

1 Introduction
The redox cycling of Fe plays a major role in the biogeochemical cycling of many elements in natural systems [9,51]. On the reductive side of the Fe redox cycle, dissimilatory microbial (enzymatic) reduction of Fe(III) oxides has a major impact on the aqueous/solid-phase geochemistry and behavior of natural and contaminant compounds in nonsulfidogenic subsurface sedimentary environments. As a result of the pivotal role of bacterial Fe(III) oxide reduction in surficial and subsurface sediment biogeochemistry, there is great interest in the factors that control the rate and long-term degree of enzymatic Fe(III) oxide reduction. The rate and extent of Fe(III) oxide reduction are governed by complex surface-chemical and physiological interactions which are as yet only poorly characterized [44]. Development of a mechanistic understanding the geochemical and microbiological controls on microbial reduction of soluble and solid-phase metals is a prerequisite for development of reactive transport models of inorganic contaminants and radionuclides in saturated subsurface environments [8]. The studies summarized here explored how the surface chemical (e.g., specific surface area) and thermodynamic properties (e.g., oxide reduction potential) of different Fe(III) oxides influence the rate and extent of bacterial (versus chemical) Fe(III) oxide reduction. The results permit development of conceptual and quantitative models of enzymatic Fe(III) oxide reduction at circumneutral pH that can be compared and contrasted with models of abiotic mineral dissolution and enzymatic soluble metal reduction.
2 Materials and methods
2.1 Fe(III) oxide phases
The Fe(III) oxides employed in this study included a variety of pure synthetic phases [40] as well as three previously characterized Fe(III) oxide-bearing subsoil or subsurface materials (HC, CP, and Oyster). The synthetic oxides were prepared according to standard methods [50]. The HC and CP natural materials are Fe(III) oxide/layered silicate mixtures obtained from Ultisols in Tennessee and North Carolina, respectively. The Oyster material is Fe(III) oxide-coated sand from Pleistocene Age Atlantic Coastal Plain sediments. More detailed descriptions of the properties of these materials are available elsewhere [42,54,55]. The synthetic Fe(III) oxides were freeze-dried and passed through a 100-μm sieve, and their specific surface area determined by multipoint BET N2 adsorption. The natural materials were air dried and passed through a 2-mm sieve prior to use in experiments.
2.2 Fe(III)-reducing microorganisms
Two well-characterized dissimilatory Fe(III)-reducing bacteria (FeRB) were employed in Fe(III) reduction experiments, Shewanella putrefaciens strain CN32 [14], and Geobacter sulfurreducens strain PCA [3,31]. The procedures used for growth and handling of these organisms for Fe(III) reduction experiments are described in detail in Roden [40] and Jeon et al. [19], respectively.
2.3 Bacterial reduction experiments
The synthetic and natural Fe(III) oxide-bearing solids were suspended in sterile, anaerobic Pipes buffer (10 mM, pH 6.8) contained in sealed serum vials to obtain Fe(III) concentrations of 5 to 250 mmol l−1. Other experiments were conducted with soluble Fe(III) citrate as an electron acceptor. The electron donor for metal reduction was either 100% H2 in the vial headspace, or 10 mM of either sodium lactate or sodium acetate. The medium was inoculated with ca. 108 cells ml−1 of either S. putrefaciens or Geobacter sulfurreducens cells. Samples for determination of dissolved (0.2-μm filtration and Ferrozine analysis) and total Fe(II) (0.5M HCl extraction and Ferrozine analysis) and pH were collected at 1–10-d intervals.
2.4 Ascorbate and AH2DS reduction experiments
Characteristics of synthetic Fe(III) oxides used in studies of microbial and abiotic reduction experiments. Modified with permission from Table 1 in Roden [40], with kind permission of the American Chemical Society
Caractéristiques des oxydes de fer synthétiques, utilisés pour les expériences de Fe(III) réduction bactérienne et abiotique. Modifié d'après le Tableau 1 de Roden [40], et reproduit ici avec l'aimable autorisation de l'American Chemical Society
| Oxide | Assumed morphologya | % HA–Ext Fe(III)b | Surface areac (m2 g−1) | Estimated mean particle sized (nm) | |
| Hydrous ferric oxide | sphere | 100 ± 0 | 600 | 1.3 | +1.230 |
| 2-line Ferrihydrite | sphere | 14.8 ± 0.2 | 290 | 2.6 | +0.918 |
| Feroxyhyte | plate | 3.61 ± 0.04 | 176 | 9.4 | +0.888 |
| Lepidocrocite | cylinder | 2.55 ± 0.05 | 63.6 | 26.7 | +0.902 |
| HSA Goethite 1 | cylinder | 1.75 ± 0.05 | 211 | 7.7 | +0.844 |
| HSA Goethite 2 | cylinder | 0.90 ± 0.03 | 135 | 12.1 | +0.844 |
| Goethite 7 °Cf | cylinder | 0.72 ± 0.01 | 96.3 | 16.9 | +0.811 |
| Goethite 20 °Cf | cylinder | 0.34 ± 0.01 | 73.0 | 22.4 | +0.803 |
| Goethite 30 °Cf | cylinder | 0.085 ± 0.003 | 62.0 | 26.3 | +0.804 |
| Goethite 90 °Cf | cylinder | 0.019 ± 0.001 | 37.6 | 43.4 | +0.787 |
| Mati hematite | sphere | 0.53 ± 0.11 | 18.8 | 30.3 | +0.781 |
| Fisher Hematite | sphere | 0.075 ± 0.005 | 10.3 | 55.4 | +0.774 |
a For use in estimating mean particle size from measured surface area.
b 0.25M Hydroxylamine–HCl/0.25M HCl, 1-h extraction; mean ± SD of triplicate samples.
c Determined by multi-point BET N2 adsorption, except for HFO, for which the assumed value is based on discussions and recommendations in Dzombak and Morel [11].
d
Estimated using the oxide densities listed in [6], and assuming the following proportions for plate and cylindrical and particle morphologies: plate diameter:
e
Half-cell reduction potentials (e.g., for reactions such as:
f Temperature refers to the temperature at which the mineral synthesis was conducted.
The synthetic and natural Fe(III) oxide-bearing solids were suspended in anaerobic 10 mM ascorbic acid or 10 mM AH2DS (the reduced form of AQDS, anthroquinone-2,6-disulfonate, prepared by reacting AQDS with 100% H2 gas in the presence of a palladium catalyst) in 10 mM Pipes buffer. The oxide suspensions were incubated at room temperature on a rotary shaker (250 rpm) and samples were removed with a N2-flushed plastic syringe at regular intervals. A portion of the sample was passed through a 0.2-μm filter into Ferrozine for Fe(II) analysis, and the remainder used for determination of pH. The final (after reduction ceased) dissolved Fe(II) concentration and pH values achieved in the AH2DS reduction experiments were used in conjunction with the
3 Results and discussion
3.1 Initial rates of reduction
Initial (0–3 d) surface area-specific rates of synthetic Fe(III) oxide reduction by S. putrefaciens and G. sulfurreducens did not vary systematically across a wide range of oxide surface area and
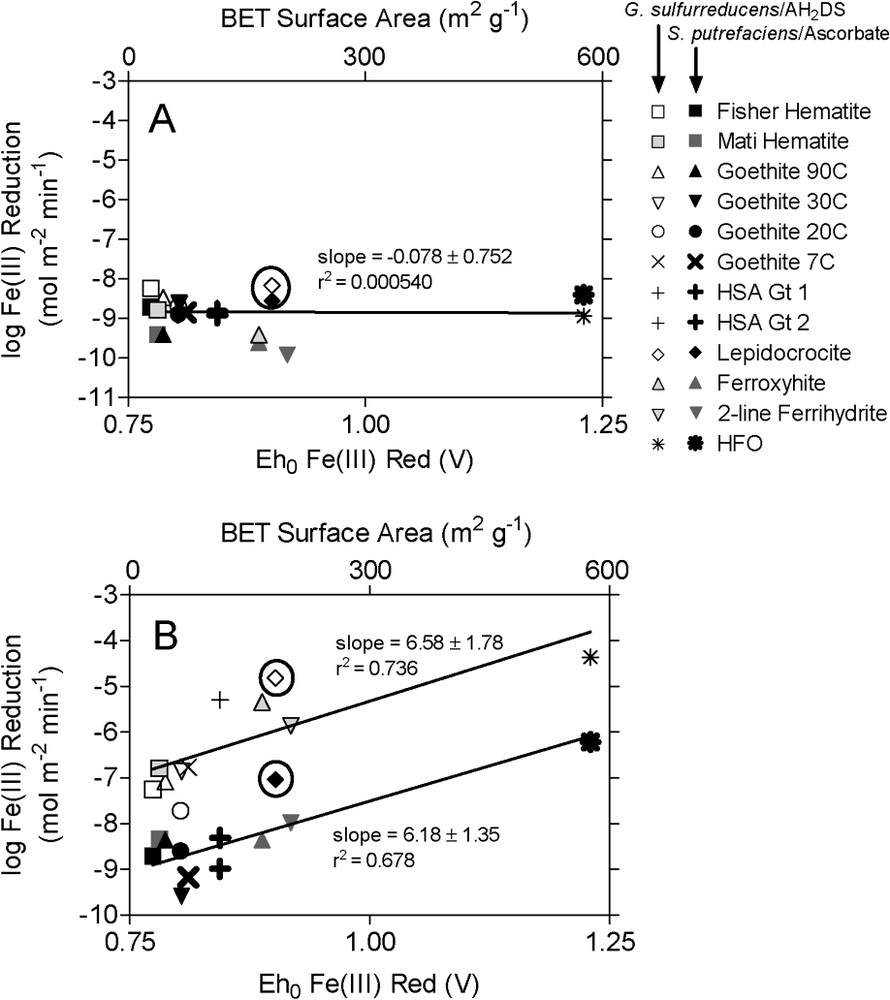
Initial (2–3-day incubation) surface area-specific rates of bacterial (A) and abiotic (B) reduction of synthetic Fe(III) oxides. Different symbols correspond to different Fe(III) oxide phases and reduction systems as indicated in the legend. The temperatures listed with the goethite phases correspond to the temperature at which the synthesis (from ferric nitrate) took place. Starting Fe(III) concentrations in the biotic and abiotic reduction experiments were 10 and 5 mmol l−1, respectively. S. putrefaciens and ascorbate reduction data are reproduced with permission from Fig. 4 in Roden [40]; G. sulfurreducens data are from Roden [41]. Masquer
Initial (2–3-day incubation) surface area-specific rates of bacterial (A) and abiotic (B) reduction of synthetic Fe(III) oxides. Different symbols correspond to different Fe(III) oxide phases and reduction systems as indicated in the legend. The temperatures listed with the goethite phases ... Lire la suite
Vitesses initiales (incubations pendant 2–3 jours) rapportées à la surface spécifique de la réduction bactérienne d'oxydes ferriques de synthèse. Les différents symboles correspondent aux différents types d'oxydes et de modalités de réduction indiqués sur la figure. Les températures relatives aux goethites correspondent à leurs températures de synthèse (aux dépens de nitrate ferrique). Les concentrations initiales en FeIII dans les expériences de réduction bactérienne et abiotique étaient de 10 et de 5 mmol l−1 respectivement. Les données relatives aux expériences de réduction en présence de S. putrefaciens et d'ascorbate sont issues de la Fig. 4 de Roden [40], les données relatives à G. sulfurreducens de Roden [41]. Masquer
Vitesses initiales (incubations pendant 2–3 jours) rapportées à la surface spécifique de la réduction bactérienne d'oxydes ferriques de synthèse. Les différents symboles correspondent aux différents types d'oxydes et de modalités de réduction indiqués sur la figure. Les températures relatives aux ... Lire la suite
Since detachment of a metal ion from an oxide surface site is generally viewed as the rate-limiting step in oxide mineral dissolution [52], it could be argued that because of the tendency for Fe(II) to reassociate (or never become detached in the first place) with oxide surfaces during enzymatic reduction at circumneutral pH (discussed in detail in Roden and Urrutia [47]), the kinetics of the enzymatic reduction system is not controlled by the presence of an obvious leaving group for which the detachment energy (related to the energy required for metal-ligand bond formation and breaking processes [1,47]) is affected by the thermodynamic properties of the oxide phase. However, during the bacterial Fe(III) oxide reduction experiments conducted under simplified aqueous geochemical conditions (i.e. in the absence of ions such as
Ongoing studies with Shewanella [10,12,32–35] and recent studies with Geobacter [24,29,30] have provided direct evidence that low redox potential, outer membrane-associated c-type cytochromes are involved in electron transfer from FeRB to Fe(III) oxides. In addition, a recent atomic force microscopy study by Lower et al. [28] demonstrated apparent molecular “recognition” of Fe(III) oxide surface sites by a putative ca. 150 kDa outer membrane protein of the dissimilatory FeRB Shewanella oneidensis (formerly S. putrefaciens strain MR-1), a close relative of the organism used in this study. Together these findings suggest the possibility that the similarity of surface area normalized electron transfer rates across a broad range of oxide minerals results from the fact that dissimilatory FeRB ‘recognize’ different Fe(III) oxide surfaces more or less equally independent of the underlying crystal structure, such that initial rates of electron transfer subsequent to recognition are not strongly dependent on crystal structure. This suggestion is consistent with an argument presented by Fischer [13] to account for the relatively minor influence of oxide solubility on rates of synthetic Fe(III) oxide reduction by Corynebacteria at pH 7. Fischer [13] reasoned that if the redox potential of the bacterial cells (i.e., their outer membrane c-type cytochromes) is sufficiently negative for reduction of well-crystallized oxide phases like hematite or goethite, each collision (or ‘recognition’) of a FeRB cell with an oxide particle will trigger reduction of a Fe(III) surface site. Therefore, the amount of Fe(II) produced during each collision event will not increase markedly with increasing oxide solubility.
3.2 Long-term extent of reduction
Similar patterns of Fe(II) accumulation were observed during long-term (3-week incubation) reduction of the various synthetic Fe(III) oxides by G. sulfurreducens (Fig. 2A): after an initial period of rapid Fe(II) accumulation, rates of reduction decreased and Fe(II) levels approached an asymptote. pH values correlated directly with aqueous Fe(II) concentrations during Fe(III) oxide reduction (Fig. 2B), as expected from the stoichiometry of Fe(III) oxide reduction coupled to H2 oxidation:
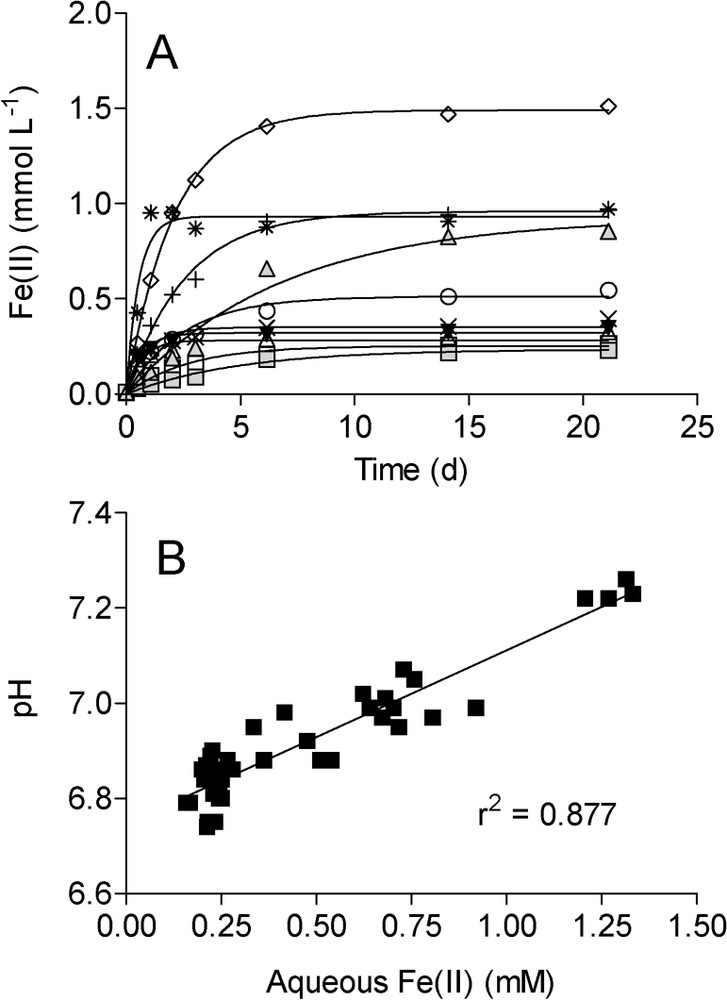
Total (0.5 M HCl extraction) Fe(II) production (A) and relationship between pH and aqueous Fe(II) accumulation (B) during reduction of synthetic Fe(III) oxides by G. sulfurreducens (data from Roden [41]). Symbols as in Fig. 1. Lines in panel A show nonlinear regression fits of the data to the equation depicting the accumulation of end-product of a first-order reaction. Line in panel shows result of a linear least-square regression analysis.
Évolution des concentrations en FeII total (extrait par HCl 0,5 M) en fonction du temps (A) et relation entre pH et FeII total extrait (B) lors de la réduction d'oxydes de fer de synthèse par G. sulfurreducens [41]. Les symboles sont identiques à ceux de la Fig. 1. Dans la Fig. 2A, les traits pleins montrent l'ajustement des données de régression non linéaire à des équations décrivant l'accumulation des produits de la réaction selon une loi cinétique d'ordre 1. Le trait plein de la Fig. 2B résulte d'une analyse de régression linéaire par la méthode des moindres carrés. Masquer
Évolution des concentrations en FeII total (extrait par HCl 0,5 M) en fonction du temps (A) et relation entre pH et FeII total extrait (B) lors de la réduction d'oxydes de fer de synthèse par G. sulfurreducens [41]. ... Lire la suite
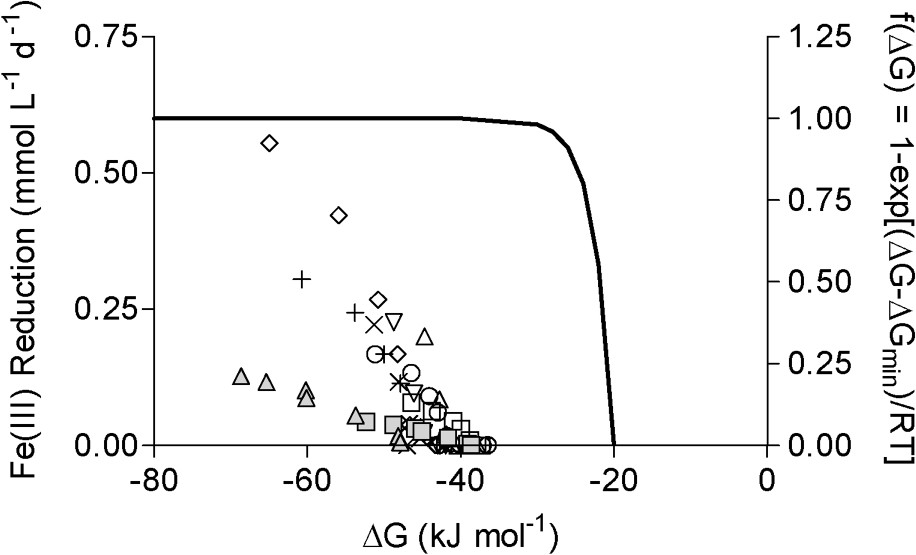
Rate of Fe(III) reduction as a function of the estimated free energy of reaction during long-term G. sulfurreducens experiments (data from Roden [41]). Fe(III) reduction rates were computed from the nonlinear curve fits shown in Fig. 2. Symbols as in Fig. 1. The solid line shows the relationship between reaction rate and energetics predicted based on transition state theory [23], modified to account for the minimum free requirement for biological energy conservation, as described in Liu et al. [26]. Masquer
Rate of Fe(III) reduction as a function of the estimated free energy of reaction during long-term G. sulfurreducens experiments (data from Roden [41]). Fe(III) reduction rates were computed from the nonlinear curve fits shown in Fig. 2. ... Lire la suite
Vitesses de réduction du FeIII en fonction de l'énergie libre estimée de la réaction lors d'expériences de longue durée utilisant G. sulfurreducens. Les vitesses de réduction du FeIII sont calculées aux dépens des courbes présentées sur la Fig. 2 et les symboles sont ceux de la Fig. 1. Le trait plein montre la relation entre vitesse de réduction et les données énergétiques prédites par la théorie de l'état de transition [23], modifiée pour tenir compte de l'exigence minimale en énergie nécessaire pour la conservation de l'énergie biologique [26]. Masquer
Vitesses de réduction du FeIII en fonction de l'énergie libre estimée de la réaction lors d'expériences de longue durée utilisant G. sulfurreducens. Les vitesses de réduction du FeIII sont calculées aux dépens des courbes présentées sur la Fig. ... Lire la suite
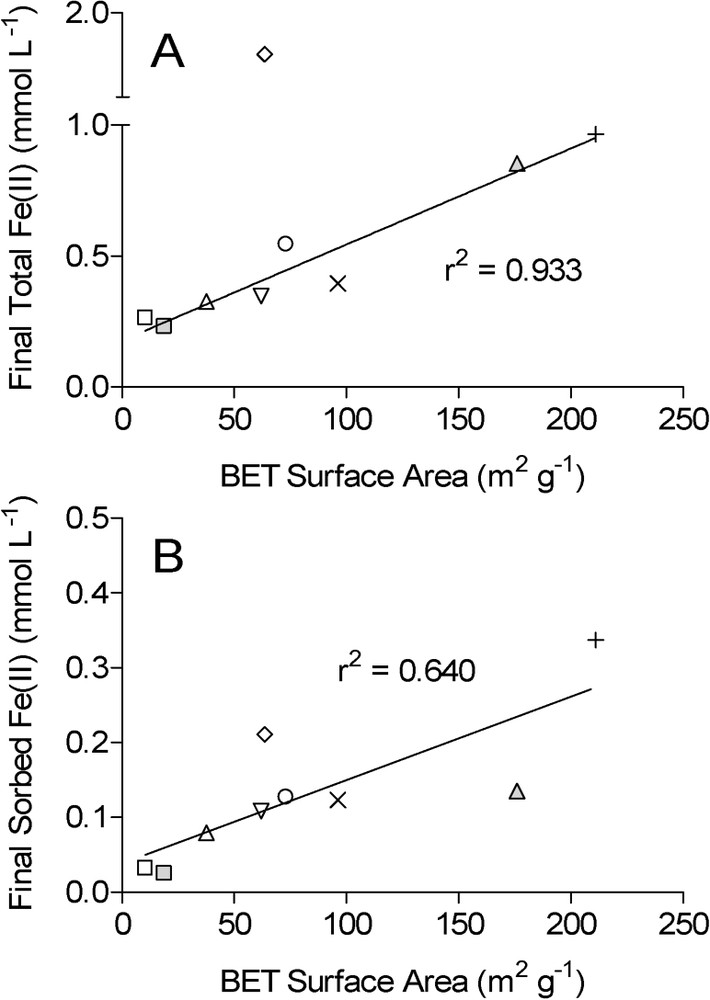
Correlation between oxide mineral surface area and the final concentration of total HCl-extractable Fe(II) (A) and surface-associated (‘sorbed’) Fe(II) (B) during long-term G. sulfurreducens experiments (data from Roden [41]). Symbols as in Fig. 1. Data for the HFO reduction system are omitted, since the mineral was transformed magnetite, which is resistant to further reduction for clearly defined thermodynamic reasons [14,20]. Lines show linear least-square regression fits of the data; data for lepidocrocite (filled diamonds) were omitted in regression in panel A. Masquer
Correlation between oxide mineral surface area and the final concentration of total HCl-extractable Fe(II) (A) and surface-associated (‘sorbed’) Fe(II) (B) during long-term G. sulfurreducens experiments (data from Roden [41]). Symbols as in Fig. 1. Data for the ... Lire la suite
Corrélation entre la surface spécifique des oxydes et la concentration finale en FeII extrait par HCl (A) ou la concentration en FeII adsorbé (B) lors d'expériences de longue durée utilisant G. sulfurreducens. Les symboles sont ceux de la Fig. 1. Les données relatives au système HFO sont omises, car ce minéral se transforme en magnétite, qui résiste à toute réduction ultérieure pour des raisons thermodynamiques bien établies [14,20]. Les traits pleins montrent les ajustements dérivant de régressions linéaires utilisant la méthode des moindres carrés. Les données relatives à la lépidocrocite (losange) ne sont pas utilisées pour tracer la droite de régression apparaissant sur la Fig. 4A. Masquer
Corrélation entre la surface spécifique des oxydes et la concentration finale en FeII extrait par HCl (A) ou la concentration en FeII adsorbé (B) lors d'expériences de longue durée utilisant G. sulfurreducens. Les symboles sont ceux de la Lire la suite
3.3 Role of amorphous Fe(III) oxide impurities?
It could be argued that the similar surface area-specific rates of reduction observed for the different crystalline Fe(III) oxide phases (Fig. 1A), as well as the correlation between oxide surface area and extent of reduction (Fig. 4), was due to the presence of easily reducible amorphous Fe(III) oxide impurities in the synthetic crystalline Fe(III) oxides, whose abundance could have scaled with oxide surface area. If this were the case, the arguments put forward here and in other recent papers [39,40] regarding surface area vs. thermodynamic control of crystalline Fe(III) oxide reduction would be invalid. Data on initial rates of ascorbate-catalyzed Fe(III) oxide reduction were used to evaluate this question.
Fig. 5 shows time course data for reductive dissolution of hematite, two goethite phases (‘Goethite 30C’ and ‘High Surface Area’ (HSA) goethite), and HFO in the presence of excess (10 mM) of ascorbic acid at pH 3. HFO was fully dissolved within 4 h, whereas much smaller amounts of the crystalline Fe(III) oxide phases were dissolved over a 26-h period. As a result of the much faster kinetics of HFO dissolution compared to the other oxides, it was possible to use the time course data to quantitatively estimate the amount of amorphous HFO impurities present in the crystalline solids. The time course data for the crystalline phases indicated a relatively rapid initial accumulation of Fe(II) during the first few hours of reaction, followed by constant rate of oxide dissolution between 4 and 26 h. This initial rapid Fe(II) production can be attributed to dissolution of HFO impurities. The overall accumulation of Fe(II) during the initial period thus represents a combination of crystalline oxide and HFO impurity dissolution. A simple mathematical model that simulated parallel dissolution of a mixture of crystalline oxide and HFO was developed to estimate the abundance of the HFO impurity. The rate of crystalline oxide dissolution (in % total Fe(III) per hr) was set equal to the linear rate observed between 4 and 26 h, as defined by the least-squares regression analyses shown in Fig. 5A–C:
| (1) |

Reductive dissolution of Fisher hematite, goethite 30C, HSA goethite, and HFO in the presence of 10 mM ascorbic acid at pH 3. Total Fe(III) oxide concentrations were 6.4, 5.8, 5.7, and 1.1 mmol l−1 for the four oxide phases, respectively. Solid lines in panels A–C show the results of linear least-square regression analyses. Solid line in panel D shows the result of nonlinear least-square regression fit of the data to an equation depicting the accumulation of end-product from a first-order reaction (
Reductive dissolution of Fisher hematite, goethite 30C, HSA goethite, and HFO in the presence of 10 mM ascorbic acid at pH 3. Total Fe(III) oxide concentrations were 6.4, 5.8, 5.7, and 1.1 mmol l−1 for the four oxide phases, respectively. Solid ... Lire la suite
Dissolution par réduction de l'hématite Fisher, de la goethite 30 °C, de la goethite HSA et du HFO en présence d'acide ascorbique 10 mM à pH 3. Les concentrations initiales en FeIII étaient de 6,4, 5,8, 5,7 et 1,1 mmol l−1 pour les différents oxydes. Les traits pleins dans les figures A à C correspondent aux résultats d'analyses de régression linéaire utilisant la méthode de moindres carrés. Le trait plein dans la figure D résulte de l'ajustement des données d'une régression non linéaire à une équation décrivant l'accumulation des produits de réaction selon une loi cinétique d'ordre l, c'est-à-dire :
Dissolution par réduction de l'hématite Fisher, de la goethite 30 °C, de la goethite HSA et du HFO en présence d'acide ascorbique 10 mM à pH 3. Les concentrations initiales en FeIII étaient de 6,4, 5,8, 5,7 et 1,1 mmol l−1 pour ... Lire la suite
The rate of HFO dissolution was modeled as a first-order reaction, using the rate constant (1.37 h−1) derived from the nonlinear least-square regression analysis shown in Fig. 5D:
| (2) |
Eqs. (1) and (2) were integrated numerically using a standard Runge–Kutta algorithm [38], and total % Fe(III) dissolution was computed as the cumulative sum of crystalline oxide and HFO reduction. The initial fractional abundance of HFO was adjusted to achieve agreement between the observed and simulated % Fe(III) dissolution vs. time data. The results of the analysis are illustrated by the dashed lines in Fig. 5A–C, which reflect estimated HFO impurities of 0.04, 0.10, and 0.32% of the total Fe(III) content of hematite, ‘goethite 30C’, and ‘HSA goethite’, respectively.
The estimated relative abundance of HFO impurities in the crystalline Fe(III) oxides can be compared to the amount of Fe(III) oxide reduction that took place in the G. sulfurreducens reduction experiments in order to constrain the potential contribution of such impurities to the observed Fe(III) reduction activity. The percent of Fe(III) subject to enzymatic reduction ranged from 2.3 to 9.7% for the crystalline hematite and goethite phases; these values are 5- to 95-fold higher than the estimated abundance of HFO impurity in the respective oxide phases. These results verify that enzymatic reduction of crystalline Fe(III) oxide surfaces – rather than reduction of amorphous Fe(III) oxide impurities – was the dominant mode of Fe(II) production in the cultures. The conclusions reached above regarding thermodynamic vs. surface-bound Fe(II) control of long-term crystalline Fe(III) oxide reduction are therefore valid, and they suggest that thermodynamic calculations should be used with caution when interpreting controls on enzymatic Fe(III) oxide reduction in soils and sediments.
3.4 Reduction of natural Fe(III) oxide phases
Data from long-term experiments on bacterial and abiotic reduction of natural Fe(III) oxides (Fig. 6) were interpreted in relation to a standard a generalized rate law for mineral dissolution [22,36]:
| (3) |
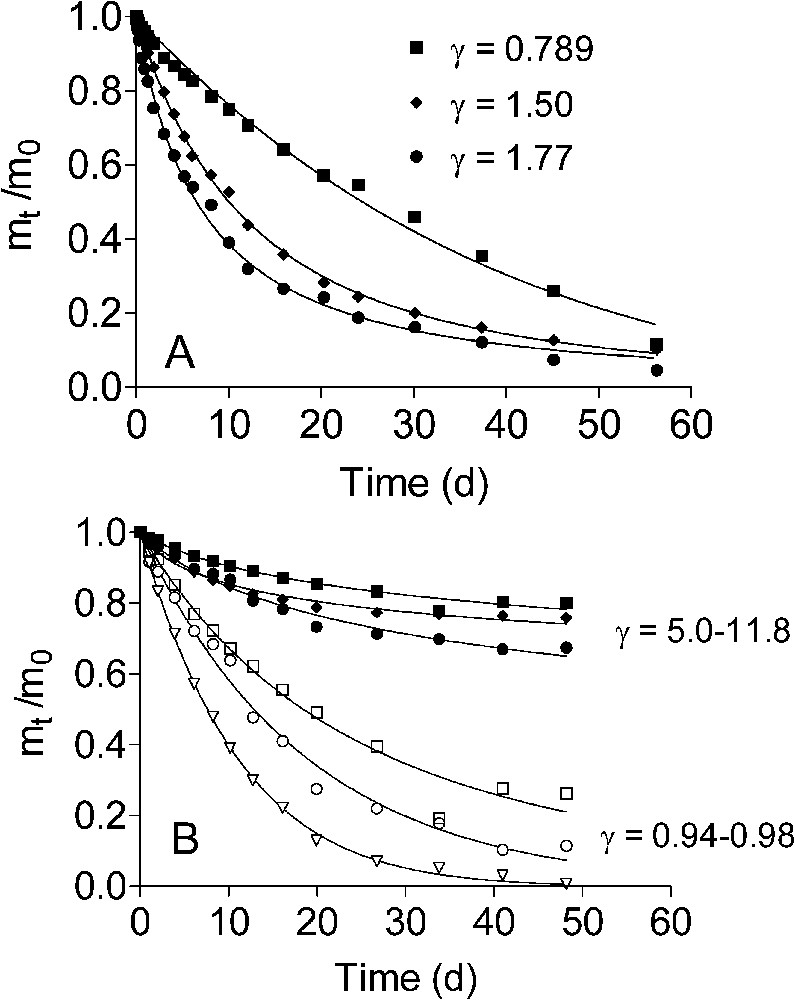
Kinetics of ascorbate (A) and bacterial (B) reduction of natural Fe(III) oxide-bearing materials. Data are reproduced with permission from Fig. 2 in Roden [42]. Squares, diamonds, and triangles represent the HC, CP, and Oyster materials, respectively. Open symbols in panel B refer to reduction of the ‘microbially available’ fraction of the oxide pool. The terms
Kinetics of ascorbate (A) and bacterial (B) reduction of natural Fe(III) oxide-bearing materials. Data are reproduced with permission from Fig. 2 in Roden [42]. Squares, diamonds, and triangles represent the HC, CP, and Oyster materials, respectively. Open symbols in ... Lire la suite
Cinétique de la réduction d'oxydes de fer naturels par l'ascorbate (A) et des bactéries (B). Les données proviennent de Roden [42]. Les carrés, les losanges et les triangles sont relatifs aux résultats obtenus avec les matériaux dénommés HC, CP et Oyster. Dans la Fig. 6B, les symboles ouverts font référence à la réduction de la fraction d'oxyde définie comme « microbiologiquement disponible». Les termes
Cinétique de la réduction d'oxydes de fer naturels par l'ascorbate (A) et des bactéries (B). Les données proviennent de Roden [42]. Les carrés, les losanges et les triangles sont relatifs aux résultats obtenus avec les matériaux dénommés HC, CP ... Lire la suite
Kinetic and thermodynamic considerations indicated that neither the abundance of electron donor (lactate) nor the accumulation of aqueous end-products of oxide reduction (Fe(II), acetate, dissolved inorganic carbon) are likely to have posed significant limitations on the long-term kinetics of oxide reduction [42]. Rather, accumulation of biogenic Fe(II) on residual oxide surfaces appeared to play a dominant role in governing the long-term kinetics of natural crystalline Fe(III) oxide reduction. This assertion is fully supported by the results of the synthetic Fe(III) oxide reduction experiments (Figs. 3 and 4).
3.5 General conceptual model and rate law for microbial Fe(III) oxide reduction
The experimental findings summarized here point to a conceptual model of bacterial Fe(III) oxide reduction kinetics that differs fundamentally from established models of abiotic Fe(III) oxide reductive dissolution in that oxide surface area, rather than crystal structure and thermodynamic stability, exerts primary control on both the initial rate and the long-term extent of reduction. Numerical simulations of surface area-controlled biotic vs. abiotic Fe(III) oxide reduction indicate that this conceptual model can account for the pseudo-first-order kinetics of reduction of the operationally defined ‘microbially reducible’ fraction of the sediment Fe(III) oxide pool [42]. The explicit surface area control of the initial rate and extent of oxide reduction leads to a general rate law for oxide reduction as a function of electron acceptor and FeRB abundance that differs from those for reduction of chelated Fe(III) and other soluble metal species. As illustrated for synthetic goethite in Fig. 7, initial rates of Fe(III) oxide reduction are a linear function of oxide loading up to relatively high (200 mmol l−1) bulk Fe(III) concentrations, a hyperbolic (Monod-style) function of total FeRB cell density, and a linear function of attached FeRB cell density. The latter relationships are analogous to the well-recognized dependence of abiotic reductive dissolution rate on total and surface-associated ligand concentration [16]. These relationships between oxide reduction rate and oxide/FeRB cell abundance are the opposite of those for reduction of soluble metals, as illustrated for Fe(III)-citrate in Fig. 8: rates of soluble metal reduction are a hyperbolic function of electron acceptor concentration, and a linear function of FeRB cell abundance. Similar patterns hold for reduction of other soluble metals such as U(VI), Co(III), Cr(VI), and Tc(VI) [25,27,43,45,53]. Table 2 provides a summary of rate laws that are appropriate for use in modeling solid-phase and dissolved Fe(III) (and other oxidized metals) reduction.
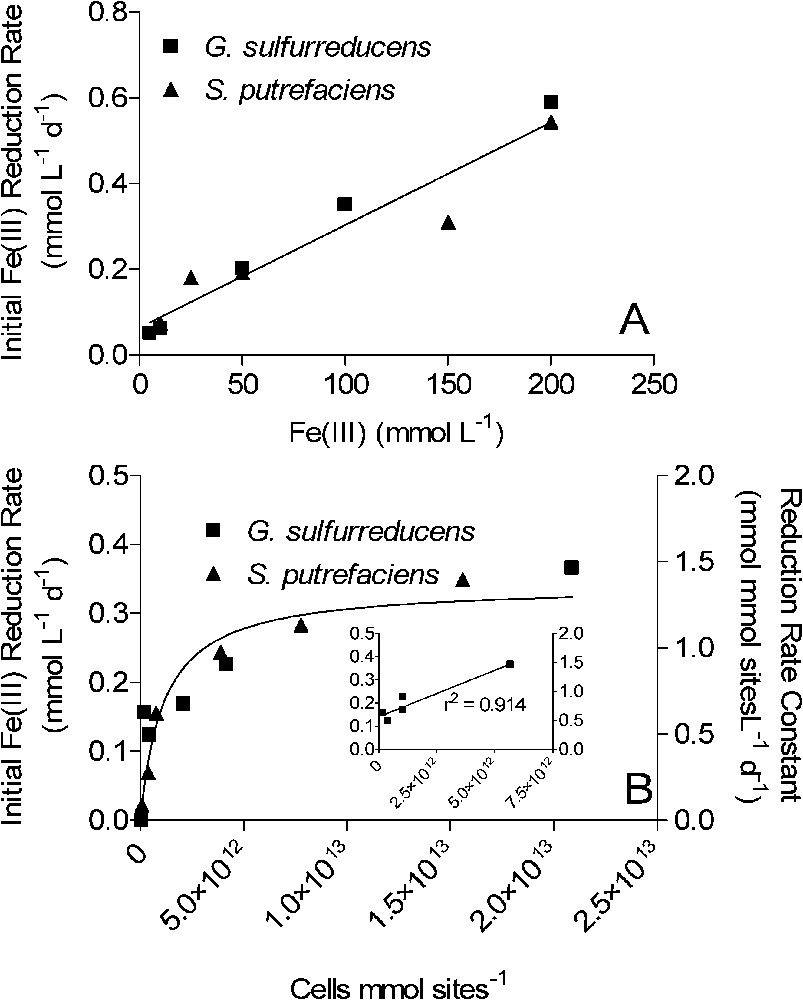
Kinetics of synthetic goethite reduction by S. putrefaciens and G. sulfurreducens as a function of oxide loading (A) and FeRB cell density (B). The concentration of cells in the experiments shown in panel A was ca. 108 cells ml−1. The concentration of surface sites used to normalize cell density in panel B was calculated based on oxide mass loading (10 mmol l−1; 89 g l−1), the measured mineral surface area (ca. 55 m2 g−1), and an assumed standard mineral surface site density of
Kinetics of synthetic goethite reduction by S. putrefaciens and G. sulfurreducens as a function of oxide loading (A) and FeRB cell density (B). The concentration of cells in the experiments shown in panel A was ca. 108 cells ml−1. ... Lire la suite
Cinétique de la réduction d'une gœthite par S. putrefaciens et G. sulfurreducens. Les pointillés de la Fig. 7B montrent la densité maximale (normalisée par rapport aux nombres de sites superficiels) de cellules de bactéries ferriréductrices (FeRB) présentes dans les expériences utilisant les différentes charges en oxydes mentionnées sur la Fig. 7A. La concentration en sites superficiels a été calculée en prenant en compte la charge en oxyde (10 mmol l−1), la surface spécifique du minéral (environ 55 m2 g−1) et une densité de sites superficiels de
Cinétique de la réduction d'une gœthite par S. putrefaciens et G. sulfurreducens. Les pointillés de la Fig. 7B montrent la densité maximale (normalisée par rapport aux nombres de sites superficiels) de cellules de bactéries ferriréductrices (FeRB) présentes ... Lire la suite
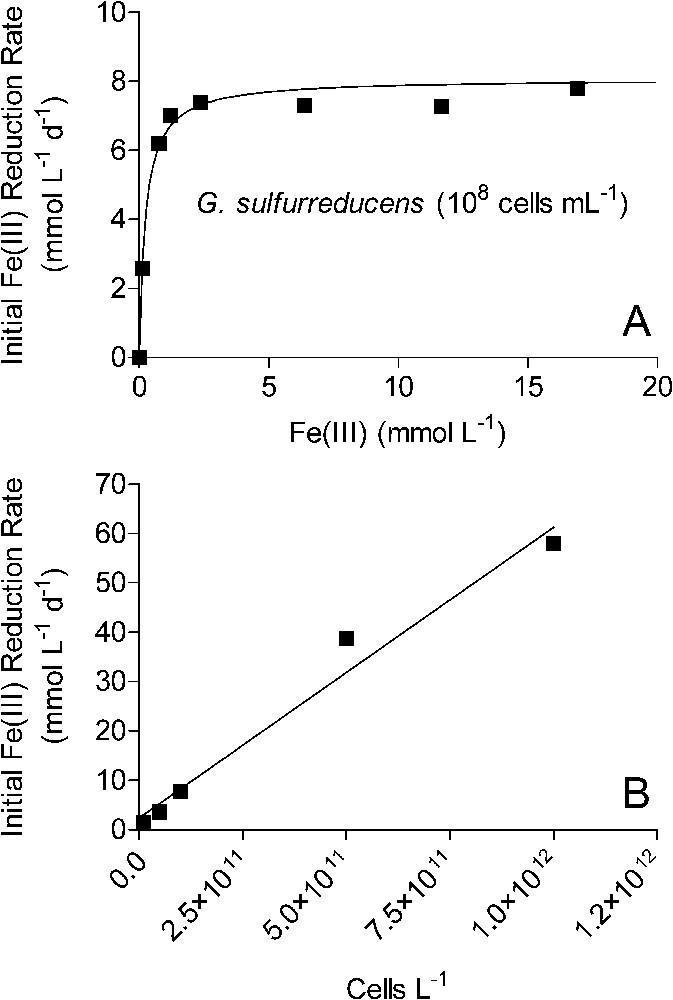
Kinetics of Fe(III)-citrate reduction by G. sulfurreducens. Solid lines in panels A and B show results of linear and non-linear least-square regression fits, respectively. The slope of the line in panel A corresponds to the parameter
Kinetics of Fe(III)-citrate reduction by G. sulfurreducens. Solid lines in panels A and B show results of linear and non-linear least-square regression fits, respectively. The slope of the line in panel A corresponds to the parameter Lire la suite
Cinétique de la réduction du citrate ferrique par G. sulfurreducens. Les traits pleins montrent les résultats d'ajustements utilisant, soit une régression linéaire (B), soit une régression non linéaire (A). La pente de la droite de la Fig. 8B correspond au paramètre
Cinétique de la réduction du citrate ferrique par G. sulfurreducens. Les traits pleins montrent les résultats d'ajustements utilisant, soit une régression linéaire (B), soit une régression non linéaire (A). La pente de la droite de la Fig. 8B ... Lire la suite
General rate laws for microbial reduction of solid-phase and aqueous Fe(III)
Lois cinétiques générales de la réduction microbienne de Fe(III) solide et du Fe(II) en solution
| Solid phase |
|
|
| where: |
| RFe(III) = bulk volumetric rate of Fe(III) oxide reduction (e.g., mmol l−1 d−1); |
| [Fe(III)fss] = bulk volumetric abundance of ‘free’ (i.e. microbially reducible) Fe(III) oxide surface sites (e.g., mmol sites l−1); |
| Vsurfmax = maximum FeRB cell density-dependent reduction rate constant (e.g., mmol (mmol sites)−1 d−1) (see Fig. 7B); |
| [FeRB]ssn = surface site-normalized FeRB cell density (e.g., cells (mmol free surface sites)−1) (see Fig. 7B); |
| KFeRB = half-saturation constant for relationship between FeRB cell density and oxide reduction rate constant (e.g., cells/mmol free surface sites) (see Fig. 7B); |
| Aqueous phase |
|
|
| where: |
| RFe(III) = bulk volumetric rate of Fe(III) oxide reduction (e.g., mmol l−1 d−1); |
| Vmax=αFeRB [FeRB]bulk (see Fig. 8A); |
| [FeRB]bulk = bulk FeRB cell density (e.g., cells l−1); |
| αFeRB = proportionality constant for relationship between bulk FeRB cell density and soluble Fe(III) reduction rate (e.g., mmol cell−1 d−1) (see Fig. 8B); |
| [Fe(III)aq] = concentration of aqueous Fe(III) (e.g., mmol l−1). |
| KFe(III) = Half-saturation constant for relationship between Fe(III)aq concentration and reduction rate (e.g., mmol/l) (see Fig. 8A). |
3.6 Implications for natural soils and sediments
Virtually all experimental work to date on bacterial synthetic Fe(III)-oxide reduction indicates that oxide mineral heterogeneity in natural soils and sediments is likely to affect initial rates of bacterial reduction (e.g., during the early stages of anaerobic metabolism following the onset of anoxic conditions) mainly via an influence on reactive surface site density. Although variations in oxide thermodynamic properties may alter rates of enzymatic reduction to some extent (as illustrated by the studies of lepidocrocite vs. goethite reduction), this is likely to represent a secondary effect in relation to the primary controlling influence of oxide surface area. Hence, the surface area of different oxides phases present in a soil or sediment will exert primary control on initial rates of enzymatic reduction.
During the later stages of bacterial Fe(III) oxide reduction in permanently reduced sediments, accumulation of aqueous and surface-bound Fe(II) is expected to exert a dominant control on apparent Fe(III) oxide reactivity toward enzymatic reduction, particularly in situations where removal of Fe(II) end-products is slow compared to the kinetics of reduction [46]. This conceptual model is consistent with a recent analysis of the kinetics of hematite reduction by S. putrefaciens strain CN32 [2,48], which showed that initial rates of reduction were under kinetic control (presumably limited by the rate of electron transfer from FeRB cells to the oxide), whereas the long-term extent of reduction was limited by mass transfer of Fe(II) away from oxide/FeRB surfaces. As discussed above (see Fig. 4), there is a general relationship between oxide surface area and long-term extent of oxide reduction in closed reaction systems, which results from the function of oxide surfaces as a repository for sorbed and/or surface-precipitated biogenic Fe(II). Although the existence of this relationship implies a connection between extent of bacterial reduction and oxide thermodynamic properties, evidence suggests that this connection is not directly related to thermodynamic properties such as
An important implication of the above findings is that inferences regarding the ability of bacterial Fe(III) oxide reduction to compete with other terminal electron accepting processes (TEAPs) in soils and sediments should be based on estimates of bulk reactive (i.e. microbially accessible) surface site density – rather than the thermodynamic properties of the oxide(s) identified as the dominant phase(s) in a particular soil or sediment. This line of reasoning leads to the provisional conclusion that recent thermodynamic explanations for the coexistence of bacterial Fe(III) oxide reduction and other TEAPs (e.g., sulfate reduction and methanogenesis) in sediments and subsurface environments [17,18,37] have produced reasonable results for mechanistically incorrect reasons. The ability of thermodynamic considerations to explain the coexistence of bacterial Fe(III) oxide reduction and other TEAPs in sediments is likely the fortuitous result of the correlation between oxide thermodynamic properties and surface properties, which, based on current experimental information, actually control the initial reduction rate and long-term availability of Fe(III) oxides as competing electron acceptors for anaerobic respiration. Experiments with a mixed culture of wetland sediment bacteria and a range of synthetic Fe(III) oxides indicate that Fe(III)-reducing bacteria can outcompete methanogens for acetate with equal effectiveness when the different oxides are present at comparable surface area loadings – despite major differences in computed ΔG values for acetate oxidation coupled to Fe(III) oxide reduction [39]. These results emphasize the need for more accurate and robust wet-chemical (e.g., Hacherl et al. [15]) and/or spectroscopic techniques for assessing the surface properties (e.g., specific surface area and reactive site density) of natural Fe(III) oxide assemblages, including ones in which the presence of sorbed or surface precipitated Fe(II) limits the potential for enzymatic electron transfer and thereby controls apparent oxide reactivity toward microbial reduction.
Acknowledgements
This research was supported by grants DE-FG07-ER6321 and DE-FG02-01ER63182 from the US Department of Energy (DOE) Environmental Management Science and Natural and Accelerate Bioremediation Programs, respectively.