

Version abrégée
1 Introduction
L'analyse précise des mécanismes d'adsorption à l'interface solide–liquide doit nécessairement prendre en compte l'hétérogénéité superficielle des minéraux. Dans le cas de solides divisés, l'approche la plus adaptée consiste à étudier les conditions d'adsorption de molécules sondes dans le domaine énergétique relatif au dépôt de la première couche. Actuellement, les approches basées sur l'adsorption des gaz apparaissent comme étant les plus performantes, car elles peuvent donner accès à des spectres continus des énergies d'adsorption. Pour un gaz donné, ces spectres peuvent être alors considérés comme étant spécifiques aux solides. Ainsi, l'utilisation de molécules sondes présentant des propriétés différentes permet de mettre en évidence des propriétés spécifiques des sites superficiels.
Cet article a pour objectif de faire une synthèse des principaux résultats obtenus à partir d'une technique d'adsorption très basse pression, récemment développée au laboratoire « Environnement et minéralurgie ». Une brève description des techniques expérimentales et des méthodes de modélisation associées sera ainsi suivie de l'application à l'étude de la morphologie des solides divisés, l'analyse des sites de forte énergie et des propriétés de mouillabilité. Enfin, l'intérêt de cette approche relativement à l'étude de l'adsorption à l'interface solide–liquide sera illustré par deux exemples.
2 Méthodes expérimentales
Le développement récent de capteurs de haute précision et de techniques à introduction continue des molécules sondes a permis d'augmenter considérablement la résolution dans le domaine relatif à l'adsorption de la première couche de gaz. Ces techniques peuvent être divisées en deux groupes :
- – les méthodes basées sur la désorption de molécules sondes, telles que la chromatographie gazeuse inverse à dilution finie [3,5], qui donne directement accès à l'isotherme de désorption, ou les méthodes de thermodésorption, telles que la thermodésorption programmée conventionnelle ou la thermodésorption isobare [69,71,73], qui, à partir du formalisme développé par l'école de Ward [32,33], donnent accès aux fonctions de distribution des énergies de désorption [75,91] ;
- – les méthodes basées sur l'adsorption de molécules sondes par volumétrie basse pression de quasi-équilibre [38,72], mesurant les effets thermiques par microcalorimétrie d'adsorption basse température [70,72] ou mesurant avec précision les quantités adsorbées dès les très faibles pressions relatives [56,57,87,89].
La volumétrie de quasi-équilibre très basse pression permet d'acquérir, par voie informatique, un très grand nombre de points expérimentaux, si bien qu'il est possible de travailler à partir de la dérivée de l'isotherme par rapport au logarithme de la pression relative [87,89]. De telles courbes font alors apparaı̂tre des pics et des vallées, qui peuvent facilement être modélisées. Ainsi, en considérant que les minéraux présentent différentes faces cristallines, il est possible d'écrire l'isotherme d'adsorption sur surface hétérogène comme une somme d'isothermes indépendantes (équation (1)) sur surfaces hétérogènes (équation (2)) ou homogènes, telles que les isothermes de Langmuir et BET, avec leurs extensions, pour prendre en compte les liaisons latérales. En considérant les dérivées premières et secondes des isothermes théoriques (équations (3)–(5)), les isothermes dérivées sont alors facilement modélisées [87] à partir d'un nombre restreint d'isothermes locales (méthode DIS) (Fig. 1). D'autres équations ont aussi été proposées pour prendre en compte l'hétérogénéité propre de chaque face [7,12,88]. Enfin, la validité de la modélisation peut être vérifiée en comparant les résultats obtenus de manière indépendante par microcalorimétrie d'adsorption basse température avec les courbes calculées à partir des paramètres de modélisation [7,28,87,88].
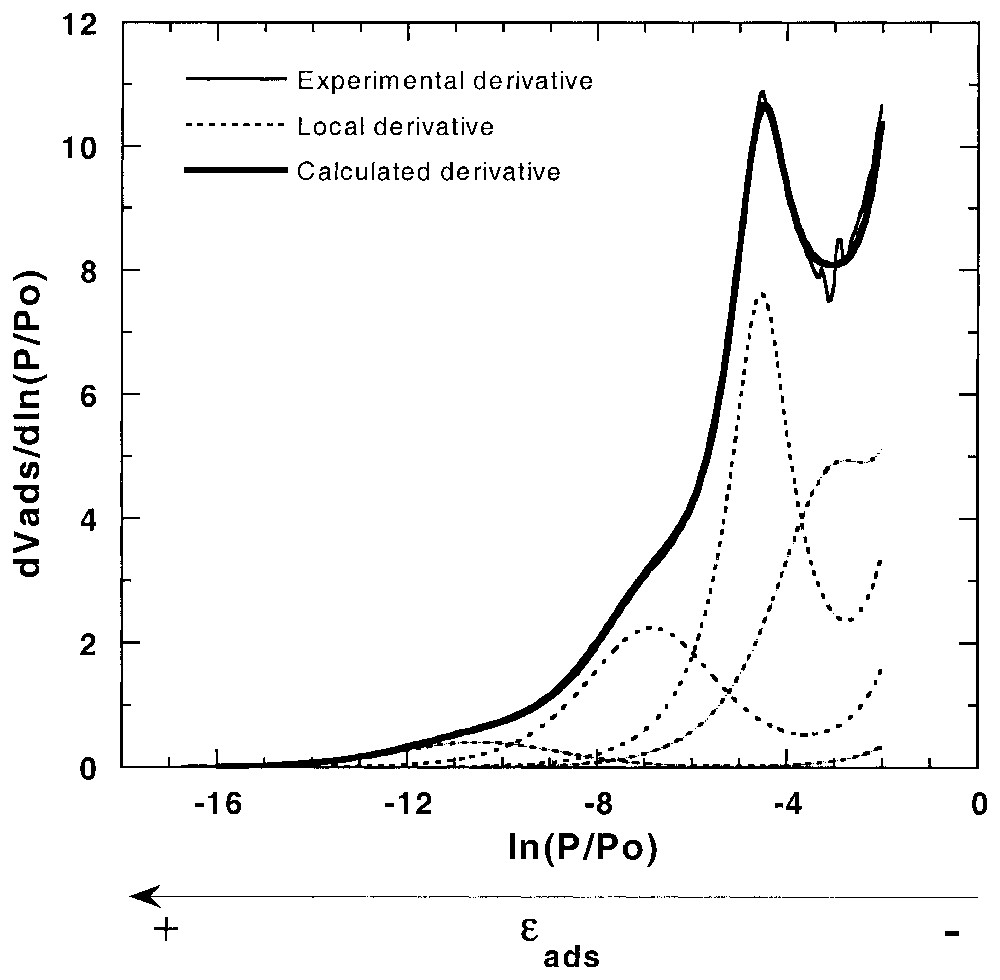
Derivative adsorption isotherm and DIS modelling for argon adsorption on illite clay mineral [7].
Isotherme dérivée et modélisation DIS pour le système argon–illite [7].
3 Propriétés texturales des solides non poreux
La détermination de la forme moyenne des solides cristallins est très importante dans de nombreux domaines. En effet, selon la nature des faces exprimées, les mécanismes et les cinétiques de sorption [9,13,14,21,23,36,40,42,44,45,47,48,50,54,66,79,84,85,95,100,101], de dissolution/précipitation [25,41,63,67,81,83,102], de croissance cristalline [62,63,96] ne seront pas les mêmes. De même, les propriétés d'usage de minéraux pulvérulents peuvent dépendre de la forme des particules et donc des conditions de broyage [10,56,97,98]. C'est typiquement le cas pour l'adhésion des charges minérales dans les polymères et des propriétés mécaniques de ces mêmes polymères chargés [56,99]. Ainsi, la quantification et l'analyse précise des mécanismes aux interfaces doivent prendre en compte les propriétés morphologiques de tels solides.
L'utilisation des isothermes d'adsorption d'argon pour déterminer la lamellarité des kaolinites a été largement discutée [16,19,87–89]. De manière générale, les isothermes dérivées obtenues sur les phyllosilicates (Fig. 1) présentent la même forme, avec un pic de faible énergie, correspondant à l'adsorption sur les faces basales, et un épaulement à moyenne pression, correspondant aux faces latérales [87,89]. Ainsi, l'approche quantitative a pu être étendue à d'autres minéraux, tels que le talc [58], l'illite et les micas [7,8,89], les saponites [61] et les silices lamellaires [34,35]. Pour les phyllosilicates chargés négativement, le cation compensateur doit être monovalent (typiquement Na+) [7,8]. Enfin, la distribution des faces cristallines de minéraux non lamellaires tels que la calcite [21,22,51,92], la gœthite [68] et TiO2 [68] semble accessible par cette méthode.
4 Analyse de sites de forte énergie
Les sites de forte énergie présents à la surface des minéraux peuvent avoir une influence non négligeable sur l'adsorption d'éléments en trace, être le siège de réactions catalytiques... Ainsi, leur quantification et leur localisation peuvent s'avérer importantes dans la compréhension des mécanismes correspondants. Dans certains cas, tels que celui du talc (Fig. 2) [58], des saponites de basse charge [61] et de certaines silices lamellaires [34,35], des sites de forte énergie sont observés avec l'argon. Ces sites, aussi accessibles à l'azote, ont une origine structurale à savoir le centre de la cavité hexagonale formée par l'arrangement des tétraèdres de silicium. Pour les autres phyllosilicates, ces sites sont accessibles à l'azote, mais pas à l'argon.
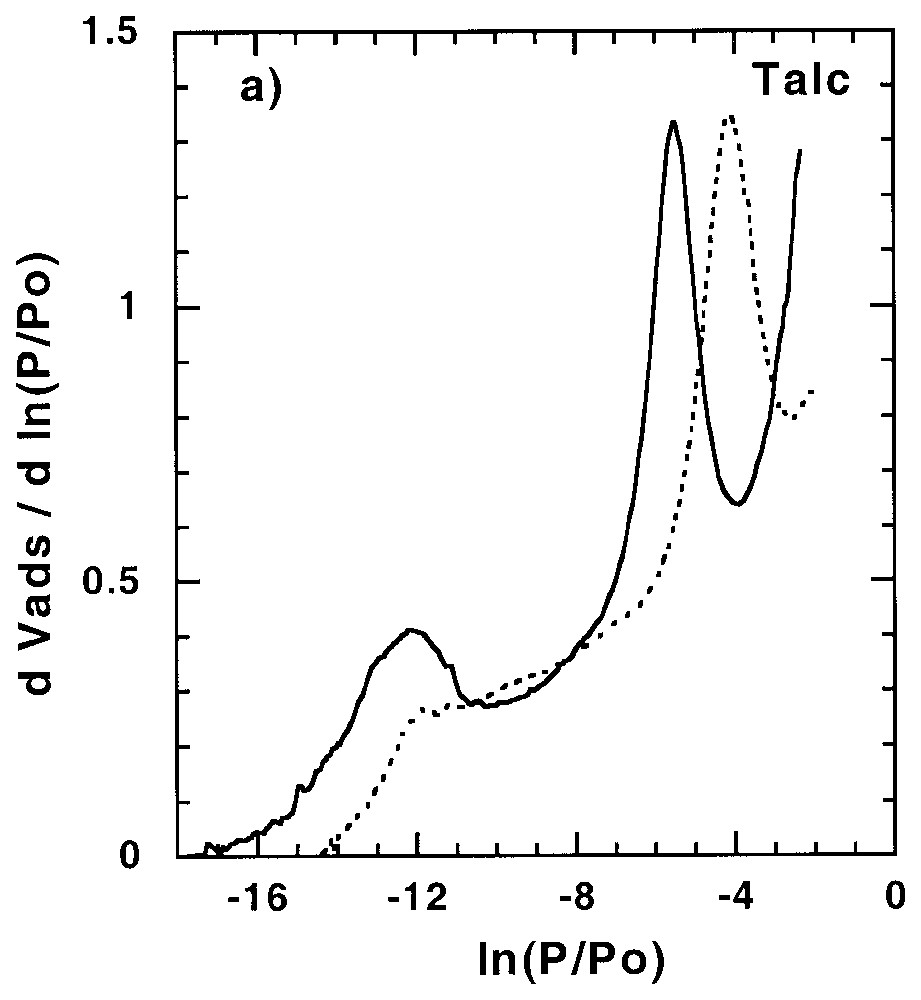
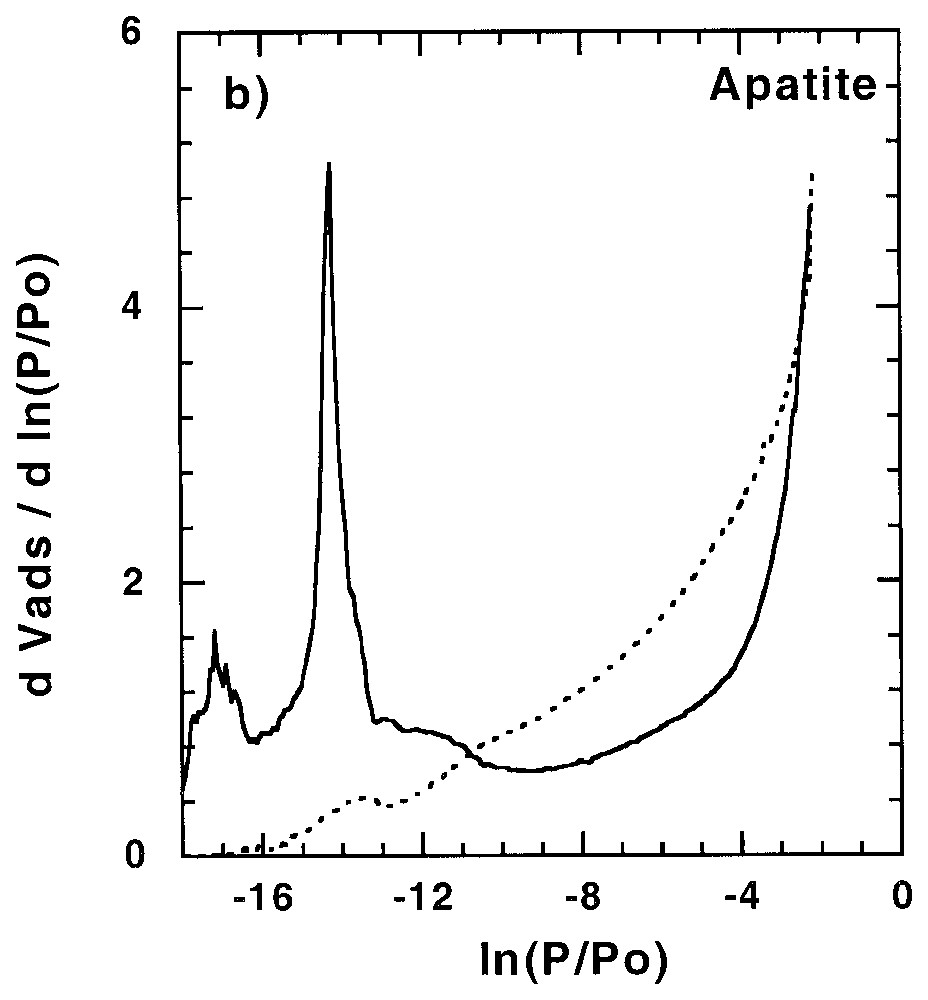
Argon and nitrogen derivative adsorption isotherms (a) on talc outgassed at 250 °C [58] and (b) apatite outgassed at 270 °C [11,91]. Dotted line: argon; plain line: nitrogen.
Isothermes dérivées d'adsorption d'argon et d'azote sur (a) un talc dégazé à 250 °C [58] et (b) une apatite dégazée à 270 °C [11,91]. Trait pointillé : argon ; trait plein : azote.
De plus, d'autres sites de forte énergie sont souvent mis en évidence avec l'azote et résultent d'interactions spécifiques entre l'azote (molécule polarisable) et des sites polaires superficiels. Pour les phyllosilicates chargés, leur distribution est fonction de la nature du cation compensateur [7,8] et de la charge du feuillet [61]. De tels sites sont aussi observés pour d'autres minéraux, avec des effets parfois très spectaculaires, comme l'illustre la Fig. 2 pour l'apatite [11,92]. Pour ce minéral, il a été démontré que ces sites correspondent aux POH superficiels, avec une distribution 1/3 et 2/3, correspondant à la distribution des plans de clivage de la face (010). Des effets de polarisation ont aussi été observés pour la calcite [21,22,51], la gœthite [68], l'anatase [68] et, dans une moindre mesure, les silices amorphes [21,68,76,90,93].
5 Hétérogénéité superficielle et mouillabilité
Les résultats présentés ci-dessus montrent que la comparaison entre azote et argon donne accès à des informations sur le caractère polaire–non polaire de la surface. Ces informations sont particulièrement importantes lorsqu'il s'agit d'évaluer la balance hydrophile/lipophile des solides, en relation avec leurs propriétés de mouillabilité, de capillarité, d'adhésion et d'agrégation. Ces aspects concernent les sols, la répartition et la rétention des fluides dans les roches réservoir de pétrole, l'altération des roches et des pierres de taille, l'adsorption des tensioactifs et autres molécules organiques [4–6,21,22,24,29–31,49,51,52,55,77,78,86]. Les propriétés hydrophiles/lipophiles peuvent être facilement analysées dans le cas de surfaces planes et de grandes extensions, par la mesure des angles de contact. Leur détermination pose cependant des problèmes lorsqu'il s'agit de solides pulvérulents et hétérogènes. Il a récemment été démontré [93] que des informations pertinentes peuvent être obtenues en comparant les isothermes dérivées d'adsorption d'argon et d'azote en fonction de lnP (Fig. 3) et non en fonction de lnP/P0. Ainsi, lorsque la surface est non (ou très peu) polaire, comme c'est le cas du talc ou d'une calcite hydrofugée, les distributions des énergies d'adsorption d'azote et d'argon sont superposées, ce qui n'est pas le cas lorsque des sites polaires sont présents sur la surface. Ce résultat est lié au fait que les paramètres Lennard–Jones de l'azote et de l'argon sont très proches, si bien qu'en l'absence de sites polaires, les deux molécules présentent le même comportement [65].


First layer Ar and N2 derivative adsorption isotherms as a function of lnP instead of lnP/P0 (a) on talc and (b) on bare (open circles) and modified calcite (black circles) [93]. Dotted line: argon; plain line: nitrogen.
Isothermes dérivées de la première couche d'adsorption d'argon et d'azote, tracées en fonction de lnP au lieu de lnP/P0 pour (a) un talc et (b) une calcite nue (ronds blancs) et modifiée (ronds noirs) [93]. Trait pointillé : argon ; trait plein : azote.
6 Application à l'étude de l'adsorption à l'interface solide–liquide
Du fait de la spécificité de chaque couple adsorbat–adsorbant, la comparaison entre adsorption aux interfaces solide–gaz et solide–liquide et n'est pas évidente. Des relations directes ont cependant pu être obtenues pour certains cas favorables, tels que ceux de l'adsorption des chlorures d'alkylamine sur les kaolinites [16], du phénol dans les charbons actifs [19,28,59] ou du chlorure de décylamine sur l'apatite [11,92]. Pour ce dernier cas, l'adsorption du tensioactif suit la même relation 1/3, 2/3 que celle observée par adsorption d'azote (Fig. 4).
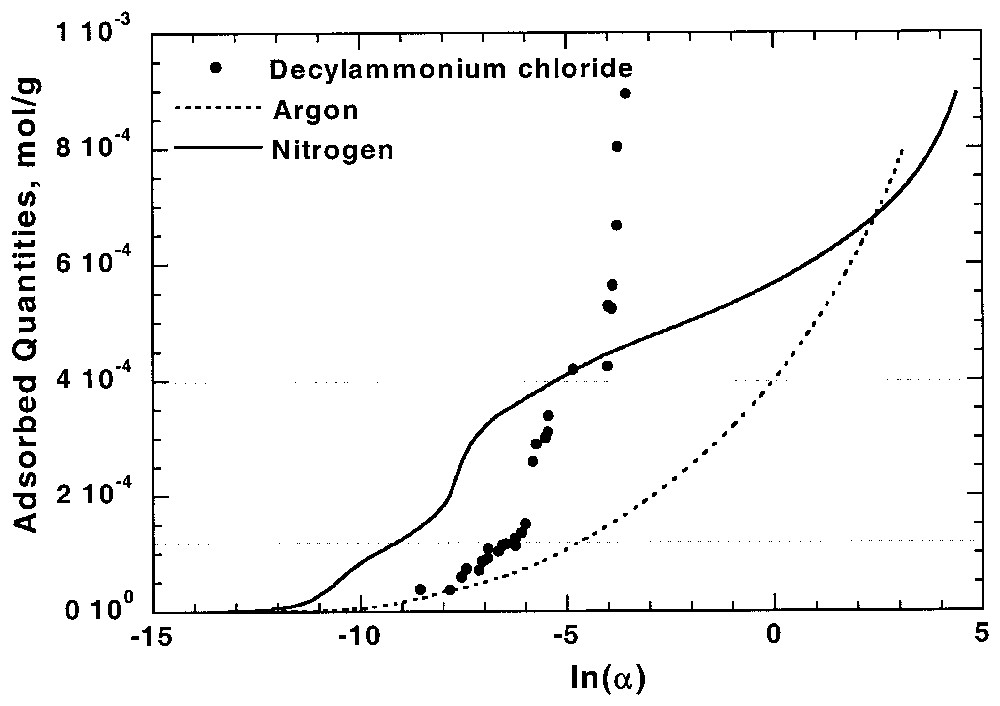
Comparison between Ar and N2 adsorption isotherms at the apatite–gas interface and decylammonium chloride adsorption isotherm at the apatite–aqueous solution interface on apatite [92]. α is the pressure for Ar and N2 and equilibrium concentration for decylammonium chloride.
Comparaison entre isothermes d'adsorption d'azote et d'argon à l'interface apatite–gaz et isotherme d'adsorption de chlorure de décylammonium à l'interface apatite–solution aqueuse [92]. α correspond à la pression pour Ar et N2 et à la concentration pour le chlorure de décylammonium.
L'étude de l'hétérogénéité à l'interface solide–gaz peut aussi s'avérer utile pour suivre les modifications superficielles des solides modifiés à l'interface solide–liquide. Ainsi, la Fig. 5 illustre l'évolution des isothermes dérivées, obtenues sur une calcite hydrofugée [21,22]. La modélisation des isothermes dérivées [21] à partir de la calcite initiale et de la calcite totalement recouverte permet (Fig. 6) de suivre l'évolution du recouvrement sur les différentes faces (argon) et les sites de forte énergie (azote) et de définir avec précision la quantité d'hydrofugeant adsorbée au niveau de la monocouche.
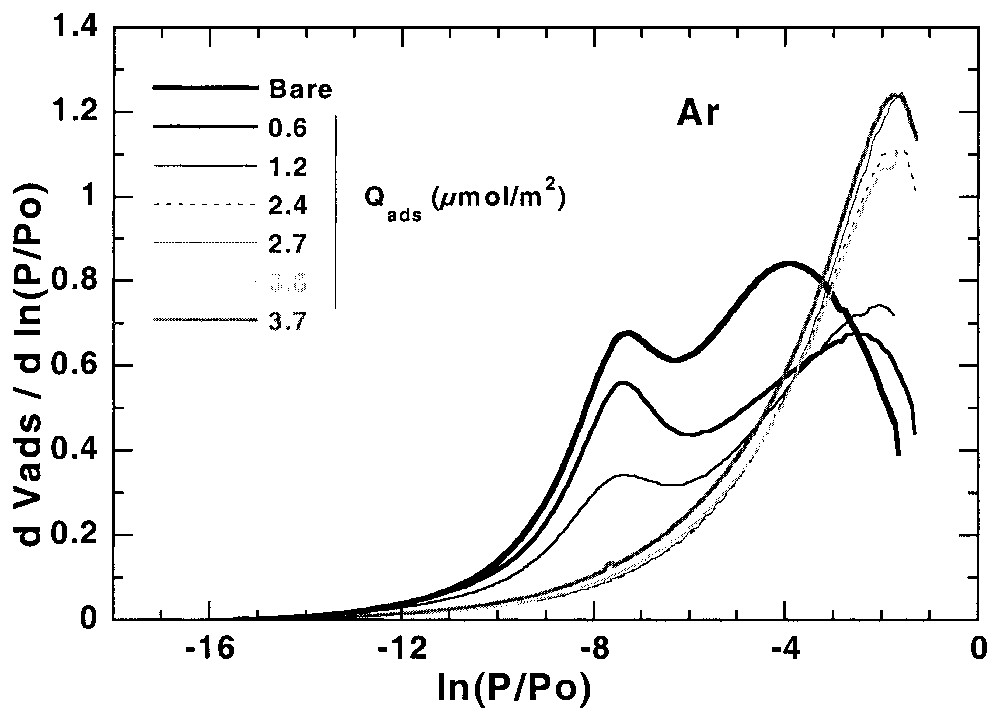
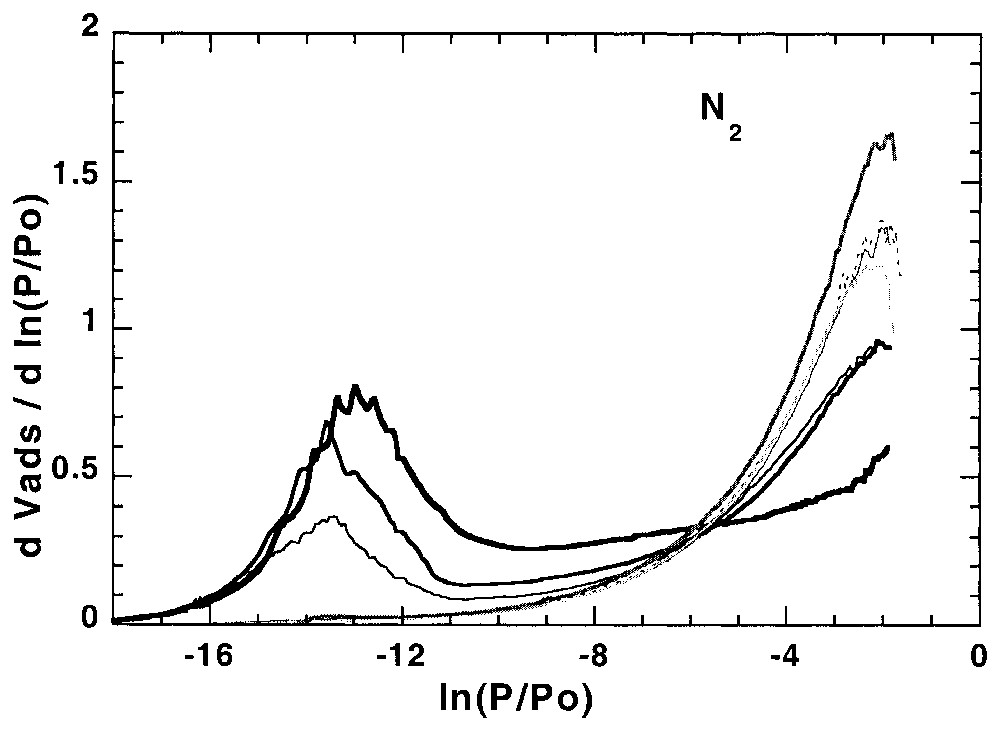
First layer Ar and N2 derivative adsorption isotherms on calcite modified with different amounts of WRM molecules [21].
Isothermes dérivées de la première couche d'adsorption d'argon et d'azote, obtenues sur des calcites recouvertes de différentes quantités de molécules hydrofugeantes [21].

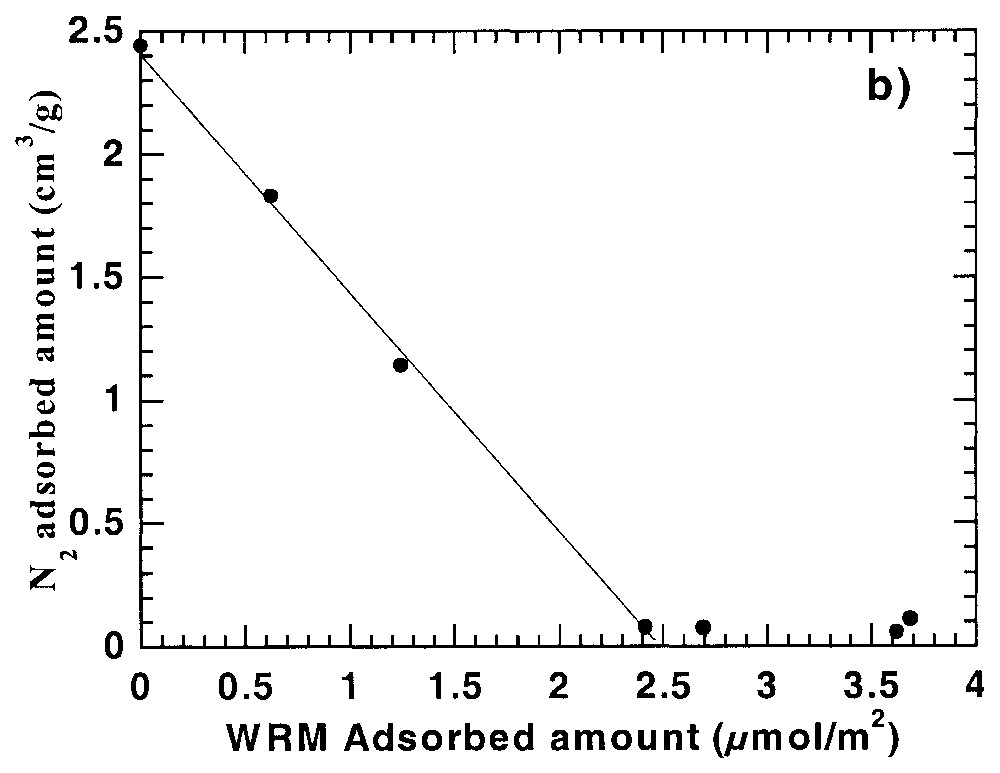
DIS quantitative analysis of derivative adsorption isotherms of Fig. 5 for (a) argon and (b) high-energy adsorption sites for nitrogen [21].
Analyse quantitative selon la méthode DIS des isothermes dérivées de la Fig. 5 pour (a) l'argon et (b) les sites de forte énergie pour l'azote [21].
7 Conclusions
L'acquisition d'isothermes précises depuis les très basses pressions, couplée à la modélisation des isothermes dérivées, donne ainsi accès à de nombreuses informations sur les propriétés énergétiques superficielles. Cette approche est actuellement en cours de développement pour d'autres sondes que l'azote et l'argon, à l'interface solide–gaz comme à l'interface solide–liquide [68,82].
L'approche développée ici trouve ainsi de nombreuses applications lorsqu'il s'agit de comprendre et de modéliser les phénomènes naturels aux interfaces, tels que l'oxydation catalytique des gaz à effets de serre dans la haute atmosphère par les aérosols et les cendres volcaniques, la dissolution/précipitation dans les sols et les sédiments, les échanges aux interfaces solides–liquides, la bioadhésion. L'analyse fine des propriétés superficielles est aussi très importante pour modéliser et prédire le comportement des minéraux et autres solides divisés, poreux ou non, lors d'opérations de broyage et de séparation (minéralurgie, remédiation de sols pollués) ou à fin d'utilisation dans les barrières géochimiques, dans les matériaux composites, en catalyse hétérogène, en traitement des eaux...
1 Introduction
The fact that the actually existing surfaces are energetically heterogeneous is now generally recognised by the scientists investigating adsorption at the solid–gas and solid–solution interfaces. This surface heterogeneity is a fundamental feature of real solid surfaces. It can be due to different crystal faces, local crystalline disorder, or the presence of impurities. It may also have its source in the presence of pores of various sizes and shapes. A large variety of experimental methods has been developed for studying more or less directly the structure of solid surfaces. In the case of clay minerals, optic and electronic microscopies provide proofs of their macroscopic heterogeneity. However, all these methods yield only auxiliary information.
In this respect, essential information about surface heterogeneity can be obtained by applying methods commonly referred to as ‘molecular probe methods’. A molecular probe method is simply a theoretical/numerical analysis of a certain adsorption quantity (characteristic) measured in the course of adsorption of a chosen adsorbate. The surface energetic heterogeneity is usually considered as a dispersion of the number of the local adsorbate–solid potential minima, called ‘adsorption sites’ among the value of the adsorbate–solid potential at these minima, taken with a reverse sign, and called ‘adsorption energy’. The majority of the methods designed to study surface heterogeneity is based on the assumption that the spectrum of adsorption energies in a given probe/solid system is sufficiently dense to be represented by a continuous function [3,43,46,53,74,94]. This function is the differential distribution of the number of adsorption sites among the corresponding values of adsorption energy.
One essential question concerns the nature of surface heterogeneity with respect to a given adsorbate molecule. Indeed, surface heterogeneity cannot be considered as an absolute feature as it is related to a given adsorption system, i.e., a given adsorbent–adsorbate system. Therefore, the nature of surface heterogeneity must always be related to the type of adsorbed molecules, i.e., to the subsequent normal adsorbate–adsorbent interactions. Different probes can then advantageously be used to reveal the specific properties of adsorption sites.
This review will show the usefulness of surface heterogeneity assessment for the study of adsorption phenomena at the solid–liquid interface. It is mainly based on adsorption data obtained at solid–gas interface by the use a of low-pressure quasi-equilibrium technique coupled to a specifically developed data analysis procedure. Results obtained on the geometrical properties of platy minerals, the determination of high-energy adsorption sites and wetting properties will then be discussed and followed by examples of relationships between solid–gas and solid–liquid adsorption on heterogeneous surfaces.
2 Experimental methods for analysing the surface heterogeneity of solid surfaces
Two main groups of techniques based on solid–gas interactions can be used to obtain precise information on energetic heterogeneity of solid surfaces:
- – methods based on desorption phenomena such as inverse gas chromatography [3,5] and thermal analysis; for this latter, desorption energy distributions can be obtained, for instance by applying the Statistical Rate Theory of Interfacial Transport developed by Ward [32,33,75]; in addition, the controlled rate thermal analysis designed by Rouquerol and co-workers [69,71,73] can be used to derive quasi-equilibrium desorption data [17,19,28,39,58,59,91];
- – methods based on quasi-equilibrium adsorption technique [38,56,57,72] by using low-temperature adsorption microcalorimetry (LTAM) [16,17,19,39,70,72] and quasi-equilibrium adsorption volumetry [17,38,89].
LTAM has rapidly proven is usefulness for the analysis of textural heterogeneity of clay minerals, to determine basal and lateral surface areas [16,19] or micropore distribution of sepiolite [39] and palygorskite [17]. The recent improvement of quasi-equilibrium adsorption volumetry, based on the use of accurate low-pressure sensors and numerical acquisition of experimental data points has been the starting point of a new and powerful data analysis approach that will be developed in the next sections.
2.1 Low pressure quasi-equilibrium adsorption volumetry
The experimental procedure has been discussed in many papers [38,56,57,72,89]. A slow, constant and continuous flow of the adsorbate is introduced into the adsorption system through a microleak. Then, from the recording of the quasi-equilibrium pressure (in the range of 10−3, 3×10−4 Pa) as a function of time, the adsorption isotherm is derived. 2000 to 3000 experimental data points are collected for relative pressures lower than 0.15. Due to the large quantity of experimental data points, the derivative of the adsorbed quantity as a function of the logarithm of relative pressure can be calculated accurately (Fig. 1) [87,89]. The lnP/P0 and lnP correspond to adsorption free energy scales graduated in unit. The derivative of adsorption isotherms is much more sensitive to surface heterogeneity as it features different peaks that can be simulated using theoretical derivative adsorption isotherms as described in the next section.
2.2 The Derivative Isotherm Simulation method
The surface of minerals can be divided into n patch-like domains corresponding to crystallographic faces (for instance, basal and lateral faces in the case of platy clay minerals), each domain being characterised by its own energy distribution. So the total adsorption isotherm θt can be expressed as follows:
| (1) |
| (2) |
In the case of the Langmuir–Temkin equation, the three parameters can easily be derived by considering the first and second derivatives [87]: (3)
| (4) |
| (5) |
A typical example of DIS modelling is given in Fig. 1 for illite [7,8]. Improvements were also proposed in order to use derivative of adsorption isotherm on heterogeneous surface for each adsorption domain (χi(ε)≠1) [12,88]. It was also demonstrated that the higher the ω parameter, the higher the surface homogeneity [7,89]. Finally, the fitting validity can be checked by comparing experimental and independent curves obtained from low temperature adsorption microcalorimetry with curves calculated from fitting parameters [7,28,87,88].
The concept of derivative of adsorption can then be used to get quick and reliable results on energetic heterogeneity, which can also be used to assess geometric properties of phyllosilicates and other finely divided solids.
3 Geometric heterogeneity of non-porous minerals
The mechanisms of sorption, of dissolution/precipitation, of crystal growth [62] depend on the nature of reacting crystal faces. For instance, many works show the role of lateral faces on surface modification of kaolinite [14,40,42,44,45,63,95] and the importance of the determination of the textural and surface properties [13,16,19,27,36,79,81,84,85,87,101]. Sorption phenomena on charged phyllosilicates like smectites [23,47,48], muscovite [63,100] and illites [45,50,66] depend on the distribution between permanent and variable charges bared by basal and lateral faces, respectively. For clay minerals, the distribution between basal and lateral surfaces is also evoked to discuss its influence on dissolution/precipitation mechanisms [41,54,63,81,102]. Many other examples can be found on the influence of surface distribution on surface properties: olivine [67], goethite [9,25,96], calcium carbonate [21,83]... For materials containing powdered minerals, use properties depend also on particle shape, then on grinding conditions and results [10,56,97,98]. This is particularly the case when mechanical properties are governed by adhesion capacity between polymer and mineral surfaces and/or by the aspect ratio of flat particles [56,59]. A determination of the mean sizes of crystalline solids is then very important to derive precise surface phenomena. The use of argon adsorption to derive geometrical properties of kaolinite has been extensively discussed elsewhere in this issue [19] and related publications [16,27,87,89]. This concept has also been applied to other clay minerals such as talc [58], illite [7,8], saponite [61], micas [89] and lamellar silica [34,35]. In the case of phyllosilicates, argon derivative isotherms generally exhibit the same features (Fig. 1) with a low-energy peak, corresponding to the adsorption on basal faces and a shoulder at higher energy, corresponding to the adsorption on lateral faces. The main differences come from the position of the low-energy peak. Indeed, the peak position depends on the nature of surface cations [7,8] but is only slightly influenced by their surface density [61]. For lamellar silica such as magadiite, kenyaite and kanemite [34,35], the peak corresponding to the adsorption on basal faces is observed at much lower energy, but its position is not sensitive to the nature of surface cation. The shape of the peaks obtained for magadiite and kenyaite indicates also that argon forms a very well organised film revealing commensurability between the surface network and the argon atoms.
The reliability of the quantitative results can be checked against electronic microscopy [34,35], surface chemical properties (as shown for kaolinite with cationic exchanged capacity [19,89]) or structural properties derived from unit cell parameters [34,35,58,61,89]. The determination of basal and lateral surface areas was rapidly obtained for uncharged clay minerals, such as kaolinite and talc. In the case of charged phyllosilicates, the influence of surface cations was checked by changing the surface cations on illites. It was then observed that reliable aspect ratio could be obtained only with monovalent cations [7,8]. Furthermore accurate basal and lateral surface areas are measured only with small cations, i.e., lithium and sodium.
In conclusion, at the moment, the DIS method seems to yield the most accurate determination of the flakiness ratio of platy materials. Encouraging results have also been obtained for goethite [68], calcite [21,22,51,92] and TiO2 [68], for which precise assignment of adsorption energies to orientation of crystallographic faces is not achieved. Finally, this approach cannot be applied when nitrogen is used as a probe due to the polarisability (quadrupolar momentum) of nitrogen molecules. Experimental evidences of polar interactions between nitrogen and surface protons of hydroxyls were given by infrared experiments of Frohmsdorff et al. [37]. However, the polarisability of nitrogen can be advantageously used as it can give information about electropositive groups of the surface as discussed in the next section.
4 High-energy adsorption sites
High-energy adsorption sites on mineral surfaces play a very important role as they can share strong interactions at very low gas pressure or chemical concentrations in liquid media. For instance, it is well admitted that, in case of high-energy interactions between solid surfaces and surfactants, the surface properties of the solid phase can be modified far bellow three-dimensional condensation concentration [15,18,20]. High-energy adsorption sites can also be involved in catalytic reactions.
At solid–gas interface, two types of high-energy adsorption sites can be observed: sites evidenced using different probes, such as argon and nitrogen and sites revealed by nitrogen only. The origin of the first ones is mostly structural. They are observed for instance in phyllosilicates and layer silica, such as magadiite and kenyaite. The second ones mainly arise from specific interactions between surface sites and nitrogen.
4.1 Structural high-energy adsorption sites: the special case of layered silicates
Such sites were evidenced for the first time in the case of talc [58]. They are observed for argon and nitrogen (Fig. 2a). The octahedral layer of talc is trioctahedral and almost purely magnesian. Consequently, the octahedral hydroxyls point directly toward the surface inside the hexagonal cavity formed by the arrangement of the silica tetrahedra of the tetrahedral layer. These OH groups could then exhibit a particular reactivity toward molecular probes, as evidenced by computer simulation of water adsorption [80]. On the basis of crystallographic parameters, it appears that each hexagon of the tetrahedral layer has an area of 25 Å2. The theoretical quantity of nitrogen adsorbed on OH groups can then be calculated by taking into account the basal surface area and an adsorption ratio of one nitrogen molecule per hydroxyl group. Calculated values, derived from argon and nitrogen adsorption experiments, appear in very good agreement with structural data [58]. Very similar results were obtained for argon adsorption on low-charge saponites [61], by considering talc-like hexagons only, and for magadiites and kenyaite [34,35]. However, for these minerals, additional adsorption sites are observed when nitrogen is used as a probe.
It can be noticed that, for most of phyllosilicates, these structural adsorption sites are always present and detected by nitrogen [7,8,61,89] while their filling by argon is only observed for trioctahedral clays without substitution. Such differences in behaviour probably come from size and/or symmetry decrease of the ditrigonal hole because of two possible reasons: (i) Al for Si tetrahedral substitutions or (ii) presence of a dioctahedral instead of trioctahedral sheet. The size influence of the hole is also illustrated by the fact that in case of talc, the adsorption of the first monolayer does not depend on the nature of the molecule adsorbed in the structural sites [58]. This means that the adsorbate is inside the structure and cannot interact with the monolayer. This does not appear to be the case for the other minerals as nitrogen adsorbed in structural sites can share attractive lateral interactions with the first monolayer, shifting adsorption on basal faces towards higher energy and changing its shape when compared to argon.
Additional high-energy adsorption sites have also been observed for argon when the surface charge of layer silicates is compensated with divalent cations [7,8,34,35]. This is interpreted as resulting from a local charging of the surface (atomic scale) because of the neutralisation of two negative charges by one cation. As a consequence, geometrical properties cannot be derived from argon adsorption isotherms. The same effect is also observed with magadiite exchanged with proton but the reason is not fully understood yet [34,35].
4.2 Specific adsorption sites
Specific adsorption of nitrogen can be observed for many minerals such as apatite [11,92], calcite [21,22,51,92], goethite [68], rutile [68] and amorphous silica [21,68,76,90]. Surface polar groups can have very spectacular effects on nitrogen derivative adsorption isotherms, compared to argon ones. This is illustrated in Fig. 2b for hydroxyapatite. In this case, polar sites are distributed in two families corresponding to 1/3 and 2/3 of the total high-energy adsorption sites, as derived from DIS modelling. The total amount of polar sites compares very well with the amount of surface OH groups. Furthermore, the existence of two sites was assigned to two different monohydrogenophosphate surface groups, which can be observed on the three equiprobable atomic planes of the (010) faces of hydroxyapatite crystals [11,92].
For calcite (see Fig. 3) and goethite, specific interactions with nitrogen probably occur with surface calcium and surface FeOH groups, respectively. However, due to non-equivalent crystallographic faces and high-energy adsorption sites distributions, precise correspondence is not known at present. In the case of silica, the origin of polar sites should be confirmed, as their assignment to surface SiOH groups remains uncertain.
5 Solid–gas interactions and wetting properties at solid–liquid interface
The results presented in the previous section show that comparison between nitrogen and argon adsorption isotherms gives access to information on polar/non-polar properties of solid surfaces. Such information is particularly worthwhile to evaluate hydrophile/lipophile properties of solids as these properties govern wetting, capillary invasion, adhesion, and aggregation phenomena. The hydrophile/lipophile balance of solid surfaces concerns then many fields in earth science: water interactions in soils, oil-brine distribution in porous rocks, surface alteration and protection of historic buildings, surfactant and other organic pollutant adsorption and transport in soils, sediments and rivers [4–6,21,22,24,29–31,49,51,52,55,77,78,86]. In the case of large surfaces, the hydrophile/lipophile balance is easily determined by using the techniques based on contact angle measurements. However, such techniques cannot be applied to determine wetting properties of heterogeneous powders.
In principle, the distributions of adsorption energies allow to calculate the surface free energy of the solid surfaces. In reality, such assignment was not achieved yet, due to difficulty in determining precisely the exact correspondence between adsorption energy and crystallographic faces, and in defining the exact organisation of the adsorbed film. Indeed, this organisation depends on distances between adsorbing centres acting on the density of the adsorbed phase and, as a consequence, on the intensity of lateral interactions. On real surfaces, atomic organisation, and then distances between adsorption centres, can differ considerably from theoretical atomic organisation on cleavage planes. Thus, adsorption energies deduced from molecular dynamic and Monte Carlo simulation are generally far from reality [1,2].
The previous section has shown that very different adsorption energy distributions can be obtained when apolar (argon) and polar (nitrogen) probes are used. Therefore, the comparison between argon and nitrogen adsorption energy distributions should bring valuable information on apolar/polar properties of powders. For this purpose, the adsorption energy distributions of the first monolayer were plotted as a function of lnP instead of the generally used lnP/P0, as illustrated in Fig. 3 for talc, calcite and hydrophobised calcite [93]. Calcite presents polar surfaces, whereas talc and hydrophobised calcite have rather apolar surfaces. From the obtained curves, it can be concluded that:
- – on apolar surfaces, argon and nitrogen adsorb through low energy interactions with surface adsorption sites, as derived from Lennard–Jones interaction potentials; as these potentials are similar for both probes [65], adsorption energy distributions plotted as a function of lnP superimpose; furthermore, a clear relationship between the surface apolar free energy and argon adsorption energy has been observed by Bardot et al. [8] from the comparison between argon adsorption on illites and contact angle measurements on laponites [64] exchanged with the same cations;
- – when polar adsorption sites are present on the surface, argon adsorbs through apolar interactions, as derived from Lennard–Jones interaction potentials, whereas additional and specific interactions exist between polar sites and nitrogen; on a given surface, nitrogen molecules then first adsorb on polar surface sites at higher energy than on apolar ones; these first adsorbed molecules will then participate to the adsorption on apolar sites through cooperative effects generated by attractive nitrogen–nitrogen interactions (; adsorption on apolar sites is thus also shifted towards higher energy, totally modifying the shape of N2 adsorption peaks compared to argon ones.
The comparison between argon and nitrogen adsorption energy distributions derived from low-pressure adsorption experiments then appears as a reliable diagnosis to assess the polar–apolar nature of a mineral surface. In addition, the extent of the difference in position between argon and nitrogen adsorption peaks is an indicator of the magnitude of surface polarity.
6 Solid–gas interactions and adsorption at solid–liquid interface
Although adsorption behaviours depend on adsorbate–adsorbent couples, the information derived from low-pressure gas adsorption could be used to predict or to interpret adsorption phenomena at solid–liquid interface. The case of non-polar molecules adsorption in microporous solids has already been extensively discussed [19,28,59], showing that correlation can be obtained between argon and phenol adsorption on activated carbon. For polar surfaces, the presence of high-energy adsorption sites for nitrogen suggests that, for a correctly chosen probe, these sites could be involved at the solid–liquid interface. In the case of long chain ionic surfactants adsorption on polar surfaces with opposite charge, the first layer adsorption isotherm yields directly the adsorption energy distribution function as strong lateral interactions allow applying the 0–1 approximation of the two-dimensional condensation [15,18,20]. For kaolinites, it was then demonstrated that the basal to lateral surface areas ratio could be calculated from argon low-temperature adsorption microcalorimetry at solid–gas interface and from alkyldodecylammonium adsorption at solid–liquid interface [16]. Comparison between adsorption at solid–gas and solid–liquid interfaces is also particularly well illustrated in the system apatite–decylammonium chloride (Fig. 4) [11,92]. Indeed, the corresponding isotherm obtained from water shows two steps during the first layer adsorption. These two steps indicate the presence of two different adsorption sites with 1/3 and 2/3 contributions, as observed from nitrogen adsorption. A similar, but less obvious, conclusion was obtained from the comparison between adsorption behaviours of nitrogen and benzoic acid from toluene on calcite surface [92].
In a recent work of Chamerois [21,22] the low-pressure gas adsorption technique was used to characterise carbonate surfaces modified by a water repellent molecule (WRM) along its adsorption isotherm from cyclohexane. The main difficulty was that all the classical techniques such as adsorption isotherms, infrared spectroscopy study of the adsorbed phase and wetting properties of the modified solids yielded inaccurate estimates of the WRM monolayer [21,22]. Upon adsorption of the WRM molecule, the derivative adsorption isotherms obtained for the modified surfaces gradually change as the signature of calcite surface sites disappears and is replaced by a low energy adsorption distribution corresponding to adsorption on WRM surface (Fig. 5). For adsorbed WRM amounts higher than 2.4 μmol m−2, the shapes of Ar and N2 derivative adsorption isotherms remain unchanged. Using the DIS modelling technique, the intermediary derivatives were then modelled using the peaks found for bare calcite and calcite covered with 2.4 μmol m−2. The results obtained [21] with argon (Fig. 6) show that carbonate surface decreases and WRM surface increases linearly with WRM pre-coverage. In the same way, the amount of nitrogen high-energy adsorption sites, corresponding to surface calcium, decreases linearly with WRM pre-coverage. With both probes it is then possible to determine precisely the WRM monolayer at 2.5 μmol m−2. Furthermore, it was demonstrated that the two main Ar domains, corresponding probably to two different crystallographic faces, are not covered concomitantly by WRM: WRM adsorbs first partially on low energy faces up to 1/3 of the total WRM monolayer; further WRM adsorption occurs on both faces.
As a conclusion, the analysis of modified carbonate samples by using low-pressure gas adsorption coupled to DIS modelling appears as a complementary tool to study complex adsorption mechanisms on a heterogeneous surface at solid–liquid interface.
7 Conclusions
This review shows that the precise determination of low-pressure adsorption isotherms at solid–gas interface can be very powerful to analyse surface properties of solid surfaces. By using argon as a probe, such isotherms allow to go deeper in the geometrical description of particles, as the conventional BET specific surface area can be fractionated according to the distribution of crystallographic faces. The same approach can also be used to analyse the pore size distribution and adsorption phenomena for microporous samples [26,28,59,60,87–90]. In addition, it is possible to extract valuable information on surface energy and surface reactivity. The combination of non-polar and polar probes appears particularly suitable for that purpose. In order to study the effect of surface heterogeneity on adsorption mechanisms of other probes, the present experimental approach has recently been extended to water vapour adsorption gravimetry and proton adsorption potentiometry [68,82] at the solid–gas and solid–liquid interfaces, respectively.
This approach can be used in many fields to understand interfacial phenomena on heterogeneous solid surfaces: chemical exchange, dissolution/precipitation, and crystal growth at solid–liquid interfaces in rivers, soils, sedimentary rocks, oil reservoirs; distribution of fluids in porous materials such as reservoir rocks and soils; gas adsorption and catalytic phenomena on atmospheric aerosols and volcanic ashes. Knowledge on surface heterogeneity of minerals is also useful for many applied problems concerning the design of geochemical barriers for organic and radioactive wastes; the optimisation of grinding and flotation in mineral processing; the design of mineral fillers in material science; the design of adsorbents or catalysts for gas and water treatment...