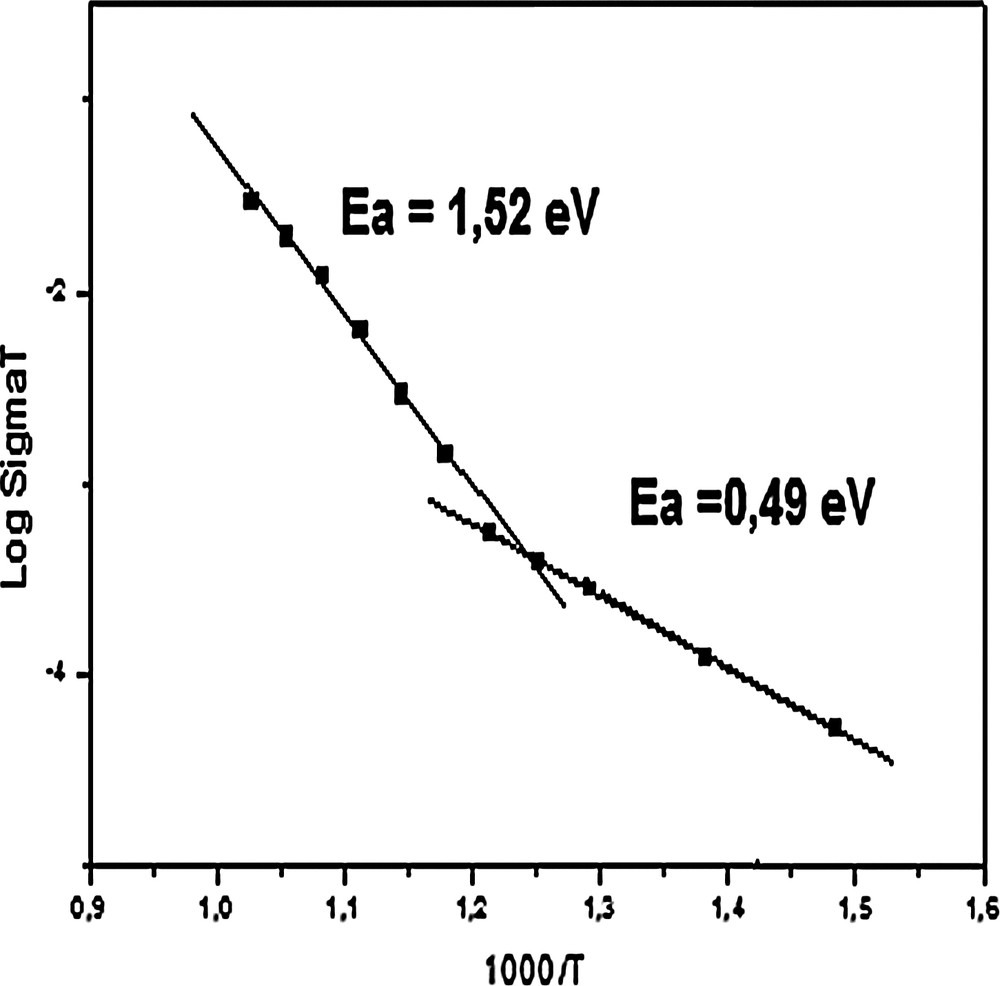
Courbes d’Arrhenius de l’échantillon Sr7La3F2.
Une rupture de pente sur la courbe d’Arrhenius est généralement liée, soit à une transition de phase [43], soit à un changement du mécanisme de conduction [44]. Dans le cas présent, les ions F– sont les seuls porteurs de charges. De plus, aucune transition de phase n’a été observée pour ce type de composé. Il est alors peu probable pour que le changement de pente soit associé à l’un ou l’autre des deux phénomènes précédents. Ainsi, comme dans la fluorapatite, le mécanisme de conduction est probablement lié au saut des ions F–, le long de l’axe c, des sites normalement occupés vers des sites interstitiels puis vers des sites normaux [45]. En étudiant les propriétés électriques des fluorapatites (M10(PO4)6F2) avec M = Ca, Pb, Ba, Laghzizil et al. n’ont observé de rupture de pente sur le diagramme d’Arrhenius que pour l’apatite au plomb [21]. Ils ont expliqué cette rupture par le caractère pseudo-ionique de la liaison Pb–F, et plus particulièrement par la forte polarisabilité du plomb. De la même manière, il est possible d’attribuer le changement de pente dans le cas présent à la liaison Sr/La–F. En effet, comme il a été indiqué plus haut, la courbe d’Arrhenius de la fluorapatite calcique ou strontique est constituée d’une seule droite [2]. Il en est de même pour les apatites, alors que celle du composé Ca6La4(PO4)2(SiO4)4F2 [46] est formée de deux branches. En revanche, la courbe de Ca6La4(PO4)2(SiO4)4O [47] ne comporte pas de cassure, bien que le lanthane soit aussi présent. Ceci conforte l’idée que la rupture de pente est essentiellement due à la nature de la liaison Sr/La–F.
4 Conclusion
Des apatites de formule générale Sr7−xCaxLa3(PO4)3(SiO4)3F2, avec x = 0, 1 et 2, ont été préparées par mécanosynthèse. De même, le composé pour x = 0 a été aussi obtenu par traitement thermique à haute température. Des différentes analyses et caractérisations réalisées, il se dégage que :
- • par rapport à la synthèse par réaction à haute température, la mécanosynthèse a conduit aussi à des apatites pures, mais carbonatées, après un broyage de 25 h. Des réactifs non carbonatés et une atmosphère de travail exempte de dioxyde de carbone permettraient l’obtention d’apatites ne contenant pas ce type d’ions ;
- • si des phases secondaires n’ont pas été détectées par la diffraction des rayons X et la spectroscopie d’absorption infrarouge, il n’est pas exclu que les poudres obtenues renferment des impuretés. Mais, en raison de leurs faibles quantités, elles n’ont pas pu être mises en évidence ;
- • la distribution des cations entre les deux sites cationiques est en étroite relation avec la méthode de synthèse adoptée ;
- • la conductivité ionique de ce type de composé est thermique activée. La rupture de pente observée sur le diagramme d’Arrhenius a été reliée à la nature de la liaison Sr/La–F.