


 CC-BY 4.0
CC-BY 4.0
1. The historical emergence of the stereochemistry concept
For centuries humans have used botanical extracts as a medical source. Only during the past century, with the consolidation of a vigorous pharmaceutical industry, the development of drugs by industrial synthesis prevailed over natural product discoveries. Notwithstanding, up to our days, the proper stereoselective synthesis of morphine, the golden standard opioid analgesic, is too laborious and expensive, so it is obtained from poppies; vast plantations are cultivated in eastern Asia to produce opium, from where morphine is extracted and crystallised. This opiate has five chiral centres; but the poppy plant synthesises only one of the possible stereochemical isomers, that with high affinity for the opiate receptors. Knowing the spatial configuration of a drug molecule is an essential step to understand drug interactions with biological receptors. Pasteur, through his brilliant and laborious investigation on tartaric acids over 170 years ago, paved the road to reveal the 3D spatial configuration of molecules: an essential code of the architecture of living molecules. This principle is a crucial concept critical for drug design. It is no overstatement that pain-relieving, hypertension, asthma, mental diseases and infectious diseases, to name some clinical conditions, are massively treated with optically active drugs, highlighting the influence of Pasteur’s contributions to modern pharmacodynamics.
1.1. Pasteur, a pioneer biochemist and a modern biotechnologist
Pasteur was born in Dole (Jura, France) in 1822. He began his scientific formation as a physicist and a chemist; along his career he succeeded in studying numerous topics in all of which he demonstrated creativity, innovation and provided scientific answers as a prototype of a modern biotechnologist. Early in his career he played an essential role in the understanding of molecular asymmetry providing the scaffold for the understanding of life’s spatial molecular structures [1, 2, 3]. Years later, Pasteur became interested in the fermentation of spirits and wines and discovered the involvement of yeast and microorganisms in the fermentation process [4, 5]. These observations questioned the dominant theory of spontaneous generation, a concept passionately defended by L. Spallanzani (1729–1799), which prevailed to his days. He meticulously demonstrated that killing environmental germs, which he latter associated with wine fermentation, stopped food spoilage [4]. A variant allows to preserve milk from acidification, inventing a heating process that keeps his name; this procedure is known worldwide as milk pasteurisation, a procedure currently used by the dairy industry to prolong the half-life of milk and its derivatives. Moreover, and based on his increasing interest on microorganisms and human disease, Pasteur succeeded in creating vaccines for cholera, rabies and anthrax [6, 7, 8], highlighting contributions to microbiology and modern biotechnology.
Notwithstanding the multiple contributions of Pasteur to modern medicine, this essay will focus on his influence on the understanding of the spatial architecture of living molecules and his decisive inspiration to the fundamentals of molecular pharmacology development. This essay reviews the development of optically active adrenergic agonists and antagonists as a heritage to modern therapeutics.
1.2. From crystals to the 3D spatial configuration, Pasteur investigated the optical activity of molecules, laying the foundations of stereochemistry and chirality
Light polarisation is an essential concept to understand the bases of optically active isomers. Quartz was first observed to deviate light; by 1809, a French physicist Malus (1775–1812) discovered the light polarisation principle [9]. An exciting debate ensued about the nature of this effect as applied to the study of chemicals in solution. Biot (1774–1862), a French physicist, affirmed that the optical activity of a chemical can be neutralised by an opposed activity [10], inferring correctly that the optical activity of a compound in solution depends on its crystalline structure [11, 12]. Decades thereafter, Mitscherlich described crystal isomorphisms while investigating why salt solutions of tartaric and para tartaric acids (C4H6O6) differ in optical activity. As a mineralogist, he compared the crystal forms of the corresponding salts and noted differences in their morphologies, a finding that was not further investigated. The acid solutions of these chemicals, although apparently identical based on the same and equal proportion of atomic composition, differed in optical activity since only tartaric acid was optically active while para tartaric was not [13].
Pasteur manually separated tartaric acid crystals. Left panel shows an etching portrait of Pasteur observing under the microscope tartaric acid salt crystals obtained during the wine fermentation process. Right panel shows the tartaric acid stereoisomers: dextrorotatory (from the latin dexter, right) and levorotatory (from latin laevus, left), referring to the polarised light rotation. Pasteur demonstrated that para tartaric acid was the racemic mixture of both dextro- and levorotatory crystals, in equal quantities, explaining why para tartaric acid was optically inactive.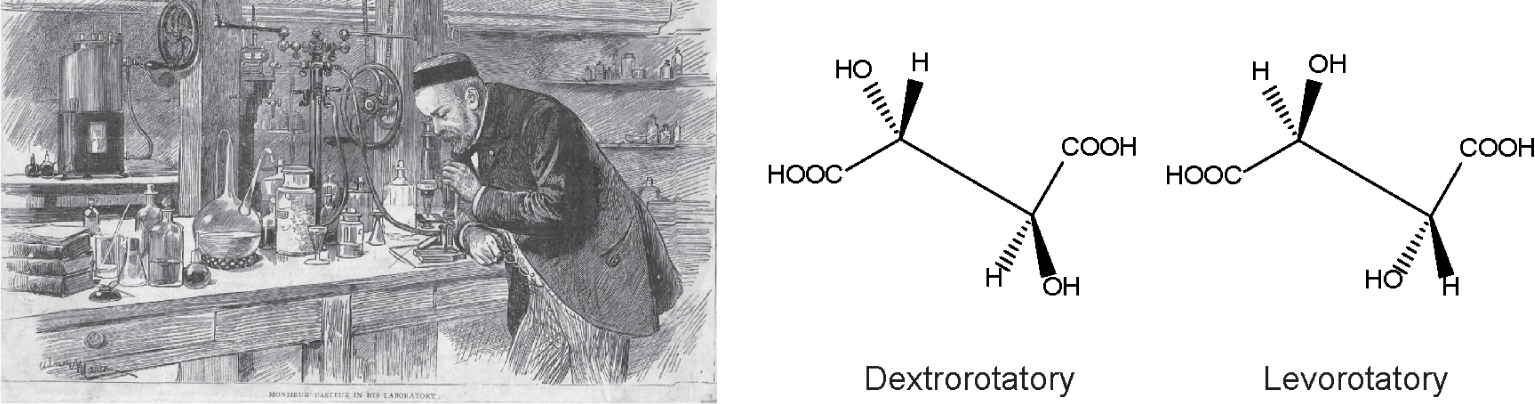
This unexplained observation set a paradox that attracted young Pasteur’s imagination. He wanted to understand why two apparently identical compounds, tartaric and para tartaric acids, differed in optical activity. Based on Mitscherlich’s observations [13], plus the notion that optical activity depends on crystalline structure, he reasoned that para tartaric acid crystals must be a mixture of compounds which annulled each other’s optical activity. To prove his thesis, Pasteur used a microscope to observe the crystals of para tartaric and tartaric acid; he noted two different crystal morphologies [1, 2, 3] as reported previously by Mitscherlich. Manually, he undertook the painstaking job of separating these crystals (Figure 1) and analysed the optical activity of each crystal type distinctly. He observed that the two types of para tartaric acid crystals had the same optical activity but of opposite signs; he concluded that para tartaric acid solutions were optically inactive because the activity of one type of molecules cancelled the activity of the other. In contrast, tartaric acid was optically active and was composed of a single crystal population. This observation explained and accounted for the observation that para tartaric acid was optically inactive, resolving the inconsistency. This result allowed to further infer that the optical activity of asymmetric molecules is due to their spatial atomic arrangements, as summarised by [11, 12]. Furthermore, this critical observation illumined a prepared mind to predict the relevance of optical activity for living molecules. In his dissertation memoire [1], he wrote: “la dissymétrie paraît être une nécessité de la constitution des molécules qui se sont édifiées sous l’influence de la vie”, which in English reads, dissymmetry seems to be a necessity of the constitution of the molecules which were built under the influence of life.
Although Pasteur’s discovery solved the para tartaric paradox, a complete understanding of Pasteur’s discovery in terms of spatial molecular configurations required a deeper grounding in spatial molecular geometry, a proposal that evolved only 25 years later. In 1874, two independent publications by Van’t Hoff [14] and Le Bel [15] raised the theory of stereoisomerism which explained the spatial configuration of the C atom, raising the notion of optical isomers based on different C atom substituents. This proposal vigorously boosted the modern view that the 3D geometry of asymmetric carbon atoms, as in tartaric acids, is represented as a tetrahedron, with four different radical substituents [16]. Some decades later, Thomson, also known as Lord Kelvin [17], coined the term chiral centres (from the Greek, chiral for hand) referring to asymmetric atoms which have different atomic substitutes, deciphering that stereoisomers cannot be superposed, mimicking mirror images. The notion of optical asymmetry known to Pasteur has been substituted by chirality, a notion that gave a powerful 3D structural support to stereochemistry, a view which prevails nowadays.
The notion of chiral centres expanded over the years from the C atom to include atoms such as S or N or even those in metal complexes. Werner realised early during the XX century that optical activity also occurred among inorganic complexes [18]. In fact, the octahedral conformation of transition metal coordinates was demonstrated to be optically active. These findings originated octahedral molecular geometry giving rise to the theory of metal coordination, for which Werner was awarded the Nobel Prize in Chemistry in 1913. Cr(III), Co(III) or even Pt(IV) under special conditions, form optically active chiral complexes [19]. Based on these fundamentals, it is no surprise that drugs based on S or N chiral centres are currently used, such as omeprazole and its active isomer, esomeprazole, which is the prototype of modern “prazoles” successfully used for ulcers treatment based on selective H+/K+ pump inhibition. Similarly, N-based synthetic opiates with chiral centres give rise to clinical relevant optimal isomers [16].
2. Principles of stereochemistry and its application to adrenergic pharmacology: the case of natural and synthetic drugs
2.1. Definition
Stereochemistry (from the Greek stereos, space) deals with the spatial conformation of compounds with the same molecular formula but differing in spatial structure; these compounds are referred as stereoisomers (from the Greek isos, equal and meros, part). Stereoisomers contain the same number and types of atoms, but the atoms are spatially oriented in differing ways. Two types of stereoisomers are known: conformation isomers—the same molecule but in different spatial arrangements; these isomers have identical chemical and physical properties; they are difficult to separate from each other except for interactions with another stereochemical isomer; and configuration isomers—distinct molecules with different physical and chemical properties, despite having identical atomic composition.
2.2. Differentiating enantiomers from diastereoisomers; the art of chirality
Enantiomers (from the Greek enantio, meaning opposite and meros, parts) are configuration stereoisomers, which cannot be superposed in the mirror; therefore, these compounds are mirror images of each other. This property is called chirality (from the Greek hand). A molecule with a chiral centre does not have a symmetry plane. As an example of a prominent psychoactive drug with a chiral centre, amphetamine (a family member of potent brain stimulants) and its enantiomers are shown in Figure 2. The dextro isomer is a central nervous system stimulant widely used as a recreational drug nowadays, but in the past, it was used by soldiers to endure physical strength, while its enantiomer is markedly less active. Enantiomers have identical chemical and physical properties, apart from the power of rotating polarised light in opposite directions: one is positive while the other is a negative polarised light rotator. The isomer that rotates polarised light to the right side is also known as dextrorotatory D (+), whereas the other, which turns polarised light to the left side, is known as levorotatory L (-), a notation created by Fischer, based on Le Bel and Van’t Hoff proposal of stereoisomerism. Fisher was awarded the Nobel Prize in Chemistry (1902) for discovering, among other issues, the absolute configuration of D (+)-glucose, a main carbohydrate energy source for most cells from bacteria to humans. Moreover, based on spatial chemical configurations and on sugars’ interaction with proteins, he proposed the “lock and key” principle [20], which prevailed for over 70 years. This theory was later modified by Koshland [21], who reviewed the salient features of the lock and key versus the induced fit theory, stressing that in the interaction of ligands with proteins, each induces a reciprocal conformational change to allow meaningful biological responses.
Spatial configuration of amphetamine and methamphetamine, two potent psycho stimulants. Amphetamine enantiomers: on the left is its levorotatory form, corresponding to (S)-amphetamine configuration; on the right the dextrorotatory form, corresponding to (R)-amphetamine. The tetrahedral representation of each molecule is shown in the middle figure panel. The major stimulant amphetamine effect is due to the R-isomer which is 4–10 times more potent than the L-enantiomer. Bottom part shows methamphetamine, using Cahn and Ingold’s rules: the priority group is the secondary amine group, followed by the aromatic-C7H7 and finally the methyl group. According to the disposition of each substituent, the rotation is not the same. To the left, the rotation is clockwise, (S) conformation, to the right, anticlockwise rotation (R) conformation.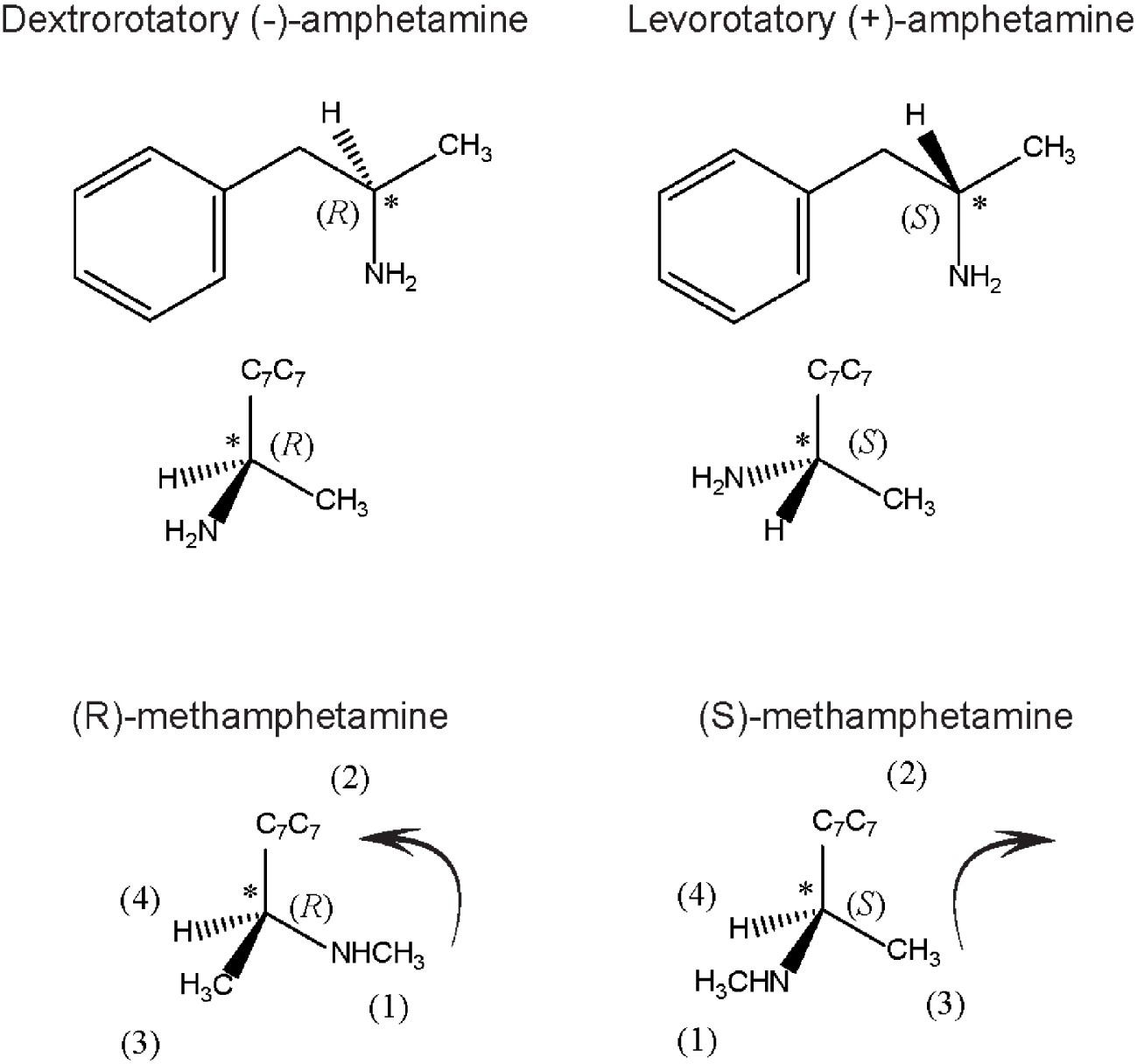
Later studies concluded that the power of polarised light rotation depends on chiral carbon substitutes. Amino acids, the building blocks of proteins (from bacteria to humans), belong to the L (-) series, with only minor exceptions; racemases convert L-amino acids to their D (+) stereoisomer in those very exceptional cases. Since proteins are constructed with L-amino acids, these macromolecules are asymmetric in nature, allowing stereoselective ligand binding—a principal pillar of life [22]. Enantiomers may have one or more than one chiral centre allowing differentiation between two absolute conformations: R for rectus, or S for the sinister conformation. This absolute conformation depends on the priority order of the substituents of the chiral centre, a nomenclature established by Cahn et al. [23].
Racemic mixtures have equal proportions of the (+) and (-) optical isomers; racemic solutions lack optical activity as with Pasteur’s para tartaric crystals [16]. Racemic mixtures are relevant to medicine since many chiral drugs are commercialised as racemates; with few exceptions stereoisomers are formulated separately as will be discussed. Usually, only one of the enantiomers carries the pharmacological activity, while the other is inactive, or even may cause non-specific side effects [22].
Representation of enantiomers and diastereoisomers of the natural product ephedrine, the popular Chinese Ma Huang plant. Only one of the isomers is synthetised by the plant and used as a medicinal component of decongestants used to alleviate common cold. The configuration of enantiomers and diastereoisomers are shown diagrammatically.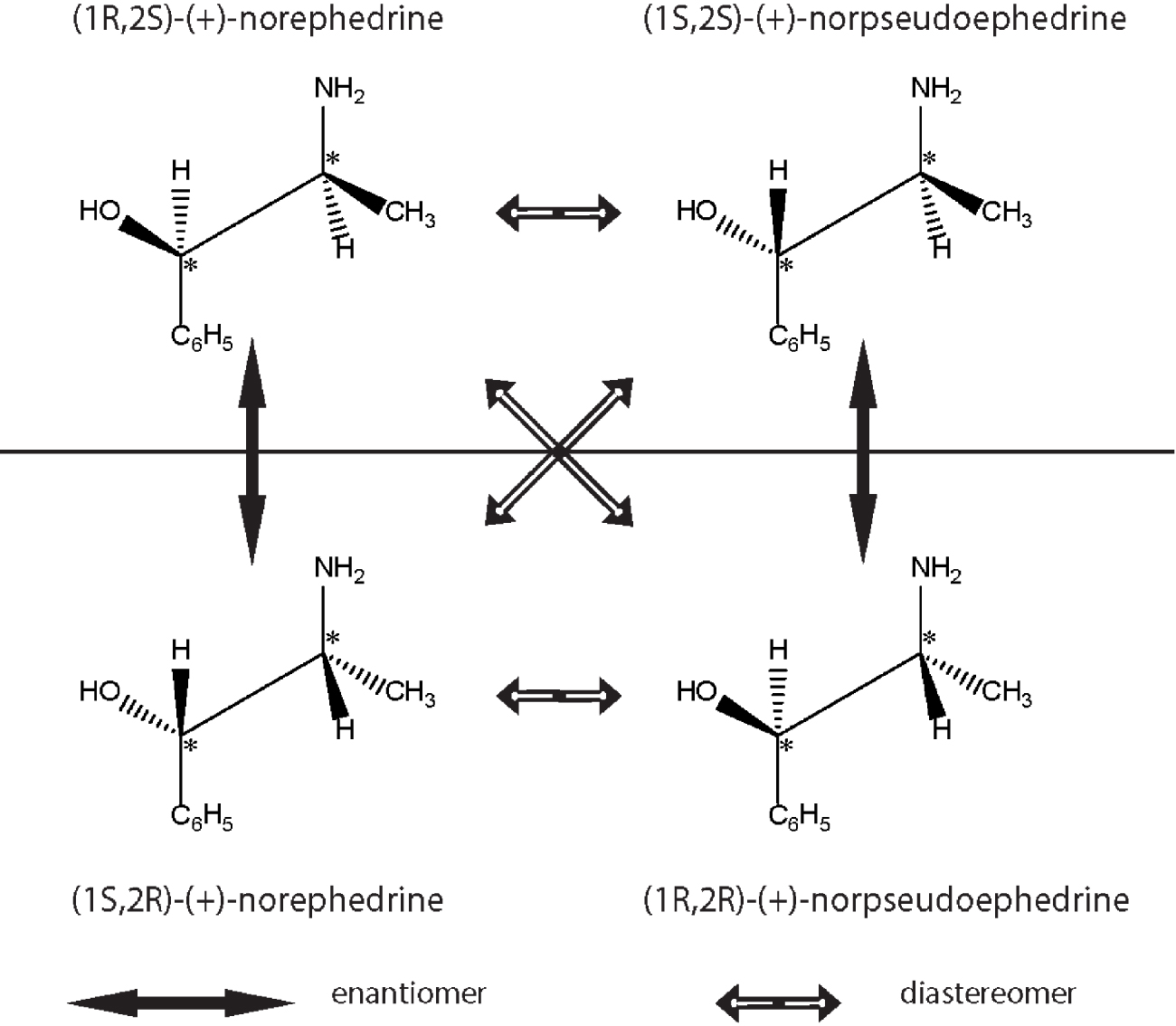
Diastereomers (from the Greek dia, through or apart, stereos, space and meros, parts) are isomers that have more than one stereocentre and can be chemically separated. Ephedrine (drug used as a mild bronchodilator, component of several decongestant mixtures) has two chiral centres and therefore four isomers, only one of which has therapeutic activity. Ephedrine’s enantiomers and diastereomers are shown in Figure 3. A pair of diastereomers, as opposite to enantiomers, have distinct physical properties and similar, but not identical, chemical properties. Compounds with a carbon-carbon double bond are famous for their cis (latin on this side) and trans (latin for across) configurations which are geometric diastereomers; an interesting example for biology is the process of vision which depends on light-governed cis-trans retinaldehyde transitions [24].
3. Stereochemistry and autonomic pharmacology
3.1. Applications of stereochemistry to autonomic pharmacology
It took over half a century before optical activity enlightened drug action principles, or in other words, before the Pasteur principles found a solid pharmacological application. In 1904, Cushny, a British chemist and pharmacist, published a series of seven papers describing the pharmacology of levo-hyoscyamine (levo-atropine) or levo-hyoscine (scopolamine) compared to racemic hyoscyamine (atropine) [25, 26]. These drugs evidenced different potencies as antimuscarinic agents in the frog heart but differed in central stimulant potencies, evidencing for the first time that optical isomers differed in pharmacological potencies, although a sound explanation was lacking [27, 28]. Years later, Ahlquist (1948) proposed that the actions of natural noradrenaline and adrenaline are mediated by adrenoceptors, the classic 𝛼- and 𝛽-receptors [29]. Natural noradrenaline and adrenaline are levorotatory, 100–300 times more potent than the corresponding dextrorotatory isomers as 𝛼- or β-adrenoceptor agonists, respectively. These seminal observations, published about 100 years after Pasteur’s proposal, led the way to the molecular aspects of autonomic pharmacology and optical activity in particular [28].
Dissociation constants (Kd), an indication of relative receptor affinities, for human 𝛽1- and 𝛽2- adrenoceptors
| Drug | Active form | Dissociation constant (𝛽1) | Dissociation constant (𝛽2) |
|---|---|---|---|
| Noradrenaline | (R)-adrenaline | −5.74 ± 0.03 | −5.41 ± 0.07 |
| Adrenaline | (R)-adrenaline | −5.15 ± 0.06 | −6.13 ± 0.05 |
| Isoproterenol | −6.06 ± 0.08 | −6.64 ± 0.09 | |
| Salbutamol | (R)-salbutamol | −4.68 ± 0.03 | −6.01 ± 0.03 |
| Propranolol | (R)-propranolol | −8.167 ± 0.08 | −9.09 ± 0.06 |
| Nebivolol | −9.06 ± 0 003 | −7.92 ± 0.04 | |
Binding energy data were obtained from the literature from Baker [30].
Almost simultaneously biochemical evidence showed the relevance of stereoisomer substrates for enzymatic reactions and protein constitution. Proteins, including enzymes, transporters, biological receptors and other relevant biomolecules, are asymmetric because of chiral amino acids. The interaction between any ligand molecule, endogenous or exogenous, and proteins is therefore stereoselective. For example, D (+) glucose is the preferred substrate of glucokinase or glucose transporters (GLUTs), among many enzymes used in carbohydrate metabolism [24].
3.2. 𝛼- and 𝛽-Adrenoceptor ligands launched optical isomers into therapeutics
Noradrenaline, the sympathetic neurotransmitter, is synthesised in sympathetic nerve endings by an enzymatic sequence that starts with L-tyrosine, an essential amino acid and ends with a 𝛽-hydroxylation that introduces a chiral centre. L or (-) noradrenaline or structural analogues are used as vasoconstrictors or as a component in the formulation of local anaesthetics. Noradrenaline binds to 𝛼- and 𝛽-adrenoceptors stereoselectively with slightly higher potency to β1- than β2-adrenoceptors; (see Table 1; for additional data consult [30]).
(-) Adrenaline has lower affinity for β2-adrenoceptors as compared to the 𝛽1-adrenoceptor (Table 1) and adjusts the body to exercise and stress, redistributing blood flow and eliciting metabolic adjustments for the “fight or flight” reaction, in response to unexpected, stressful situations. The hormone adrenaline is synthetised by a further enzymatic step through noradrenaline N-methylation. Adrenaline is used clinically in rare occasions as in anaphylactic or septic shock or other severe collapsing vasodilatations caused by life-threatening conditions. In further support of 𝛽-adrenoceptors, the isopropyl substitution of noradrenaline originated isoproterenol, a higher-affinity, non-selective β-adrenoceptor agonist (Table 1).
Prior to decoding the structural elements of the purported adrenergic receptors, Easson and Stedman [31] advanced a thought-provoking proposal to account for the biological activity of optical isomers; the proposal advocated at least a three-point attachment of ligands to the putative biological receptors. This hypothesis intended to explain the potency of drug stereoisomers by arguing that only one of the isomers met the correct receptor conformation of a C chiral centre requirement, based on the main bonds between ligands and the receptor; with at least 3 of the 4 tetrahedron bonds of the chiral centre conferring higher receptor-complex stability. In addition, this proposal provided a graphical representation of the “key and lock” principle articulated by Fischer almost 40 years earlier [20]. In this model (Figure 4), the dextrorotatory bioamine has only two dock points (phenol aromatic ring and the charged amino function) whereas its levorotatory isomer displays three binding points (aromatic ring, the charged amino function plus an H bond). The extra hydrogen bond accounts for the larger affinity of the levo isomer over the dextro isomer (Figure 4).
The three-point attachment theory applied to adrenaline isomers explains the difference in 𝛽2-adrenoceptor binding affinity for (R)- and (S)-adrenaline, based on the chiral 𝛽 hydroxyl substituent (A and B). Simplified schematic conceptualisation of the three-point attachment model proposed for the binding of adrenaline enantiomers (A), while C represents, as a graphical abstract, the interaction of adrenaline with the 𝛽2-adrenoceptor illustrating the three-point attachment at the adrenoceptor binding pocket identifying main interactions based on π-π interactions, ionic and an H bonds. Docking to the crystallised 𝛽2-adrenoceptor receptor with adrenaline positioned in its binding pocket, modified from reference [17] (D).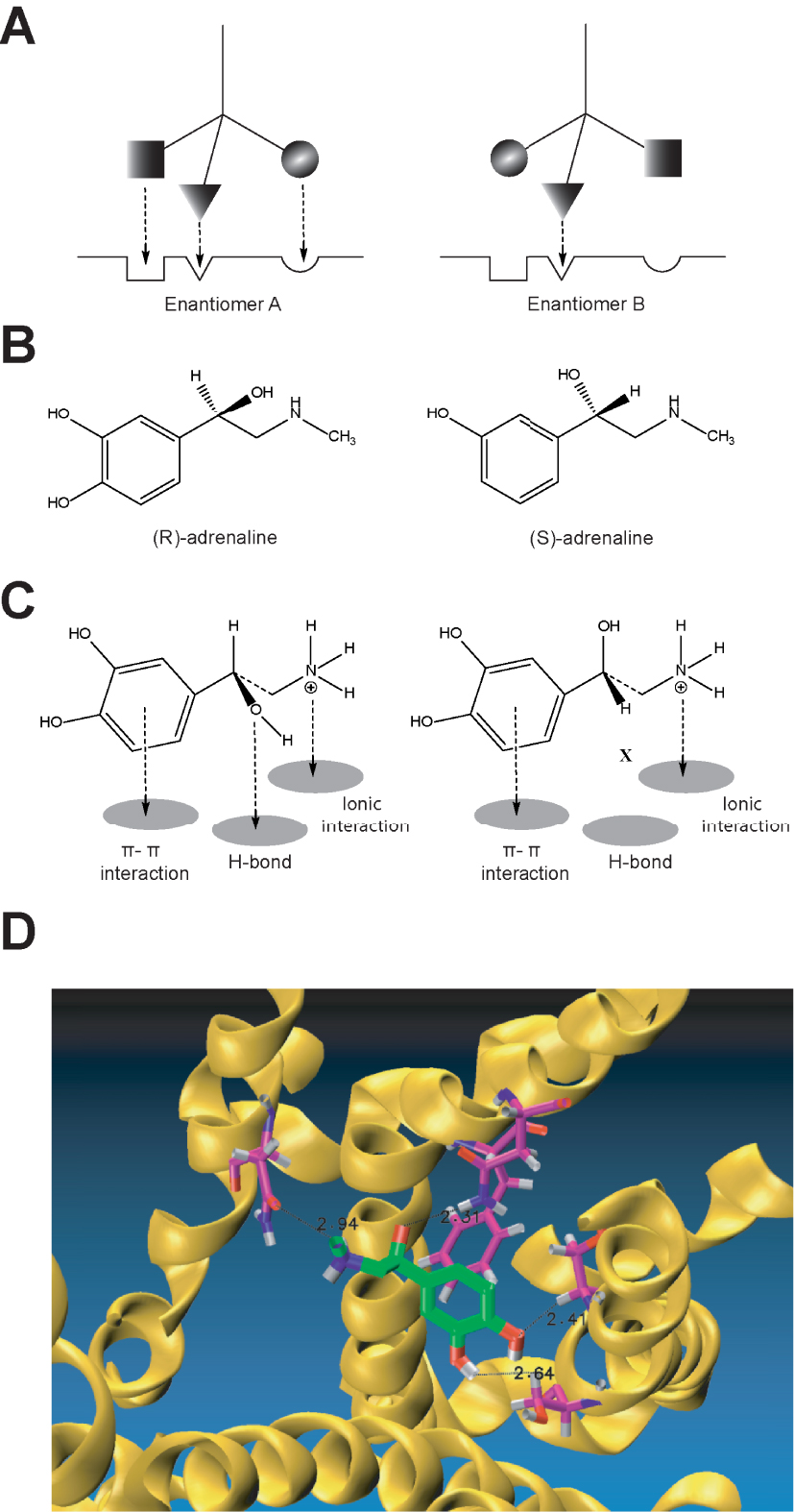
The concept of catecholamines adrenergic receptors took shape fifteen years later; the seminal classification of adrenergic receptors into 𝛼 and 𝛽 based on excitatory or inhibitory adrenaline actions in smooth muscles was published in the American J. Physiol [29] since the editors of the Pharmacology Journal found no major pharmacological relevance of this research. Ahlquist 1976 and Baker 2010 reviewed this topic [30, 32]. Years later, both the α- and β-adrenergic receptors were further sub-classified as 𝛼1- and 𝛼2- and 𝛽1- and 𝛽2-adrenoceptors [33], allowing a more precise adrenergic responses classification, nurturing clinical significance. These developments paved the development of the renowned β-adrenoceptor antagonists as the massive first-choice worldwide antihypertensive treatment. Nowadays, other medications have been launched as useful alternatives; nonetheless, β-adrenergic antagonists are still widely used to treat arrythmia, ischemic heart disease, glaucoma, cardiac failure, first stages of hyperthyroidism, performance anxiety, migraine prophylaxis, and so forth. At the time of the 𝛽-adrenergic classification [33], no notion was available related to the chemical nature of receptors, which started to mature 20 years later, linking adrenoceptors with the trimeric G proteins. Within the past 10 years, the 𝛽-adrenoceptor was crystallised with its corresponding G proteins, providing topological conformation details prior to and upon receptor occupation by agonists and antagonists [34].
4. Optically active sympathomimetic amines: synthetic and natural products
4.1. Amphetamines, a family of potent psychostimulants
In the early 30s at the School of Pharmacy of the University of California Medical Centre in San Francisco, Alles and co-workers (1928) synthetised a series of alpha methyl phenylethyl amine derivatives (chemical acronym shown in bold for amphetamine and congeners), which caused potent brain stimulation and classified as psychostimulants [35, 36, 37]. Due to structural similarity with catecholamines (Figure 4), these chemicals were thought to produce brain stimulation due to catecholamine mimicry in brain circuits. The clinical potential of amphetamines was immediately recognised, and its central stimulant action was used clinically in narcolepsy patients [38]. Decades later, these compounds, well-known for antifatigue effects, were used during World War II as military warfare and much later, during the 60s, these chemicals became popular as recreational agents. These compounds cross the blood– brain barrier better than catecholamines or derivatives. Although first used as racemates, soon the psychostimulant activity was shown to reside in the dextro (+)-isomer, a finding confirmed in many behavioural and pharmacological studies. Amphetamines also elicit a spectrum of peripheral sympathomimetic effects including metabolic adaptations. A thorough analysis of amphetamine mechanisms of actions shows that not all family members act via a unique mechanism. In addition to the presynaptic release mechanism, some members interfere with dopamine and noradrenaline transport while others combine a complex reuptake blockade with a release mechanism and even some, direct stimulation of adrenoceptors [39]. The discovery of receptors for phenylethylamine [40] as a novel amphetamine target is an emerging actively pursued topic.
Methamphetamine, the N-methylated amphet- amine derivative, crosses the blood–brain barrier more extensively and has an even stronger psychostimulant effect and causes more dependence than amphetamine itself. Methamphetamine causes a considerably long-lasting loss of sympathetic nerve endings, explaining its cytotoxic potential far beyond amphetamine. Only dextro methamphetamine is used as a recreational drug; its optical isomer is considerably less active as a stimulant.
Methylphenidate, an amphetamine variant widely used worldwide to treat children with attentional deficits or hyperactive disorder as the parent drug causes considerable addiction and is heavily abused as reviewed by Kollins et al., [41]. Methylphenidate essentially blocks dopamine transporter rather than increasing bioamine release as compared with amphetamine. Methylphenidate has two chiral centres; however, it is the (1R, 2R)-methylphenidate which is biologically active [41]. The psychostimulant action is of central origin; amphetamine crosses the blood–brain barrier to reach bioamine brain circuits. Most of the psychostimulant effect is exerted at dopaminergic or noradrenergic synapses; serotoninergic targets cannot be discarded.
4.2. Ephedrine, the Ma Huang alkaloid
Ephedrine is an alkaloid extracted from the popular Chinese Ma Huang plant, (EphedraSinica), used medicinally for over 5000 years in oriental traditional medicine. A main constituent of the plant extract was isolated in 1887; this natural product has central stimulant activity, plus peripheral sympathomimetic effects early recognised. Pharmacodynamically, the ephedrine mechanism differs from those of the amphetamines. The prevailing view is that ephedrine is a mixed adrenergic agonist combining an indirect action mediated by sympathetic nerve terminal release together with a direct, although lower affinity interaction with α- and β-adrenoceptors. The ephedrine structure was soon revealed; it has two chiral centres and all the isomers have been characterised pharmacologically (Figure 3). The natural ephedrine alkaloid is the (1R, 2S)-(-)-ephedrine, also known as pseudoephedrine, a compound endowed with decongestant effect in addition to bronchodilator 𝛽2-agonism (Figure 3), two useful clinical properties that call for pseudoephedrine as a common component of cold decongestant mixtures. From a synthetic point of view, it is considered as a precursor in the illicit synthesis of methamphetamine, a condition that is limiting its medical use. Its four ephedrine isomers are:
(1R, 2S)-ephedrine (levorotatory) is the isomer with the most pharmacological activity.
(1R, 2R)-(-)-ephedrine is an inactive alkaloid.
(1S, 2R)-ephedrine (dextrorotatory) is an inactive stereoisomer.
(1S, 2S)-pseudoephedrine (dextrorotatory) is an enantiomer with slight sympathomimetic activity; not used to treat asthma, but currently used as a mixture component of antiviral, cold decongestants.
4.3. Phenylpropanolamine, the dual acting decongestant
Phenylpropanolamine, or norephedrine, is a synthetic phenylethylamine congener. Based on its structural similarity with amphetamines and ephedrine, it causes indirect and direct sympathomimetic activity, much like amphetamine or ephedrine. This drug is generally used combined with other remedies to treat nasal and bronchial decongestion during severe cold. It is also used as an appetite suppressant. Like ephedrine, phenylpropanolamine has two chiral centres; four distinct isomers are involved in the racemic preparations: dextrorotatory (+)-norephedrine, levorotatory (-)-norephedrine, dextrorotatory (+)-pseudo- norephedrine and levorotatory (-)-pseudo-norephedrine. The therapeutic properties are:
(1R, 2S)-norephedrine (levorotatory). The commercialised form of norephedrine is the racemic mixture; levorotatory form is the only isomer with pharmacological activity.
(1S, 2R)-norephedrine (dextrorotatory). Lacks pharmacological effect, but it is not toxic, (1S, 2S)-pseudonorephedrine (dextrorotatory). (+)-Pseudonorephedrine, called cathine, belongs to the amphetamine family with central stimulant activity. It is a natural product of Catha edulis plant; less potent compared to amphetamine.
(1R, 2R)-pseudonorephedrine (levorotatory). (-)- Pseudonorephedrine is also considered an amphetamine; it elicits dopamine and noradrenaline release.
4.4. Miraculous (L-DOPA), a stereoisomer used for Parkinson’s disease treatment
L-Dopa, corresponds to the levorotatory enantiomer of DOPA, an endogenous dopamine precursor currently still used for Parkinson’s disease treatment. This chiral molecule needs to be transported to the brain substantia nigra where the dopaminergic population is progressively and markedly reduced during disease progression. The rationale to introduce this precursor in the treatment of this neurodegenerative disorder relates to the fact that L-DOPA is a substrate of the aromatic amino acid transporter allowing levo-DOPA passage through the blood–brain barrier to increase dopamine synthesis in this brain region. A serious limitation to the sole use of L-DOPA is its large liver metabolism. To bypass this caveat, treatment with L-DOPA is generally combined with a peripheral DOPA decarboxylase inhibitor, a molecule that will largely reduce its liver metabolism, increasing L-DOPA brain transport to the nigrostriatal circuitry. In view of the progressive disease nature, patients are also treated with dopamine receptor agonists or other agents to block dopamine metabolism.
4.5. Dobutamine, a semi-selective 𝛽1-adrenoceptor agonist with cardiac applications
This synthetic chiral adrenaline congener with a bulkier aromatic amino substituent has significant 𝛽1-adrenoceptors selectivity; it was introduced in clinical use for the treatment of cardiac insufficiency because of its direct interaction with heart 𝛽1-adrenergic receptors, causing positive ino- and chronotropism. In contrast to dopamine, dobutamine acts selectively on adrenergic receptors. Although commercialised as a racemic mixture, only (+)-dobutamine is a relative selective 𝛽1-adrenoceptor agonist, with residual 𝛽2-adrenoceptor activity. In contrast, the (-)-dobutamine enantiomer has low affinity as an 𝛼-adrenergic agonist.
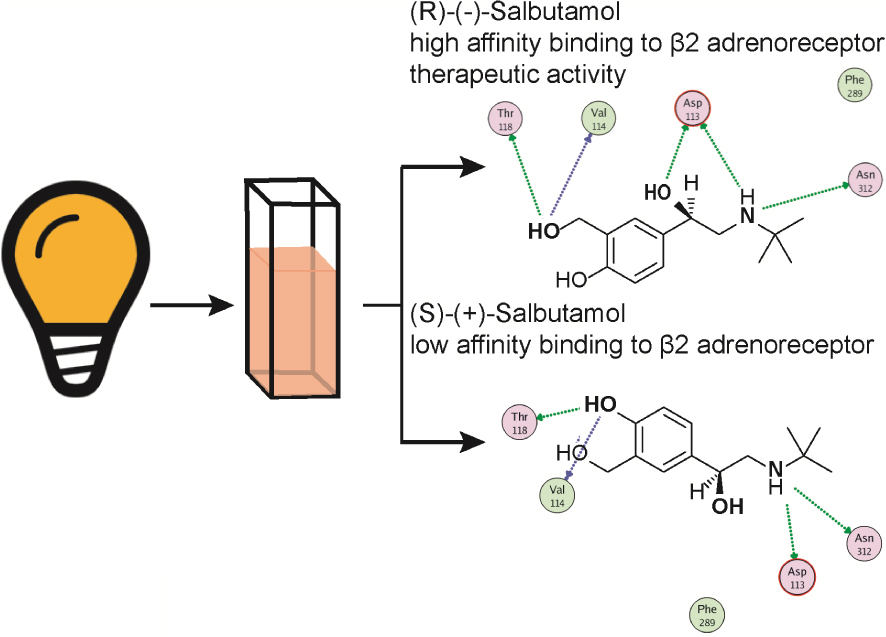
Graphical abstract illustrating the salbutamol stereochemical interaction with the 𝜷2-adrenoceptor. Representation of the two salbutamol stereoisomers, one of which is a preferred 𝛽2-adrenoceptor agonist, amply used as a bronchodilator because of its high affinity for bronchial 𝛽2-adrenoceptors. 𝛽2-Adrenoceptors are coupled to an intracellular signalling cascade including activation of G proteins to elicit cAMP formation which acts as a second messenger, which as an end result, causes bronchodilatation. Note that the secondary N group is substituted by a tert-butyl group, represented as a cross. Only the (-) salbutamol is therapeutically relevant as a bronchodilator.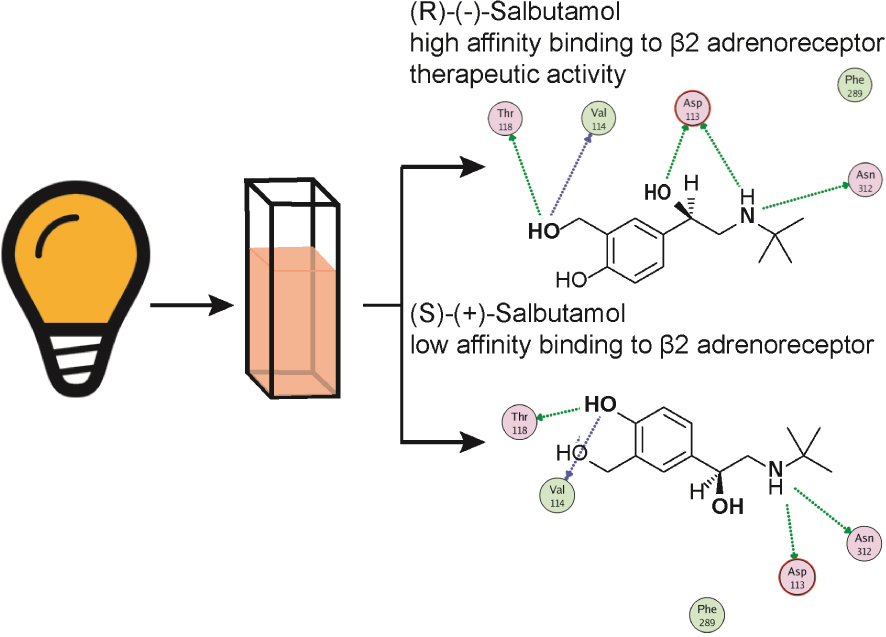
4.6. Salbutamol and structurally related derivatives, semi- selective 𝛽2-adrenoceptor agonists used for respiratory distress syndromes
A series of synthetic catechol and non-catechol adrenaline congeners were elaborated searching for potent bronchodilators. Salbutamol, also known as albuterol, became after years of clinical experience, the top bronchodilator used for acute antiasthmatic use. It is a non-catecholamine rather selective 𝛽2-adrenoceptor (Table 1) agonist bearing a chiral β hydroxyl centre and a tert-butyl amino substituent (Figure 5). This drug is effective in asthma treatments, and related respiratory syndromes, because it relaxes the bronchial tree independent of the nature of the bronchoconstrictor agent. Salbutamol is commercialised as a racemic mixture. While the (R)-levo-salbutamol bears high affinity 𝛽2-adrenoceptor activity, (S)-salbutamol, is pharmacologically inactive. The racemic mixture formulation permits to reduce the metabolisation’s velocity of the active enantiomer, tightening metabolic pathways of elimination. Salbutamol and derivatives proved relatively selective 𝛽2-adrenoceptor agonists; however, in larger doses, tachycardia, a sign of 𝛽1-adrenoceptor activity is evidenced.
5. Adrenergic antagonists
Parallel to the development of stereoisomers with agonist properties, Pasteur’s principle was extended to receptor antagonists. Although 𝛼-adrenergic antagonists, based on ergots preparations, were known by Dale [42] in the early last century, the classic “reversal of adrenaline pressor effect” response was popular in medical teaching to exemplify the dual effects of adrenaline in the vascular system. No systematic development of adrenergic blockers emerged until the mid-50s. The clinical use of phentolamine and congeners was dampened due to lack of selectivity and affinity for 𝛼-adrenoceptors. Almost simultaneous with the sub-classification of 𝛽-adrenoceptors into 𝛽1- and 𝛽2- adrenoceptors [33], the discovery of dichloroisoproterenol first, and propranolol next, as a prototype 𝛽-adrenergic antagonist became a landmark. Two major improvements were rapidly recognised. Propranolol proved efficient to control certain cardiac arrhythmias, but most relevant, it paved the way for the pharmacological management of hypertension avoiding the severe reserpine side effects. Second, propranolol displayed a remarkable high affinity for 𝛽-adrenoceptors (Table 1), allowing the daily use of 10–20 mg doses, a true innovative high standard medication for the time. Propranolol has a 𝛽 hydroxyl chiral centre; at the light of Pasteur findings only one of the stereoisomers is active, fulfilling the promise of optical isomers for clinical use with worldwide consequences.
Four generations of 𝛽-blocking agents
Propranolol belongs to the first-generation of chiral β-adrenergic blockers. This drug, as most of the group members, is used as a racemate; propranolol was followed by structurally related optical isomers with higher affinity for the 𝛽1- than the 𝛽2-adrenoreceptor subtype, compounds known as the cardio-selective 𝛽1-blockers. Some of the first- and second-generation 𝛽-adrenoceptor blocking agents have a novel and compounded pharmacological property described as intrinsic sympathomimetic activity (ISA), the nature of which has not been clarified as yet in molecular terms. A third-generation of this family of drugs combined competitive β- and α-adrenergic blocking properties. More recently, a fourth generation of structurally related chiral compounds that in addition release nitric oxide, such as carvedilol, or nebivolol (Table 1) and congeners, provide novel properties allowing heart failure treatment with 𝛽-adrenergic antagonists, a condition contraindicated for propranolol. All 𝛽-adrenoceptor antagonists developed are optical isomers but are clinically used as racemic mixtures. Note the more than 10-fold higher affinity of nebivolol for 𝛽1- over 𝛽2-adrenoceptors, the inverse of propranolol (Table 1).
5.1. (±) Propranolol
This drug replaced the catechol moiety of isoproterenol with a naphthoxy ring while keeping alkyl side chain conserving the 𝛽-hydroxyl chiral centre. (-) Propranolol, as with adrenaline, is more potent and active than its (+) enantiomer, although both isomers bind to plasma proteins and elicit local anaesthetic activity in about equal proportions. Its affinity for the 𝛽2-adrenoceptor is 10-fold larger than for the 𝛽1-adrenoceptor (Table 1).
5.2. Labetalol, combines 𝛼- and 𝛽- antagonist properties
This drug has two chiral centres, two of its diastereomers have different pharmacological properties. Labetalol reduces systemic blood pressure via a compounded heart and vascular component due to α- and 𝛽-adrenergic blockade. Its affinity for human β1-adrenoceptor is 31 and 9.7 nM for 𝛽1- and 𝛽2-adrenoceptors, respectively [30]; see Table 1. The pharmacological properties of each diastereomers are:
(1S, 2R)-labetalol: is a potent 𝛼1-antagonist.
(1R, 2R)-labetalol: also known as dilevalol, is a non-selective 𝛽1- and β2-adrenoceptor antagonist and a weak 𝛼1-antagonist.
(1R, 2S)-labetalol and (1S, 2S)-labetalol: are inactive, with no major toxicity.
5.3. Tamsulosin, an 𝛼1-selective adrenoceptor antagonist prototype
The clinically relevant isomer is (R)-tamsulosin, which has a preferential α1A∕D-adrenoceptor affinity; it is commercialised as the racemic compound. It was introduced for benign prostatic hyperplasia treatment, due to the predominant 𝛼1A-adrenoceptor subtype population in the human prostate. Blockade of this receptor subtype causes augmented urinary flow due to blockade of 𝛼1A-adrenoceptor and reduced urine flow resistance.
6. Application of optical activity to fields other than autonomic pharmacology
6.1. Thalidomide lessons
The role of inactive isomers was not well documented and frequently disregarded as an irrelevant compound. This negligence caused a severe public health problem with (±) thalidomide used as sedative agent and against pregnancy-associated morning sickness. Years later, it was identified that the (R)-enantiomer caused severe teratogenesis due to embryonic limb malformation, known as phocomelia, while (S)-thalidomide was associated with sedation [43]. Nowadays, racemic thalidomide or its structurally related chiral analogue, lenalidomide, is safely and efficaciously used against multiple myeloma cancer patients, an unforeseen medical application.
6.2. Antibiotics
The first worldwide highly successful antibacterial agents were the β-lactam antibiotics. This family of drugs which encompass at least five groups, are produced until now by semi- synthesis based on the 6-aminopenicillanic acid, the 7-aminocephalo- sporanic acid, 3-amino-4-methylmonobactamic acid or the 3-hydroxyethylcarbapenemic acids used as precursors. Since the stereochemistry of these compounds is complex with three or more chiral centres, depending on the antibiotic, the pharmaceutical industry relies on genetically engineered penicillium industrial cultures to produce these acids which are then purified and used as the building blocks for the synthesis of the corresponding commercial derivatives of 𝛽-lactams. Likewise, some macrolides, aminoglycosides or fluoroquinolones are chiral compounds, only one of which has the desired antimicrobial activity, nevertheless commercialised as the corresponding racemic mixtures.
6.3. Psychoactive and other drugs
Newer generation of benzodiazepine-like congeners or antidepressants contain chiral centres which in some cases are commercialised as the corresponding active isomer, rather than racemates. Such is the case for zopiclone, which is commercialised as eszopiclone, the active enantiomer. In the field of gastrointestinal pharmacology, esomeprazole, the active isomer of omeprazole, is commercialised and has gained popularity for gastrointestinal ulcer management. Similarly, many other fields of pharmacology have benefitted from Pasteur’s ideas on spatial chemistry and chirality developing and commercialising the biologically relevant isomer, providing safer and more efficacious medicines.
7. Conclusions
Pasteur’s contribution to the architecture of molecules has endured the test of time and provided multiple applications to medicine through molecular pharmacology developments. The brilliance of Pasteur’s separation of crystals with opposing optical activity propelled the way to understand the 3D spatial geometry of molecule conformation, a fundamental code of chemistry with deep roots in pharmacology (see graphical abstract in Figure 5). Receptor proteins recognise spatial molecular conformations with nanomolar affinities to form stereochemical ligand–receptor complexes which are therapeutically relevant. Understanding this basic chemical code has resulted in safe and efficacious drugs with applications from antibiotics to psychotropic drugs, highlighting the relevance of Pasteur’s findings to pharmaceutical chemistry and modern lifestyle. As a proof of concept, pain control through morphine, an alkaloid with three chiral centres, only one of which is synthetised by poppies, is the clinical golden standard for pain management. Likewise, chiral antihypertensive agents are still the first-choice drug treatments. It is no exaggeration that pain, hypertension as well as many infectious diseases, to mention common pathologies, are currently treated with chiral molecules, honouring Pasteur’s creativity.
Acknowledgements
The authors acknowledge Dra V. Donoso for dedicated editorial assistance and help to gather original reference citations. It is funded in part by grants FONDECYT 117-0842 and AFB 18001, CEDENNA.