

1 Introduction
Understanding and predicting the global transfer of carbon at geological timescales have stimulated the development of ever more detailed models of the geological carbon cycle (Berner, 1990; Berner et al., 1983; François and Walker, 1992; Goddéris and François, 1995; Hayes and Waldbauer, 2006). These models incorporate a large variety of processes playing roles over timescales ranging from days to millions of years, such as those affecting the oceanic lithosphere or occurring in subduction zones (Hayes and Waldbauer, 2006). This study will examine key pieces of literature from 1777 to 1847 to highlight major steps in the progressive development of the carbon cycle concept.
The global carbon cycle was not a fully developed, multi-disciplinary model in the 19th century. Rather, multiple scientists discovered different processes that would become progressively incorporated into the carbon cycle, owing to a growing concern for the stability of the atmospheric composition that was recognized as a fundamental requirement for sustaining life on Earth. This study will outline 12 major steps toward the development of the carbon cycle. These are part of the “biochemical” and “geochemical” carbon cycle (Albrecht and Ourisson, 1971; Berner, 2003), including photosynthesis, respiration, burial of carbonaceous material (CM), silicate alteration, volcanic CO2 emission.
The period from 1777 to 1847 will be the main focus of this study because it precedes the works of Jacques Joseph Ebelmen in the 1840s, a French Ingénieur des Mines, who has been recognized for his contribution to the carbon cycle research (Berner, 2012, this volume; Berner and Maasch, 1996), this volume for which he had coined the term carbon rotation (Ebelmen, 1847). In this article, we try to understand why and in what context J.J. Ebelmen wrote his seminal papers (Ebelmen, 1845, 1847). The first major discovery referred to in this study was rooted in the chemical revolution, the discovery of the compound nature of carbonic acid by Antoine Laurent Lavoisier, and the search for chemically compensating processes that would guarantee the stability of the atmospheric composition. This fundamental revolution brought about tremendous progress in the understanding of the chemical roots of natural processes, especially those related to life on Earth. The year 1847 corresponds to J.J. Ebelmen propounding of the carbon rotation theory. This came as an effort to merge the numerous scientific achievements of two broad lineages of scientists – one that corresponded to the recognition of the fundamental chemical importance of plants and animals on the Earth's biogeochemical functioning, and the other that investigated the impact of purely “geological” or “mineralogical” processes on Earth surface processes. Our contribution shows that between these two periods the construction of the carbon cycle was a collective research effort, encompassing european physiologists, chemists and geologists, who all added their respective knowledge to a topic that would emerge out of a major problem – one of the homeostatic atmospheric composition.
2 From fixed air to carbonic acid
The experiments by Jan Baptist Van Helmont (1577–1644) and the microscopic observations and sketches of plants stomata (e.g., Grew, 1682) by early physiologists – including the Italian doctor Marcello Malpighi (1628–1694) (Malpighi, 1675) and the English botanist Nehemiah Grew (1641–1712) – suggested that “air”, or “gas”, is exchanged between rocks, the atmosphere, animals and plants (Fig. 1A and B). During these exchanges air was either “fixed” or “elastic” and kept changing from one state to another. These early investigations were extensively reviewed (1774, part 1) by the French chemist Antoine Laurent Lavoisier (1743–1794). However, the discovery of the nature of these gases “fixed” in rocks and living bodies was only made possible by the British chemist Stephen Hales (1677–1761), designed and described in his book Static of Plant and animals (Hales, 1726, preface – and Fig. 1C) a machine to collect these gases (Fig. 1C).
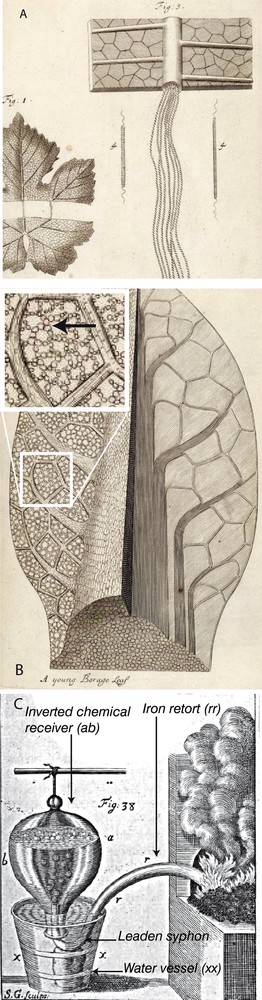
A. Drawing by N. Grew of wine leaf conducting vessels (modified from Tab 51 in Grew, 1682). B. Wine leaf drawing by N. Grew of “pass ports” for gases and fluids (stomata indicated by the black arrow in the inset) (modified from Tab 50 in Grew, 1682). C. S. Hales's illustration of his apparatus to collect gases during combustion and calcinations experiments (modified from Plate 17 Fig. 38 in Hales, 1726): “Having first put the matter to be distilled (vegetable matter, minerals etc., see Fig. 2) into the iron retort (rr) (Fig. 1C), which was made of a musket barrel, I then fixed a leaden siphon to the nose of the retort, and having immersed the siphon in the vessel of water (xx) (Fig. 1C), I placed over the open end of the siphon the inverted chemical receiver (ab) which was full of water; so that the air which was raised in distillation, passed through the water up to the top of the receiver (ab), a good part of the acid spirit and sulphureous fumes were by this means intercepted and retained in the water […]” (Hales, 1726, chap. 6). Masquer
A. Drawing by N. Grew of wine leaf conducting vessels (modified from Tab 51 in Grew, 1682). B. Wine leaf drawing by N. Grew of “pass ports” for gases and fluids (stomata indicated by the black arrow in the ... Lire la suite
A. Illustration de N. Grew des vaisseaux conducteurs de feuilles de vigne (modifié d’après Grew, 1682). B. Dessin de N. Grew de la surface d’une feuille de vigne montrant les conduits (« pass ports ») de passage pour les gaz et fluides (stomates indiqués par la flèche noire dans l’image en encart) (modifié d’après Grew, 1682). C. Illustration de S. Hales représentant sa machine permettant de conserver les gaz issus de ses expériences de combustion et calcination (modifiée d’après la Fig. 38 de la Planche 17 dans Hales, 1726). Masquer
A. Illustration de N. Grew des vaisseaux conducteurs de feuilles de vigne (modifié d’après Grew, 1682). B. Dessin de N. Grew de la surface d’une feuille de vigne montrant les conduits (« pass ports ») de passage pour les gaz et ... Lire la suite
2.1 The identification of fixed air in the atmosphere, mineral and living bodies – step 1 in Fig. 2 (S1)
Despite the variety of experiments that he described, S. Hales considered that “the great quantities of air which are thus obtained from these several substances by distillation are true air”, meaning atmospheric air, whose different properties (inflammability, solubility and respirability among others) were only due to the presence of impure substances (Hales, 1726).
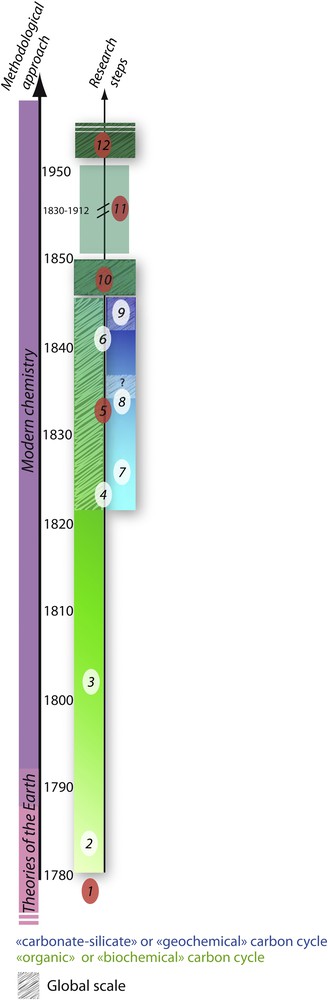
Twelve stages proposed for the historical development of the carbon cycle concept with approximate dates. Each step is detailed in the text under the symbols S1 to S12. White markers correspond to a main event, and red markers correspond to multiple events occurring during the same period. The pale green historical trend corresponds to a period dominated by investigations on the “biochemical” (Albrecht and Ourisson, 1971) or “organic” (Berner, 2003; Berner et al., 1983) carbon cycle. The progressive understanding of biogenic material cycling through photosynthesis and respiration processes as well as organic matter burial were the main findings of this period. The blue historical trend corresponds to discoveries related to the “geochemical” (Albrecht and Ourisson, 1971) or “carbonate–silicate” (Berner et al., 1983) sub-cycles associating silicate weathering on lands and carbonate precipitation in the ocean. The dark green color corresponds to the merging of both of the above-mentioned independent trends that occurred in the late 1840s. Masquer
Twelve stages proposed for the historical development of the carbon cycle concept with approximate dates. Each step is detailed in the text under the symbols S1 to S12. White markers correspond to a main event, and red markers correspond to ... Lire la suite
Les 12 étapes majeures proposées pour la construction du concept de cycle du carbone, avec une indication des dates approximatives de la formation des concepts clés, ainsi que de leur démonstration. Le détail de chaque étape est donné dans le corps du texte. Les étapes indiquées par un marqueur blanc correspondent à un unique événement, celles indiquées par un marqueur rouge correspondent à plusieurs événements majeurs se produisant à la même période. La succession d’étapes le long de la tendance verte pâle correspond à la fondation du cycle du carbone dit « biochimique » (Albrecht et Ourisson, 1971) ou « organique » (Berner, 2003; Berner et al., 1983). Cela correspond au recyclage des matériaux biogéniques à travers les processus du vivant, ou à travers des processus géologiques tels que l’enfouissement des matériaux carbonés. La succession d’étapes le long de la tendance bleue correspond à la découverte des processus du cycle « géochimique » (Albrecht et Ourisson, 1971) du carbone, autrement appelé sous-cycle des “carbonates–silicates” (Berner et al., 1983), comme l’altération des silicates sur les continents suivie de la formation de carbonates dans les océans. La couleur vert sombre indique la réunion de ces deux lignées initialement indépendantes à la fin des années 1840. Masquer
Les 12 étapes majeures proposées pour la construction du concept de cycle du carbone, avec une indication des dates approximatives de la formation des concepts clés, ainsi que de leur démonstration. Le détail de chaque étape est donné dans le corps ... Lire la suite
Unlike S. Hales but making use of his invention, Joseph Black (1728–1799) and Henry Cavendish (1731–1810) argued that among these gases, fixed air “that is extracted from alkaline substances by solution in acids, or by calcinations” (Cavendish, 1766) was different from atmospheric air (Black, 1777; Cavendish, 1766). Fixed air was only a part of the latter because quicklime “exposed to open air […] absorbs particles of water and of fixed air which come within its sphere of attraction” (Black, 1777, p. 54). More importantly, H. Cavendish (Cavendish, 1766, part III, exp. 3–5) showed that both the fermentation of plants and marbles treated with acids yielded the same gas, namely fixed air. With these detailed experiments, H. Cavendish and J. Black greatly contributed to underpin the chemical connections between the mineral and the living world. While fixed air was still considered to be a simple substance, it became called “aerial acid” by the Swedish chemist Torbern O. Bergman (1735–1784) in the first volume of his book Physical and chemical essays (Bergman, 1779, chapter 22–23). T.O. Bergman postulated that aerial acid was part of the atmosphere, but in minute quantities: “pure alkali are made mild in the open air, lose their desliquating quality, and form crystals which effervesce with acids, circumstances which can only be ascribed to their attracting fixed air. Since then, these phenomena all take place at every time, and in every part of the world, a quantity of uncombined fixed air, by no means inconsiderable, must be perpetually present in the atmosphere” (Bergman, 1779, p. 74).
However, this “aerial acid” was not yet chemically identified nor quantified.
2.2 Carbonic acid is a compound and is released by respiration – S1
Antoine Laurent Lavoisier (1743–1794) was a key contributor to the foundation of the carbon cycle. By departing from a long-held opinion among scientists, A.L. Lavoisier argued that “fixed air” (which he termed carbonic acid) was not a simple compound.
In 1777, A.L. Lavoisier presented to the French academy an “hypothesis” with which “we explain […] all the phenomena of combustion, of calcinations”, and that this hypothesis could even account for those “concerning animal respiration” (Lavoisier, 1777b).
A.L. Lavoisier set up a combustion experiment, designed with his collaborators Pierre-Simon de Laplace (1749–1827) and Jean-Baptiste Meusnier (1754–1793), to illustrate his “hypothesis”. He carried out his experiment with “very-exact apparatus and very thorough attentions”(Meusnier and Lavoisier, 1781). Great care was taken to ensure “exactness of the determination of weight” for both the solid and gas phases (Meusnier and Lavoisier, 1781, p. 270). In addition, they worked under the fundamental hypothesis that no solid, liquid or gas compounds included in the bell and involved in the chemical reactions could enter or leave the bell used for experiments, in opposition to the tenets of some contemporaneous scientists defending the Phlogiston theory (Lavoisier, 1774, 1783):
« si l’on brûle du charbon très pur sous une cloche remplie d’air vital renfermé par du mercure, une portion de l’air vital se trouve convertie en air fixe ou acide charbonneux; que si l’on absorbe, par le moyen d’alcali caustique en liqueur, la portion d’acide charbonneux qui s’est formée, le résidu est encore de l’air vital pur, dans lequel on peut brûler une nouvelle quantité de charbon; et, en répétant un certain nombre de fois cette expérience, on peut convertir la totalité de l’air vital en acide charbonneux, sans qu’il reste aucun résidu. »1 (Lavoisier, 1781).
In this paper, A.L. Lavoisier introduced the term carbonic acid (“acide charbonneux”) in place of the old terms “fixed air” or “chalky acid” (Lavoisier, 1781, p. 403), recognizing that the “substance charbonneuse” (or carbonaceous substance, later baptized carbon (de Morvaux et al., 1789)) was present in both coal, “charbon”, and carbonate rocks. A.L. Lavoisier argued that despite his admiration for S. Hales, his findings “forced him” to recognize that “carbonic acid does not exist in plants, but is a product, a result of distillation” (Lavoisier, 1786). For the French chemist “fixed air”, or carbonic “gas”2, was a compound body. He determined that it is composed of “23.4503 part of “principe charbonneux”, and “76.5497 part of ‘principe oxygine’ [see (Lavoisier, 1778)]” (Lavoisier, 1781, p. 407) (later modified to “28 parts of carbonic matter and of 72 parts of principe oxygine” (Lavoisier, 1789)). In the book Traité de nomenclature chimique 3 (de Morvaux et al., 1789), Louis-Bernard G. de Morvaux (1737–1816), A.L. Lavoisier, Claude L. Berthollet (1748–1822) and Antoine de Fourcroy (1755–1809) introduced the term carbon, which is still in use today: « au charbon principe des substances charbonneuses […] nous lui adaptons l’expression simplifiée carbone, qui indiquera le principe pur, essentiel du charbon, et qui a le mérite de spécifier par un seul mot, de manière à prévenir toute équivoque »4 (de Morvaux et al., 1789).
In two major studies (Lavoisier, 1777a, 1785), A.L. Lavoisier published a series of experiments designed to investigate changes induced by animal respiration on the amount of “vital air” contained in atmospheric air. He concluded that respiration is a “slow combustion” (Lavoisier, 1777a, 1777b; Séguin and Lavoisier, 1789) that releases carbonic acid. More importantly, Armand Séguin (1767–1835) and A.L. Lavoisier found that animals could survive longer when quicklime was added in the bell jar as a sink for carbonic acid (Séguin and Lavoisier, 1789). In short, these experiments provided evidence for two “inverse” (Lavoisier, 1777a, p. 180) or “compensating” (Lavoisier, 1785; Séguin and Lavoisier, 1789) processes that counterbalanced one another with respect to their effect on the composition of respired air.
Aware of A.L. Lavoisier's findings, Jean H. Hassenfratz (1755–1827), one of A.L. Lavoisier's young collaborator, struggled to “explain what is the fate of carbonic gas that is daily formed by combustion and respiration, and […][to find a source for] the continual replacement of oxygen gas employed in these various operations” (Hassenfratz, 1792). J.H. Hassenfratz was noting the need for compensating mechanisms that form oxygen. The same year, A.L. Lavoisier speculated on this subject and conjectured on strictly theoretical and/or esthetic grounds that: « si une substance végétale, plus de l’oxygène, forme de l’acide carbonique et de l’eau, il en résulte qu’en enlevant de l’oxygène à de l’acide carbonique et à de l’eau, on doit reformer une combinaison végétale »5.
However, « quoique l’art ne nous fournisse encore aucun moyen d’opérer cette merveille, il n’est pas sans vraisemblance, et l’analogie porte à le croire, que c’est la marche que suit la nature pour la formation des végétaux. »6 (Lavoisier, 1792).
Although this theory did not present “clear evidence”, Lavoisier mentioned the preliminary works of J. Priestley (1733–1804), J. Ingenhousz (1730–1799) and J. Sénebier (1742–1809) regarding a process “that nature surrounded with a thick veil” (Lavoisier, 1786).
Abruptly achieving his intensive researches on the chemistry of life processes, a year before his death, A.L. Lavoisier prepared one of his last manuscript entitled sur le charbon (Lavoisier, 1793) to be included in the unpublished book Dictionaire d’artillerie de l’encyclopédie méthodique of M. de Pommereul, in which he briefly outlines the ubiquity of the element carbon in nature and the importance of the carbonate reservoir:
« Les expériences modernes ne permettent plus de douter que le charbon [meaning here carbon (de Morvaux et al., 1789)] ne soit un des principes des corps et qu’il n’entre en nature dans un très grand nombre de combinaisons des trois règnes […]. Le charbon, d’après cela, doit être considéré comme un principe combustible simple, ou au moins que la chimie n’est point encore parvenue à décomposer, qui forme par sa combustion l’air fixe ou acide carbonique […] On conçoit, d’après cela, quelle immense quantité de charbon se trouve renfermée dans les entrailles de la terre puisque les marbres, les craies, les terres calcaires contiennent environ trois dixièmes et quelques fois même un tiers de leur poids d’air fixe, et que ce dernier est composé pour les 28/100 de son poids de charbon […]. Ces conséquences s’éloignent beaucoup de toutes les idées qu’on avait eues jusqu’à présent; mais elles ne sont pas moins une suite nécessaire des expériences qu’on vient de rapporter. […] Nous ne suivrons pas ici les changements de forme que prend le charbon en passant du règne minéral dans le règne végétal et dans le règne animal. Nous nous jetterions dans des discussions chimiques étrangères à l’objet de cet article »7 (Lavoisier, 1793).
The above passage illustrates that the concept of C reservoir, especially the carbonates in the crust (Des Marais, 2001; Hayes and Waldbauer, 2006), might have been sketched just before the 1800s. This aspect forms a critical component of our current understanding of the carbon cycle.
Shortly after these diverse seminal works by A.L. Lavoisier, the English chemist Humphrey Davy (1778–1829), both a fervent admirer and a critic of A.L. Lavoisier achievements, reformulated the question of the compensating mechanism for respiration in 1799: “Since the atmospheric composition is uniformly similar, we are led to inquire by what means a quantity of phosoxygen equal to that consumed by respiration and combustion is again supplied to the atmosphere.” (Davy, 1799b, p. 152). For H. Davy, this “inquiry is no less important than curious, as our existence itself depends on the equilibrium of gases in the atmosphere” (Davy, 1799b, p. 151–152).
In the following section, we discuss the discovery of the compensating mechanism on a global scale.
3 The role of life on atmospheric composition
3.1 Plants are “repairing” the respirability of atmospheric air – S2
As respiration was being discovered, several botanists and chemists – notably, Charles Bonnet (1720–1793), J. Priestley, J. Ingenhousz and J. Sénebier–showed the purifying role of plants with respect to atmospheric gases, “repairing” (Sénebier, 1800, p. 277) the “alteration that thousands of causes make on the air that we breath” (Sénebier, 1800). Extensive reviews on these early works can be found in Dumas and Boussingault (1841). In particular, the findings of J. Ingenhousz and J Sénebier benefited from the invention of the Eudiometer. The basic application of this apparatus is to evaluate the volume of a gas consumed or released by the displacement of a fluid in a jar through a reaction of the gas to be analyzed with nitrous oxide. By immersing green parts of land plants (e.g., houseleeks) into water filled vessels under various conditions of illumination (Ingenhousz, 1779) and water composition (Sénebier, 1783), J. Sénebier and J. Ingenhousz showed that the decomposition of atmospheric carbonic acid “released oxygen as a by-product that is useless for him” (Sénebier, 1783) under light. Soon after these early investigations and numerous European studies dedicated to this compensating mechanism, H. Davy published the result of a series of 9 experiments showing, among other findings, the “discovery of the production of phosoxygen by the various orders of the marine cryptogamia class of plants” (Davy, 1799b, p. 154) that has been grown in the laboratory using sea water. Thus H. Davy explicitly introduced the question of compensating processes in the ocean and used the term “carbon” that had been recently published in the chemical nomenclature (de Morvaux et al., 1789): “Animals may be considered as absorbing in their respiration phosoxygen, and as taking in their nutriment, hydrogen, carbon, nitrogen, and oxygen, and giving out perpetually these principles in new combinations; water, carbonic acid, and probably ammonia and electric fluid. On this perpetual series of changes there life appears to depend. Vegetables, on the other hand, are continually absorbing water, carbonic acid, and probably ammonia and nitrogen, and assisted by light in the exercise of their organic functions giving out phosoxygen. […] The vegetation of land plants then may be considered as the great source of the renovation of the atmosphere of land animals. […] As land vegetables are the renovators of the atmosphere of land animals, analogy led me to suppose that sea vegetables must be the preservers of the equilibrium of the atmosphere of the ocean” (Davy, 1799b, pp. 164–166).
In close association with his friend J. Sénebier, the Italian geologist and physiologist Lazzarus Spallanzani (1729–1799) was investigating the possibility of natural waters to be an alternate compensating sink for the carbonic acid that was released by respiration. L. Spallanzani concluded that “for modern physicists and chemists, there are two major means used by nature to purify the atmosphere and to free her from the impurities that should bring her the great quantity of carbonic acid produced by human and animal respiration, by fermentation and by combustion: these two means are the waters and plants. Waters produce this effect by their affinity for carbonic acid, that they absorb continually, and plants by the oxygen gas that they spread continually, when they are covered by the intense sunlight” (Spallanzani, 1809b, p. 5).
However, 16 of L. Spallanzani experiments translated and published posthumously in 1809 by Jean Sénebier (Spallanzani, 1809b) discarded the hypothesis of water acting as a carbonic acid sink. The Italian physiologist came to favor the “more powerfull method” (Spallanzani, 1809a) of plants to reproduce the portion of oxygen lost during respiration, giving much credit to the “brilliant discoveries” of J. Priestley, J. Ingenhousz and J. Sénebier. These early works reveal that the focus of these early researches were on processes acting on the composition of the atmosphere. In this context, researchers such as A.L. Lavoisier, L. Spallanzani, J. Priestley or J. Sénebier were using the terms “compensating”, “repairing”, “returning” to refer to these counterbalancing phenomena.
3.2 N.T. de Saussure's experiments on plant C fixation – S3
“The observations of MM Priestley, Sénebier, and Ingenhousz started the career that I realized, but they didn’t reach the goal that I envisioned”. These words by Nicolas T. de Saussure (1767–1845), the son of the famous Swiss physicist and alpine geologist Horace-Benedict de Saussure (1740–1799), formed the opening lines of his famous book Recherches chimiques sur la végétation (de Saussure, 1804, p. iv). After the discovery that green parts of plants immersed in acidified water were decomposing “fixed air” and releasing pure air (oxygen), N.T. de Saussure indisputably quantified the process associated with carbonic acid decomposition by plants. His experiments were performed in bell jars closed with mercury. In contrast to J. Sénebier, N.T. de Saussure used a phosphorus eudiometer to reach a “degree of precision that the nitrous gas eudiometer employed by the other authors could not attain” (de Saussure, 1804, p.iv) in the quantification of oxygen in the produced gas. He tested different experimental procedures on various plant species that he reported in his book Recherches chimiques sur la végétation with much detail (de Saussure, 1804).
To “verify the first conjectures of Percival”, (de Saussure, 1804) which reported experiments regarding the role of carbonic acid on the growth of mint leaves (Percival, 1785), N. T. de Saussure designed and carried out experiments in which fresh plants were immersed in distilled water with their leaves inside a bell jar filled with artificial atmospheric air containing between 6 and 10% of carbonic acid. The jar was closed with mercury overlaid by a “thin layer of water” to avoid reactions between the air and mercury. Using this procedure, N.T. de Saussure first showed that the composition of atmospheric air “does not contain more than 0.005% by volume of carbonic acid” (de Saussure, 1804, chap. 2). By comparing dry and calcined mass gain or loss of the plant material, as well as by analyzing the volume loss and gain of the different gases inside the jar after 5 to 18 days of experimentation, he also showed that carbonic acid was absent in the final gas and that the volume of oxygen added to the original gas was lower than that expected from the carbonic acid decomposition. Therefore he concluded from these experiments that part of the oxygen, and all of the carbon from the carbonic acid was assimilated to form the biomass (Fig. 2, S3). Consequently, he opposed the proposals of J. Sénebier (Sénebier, 1783, 1800) and H. Davy (Davy, 1799a, 1799b) regarding the fate of oxygen during the decomposition of carbonic acid by plants. N.T. de Saussure also completed H. Davy early experiments (Davy, 1799a, 1799b) by demonstrating through rigorous quantification, that water was decomposed during plant growth and that oxygen was released during the process. Finally, based on experiments regarding the fermentation of wood, he argued that atmospheric oxygen removes carbon from plant debris and transforms it to carbonic acid, thereby augmenting the proportion of carbon in the ligneous residue (de Saussure, 1804, p. 149). This is an important contribution to the understanding of CM oxidation as a part of global carbon circulation.
The works of N.T. de Saussure were recognized by the subsequent generation of geologists for describing a global “rotation” of carbon, an “admirable law of nature” (Ebelmen, 1845) by which carbon was transferred from the atmosphere to the plants and returned back to the atmosphere through respiration or oxidation, although N. T. de Saussure himself did not explicitly refer to the terms “compensating” or “repairing” processes.
3.3 Adolphe Brongniard, CM burial and the origin of coal – S4
The aforementioned advances laid the groundwork for further developments towards the understanding of carbon transfer on a global scale.
An attempt to synthesize the most recent findings in a global perspective occurred in the 1820s (Fig. 2, S4) by the French geologist and botanist Adolphe Brongniard (1801–1876), the son of the renowned geologist Alexandre Brongniard (1770–1847). One of Adolphe Brongniard's tenet was that “geologists should also make use of botany”, as this field might provide information on the state of the Earth during ancient times (Brongniard, 1828). By 1828, he recognized the global “purifying” effect of vegetation, and suggested that atmospheric air could have contained more carbonic acid in the past. His reasoning was based on the discovery of ancient plant remains in coal-rich geological formations from Germany, Switzerland and France (e.g. deposited during Carboniferous times). Adolphe Brongniard proposed that the “accumulation of carbon” (Brongniard, 1828) in the form of “coal, lignite and bitumen” in the sedimentary strata could have pumped carbonic acid from the atmosphere and purified it to eventually favor the emergence of animals that require oxygen (Brongniard, 1828). Although the biogenic origin of coal was clear to Adolphe Brongniard in 1828, the question of the biogenic (or organic) vs. inorganic origin of coal remained fiercely debated throughout the 18th century (Stevenson, 1911). The biogenic origin of coal was defended by De Buffon (1707–1788) in France and Franz von Beroldingen (1740–1798) in Switzerland (Scott, 1998; Stevenson, 1911), and it became accepted throughout continental Europe by 1780. Coal's biogenic origin was first accepted in Britain by James Hutton (1726–1797), and even became a cornerstone of his theory of the Earth by 1795 (Allchin, 1997; Hutton, 1795). Opposed to many of Hutton's propositions, the geologist and mineralogist Joseph Kirwan (1796–1849) challenged this hypothesis which he recognized as the “most prevailing opinion”, and developed his own hypothesis based on the “important discovery” made by A.L. Lavoisier that “[carbon] was a constituent part of fixed air. […] This knowledge being gained, we must, of course, allow that carbonic substances are of equal antiquity with fixed air, and this air being a component part of many of the solid substances [i.e. carbonates] which form the great mass of the globe, it is equally evident that it must be coeval with those substances, and, consequently, must have preceded the existence of animals and vegetables” (Kirwan, 1799, pp. 315–316). He grounded this “abiotic” theory (would currently call it) for the origin of coal strata on the fact that granitic rocks “sometimes contain small veins of coal” (Kirwan, 1799). Nevertheless, he also admitted that some coal, such as those “found under traps” were of vegetable origins formed during a major flooding event (Kirwan, 1799, pp. 324, pp. 340–341).
By the time Adolphe Brongniard published his work, the biogenic origin of coal was largely admitted, even though the details of its formation were not fully understood (drift versus autochtonous origin; see Stevenson (1911) and Scott (1998)). However, by the 1820s, a clear connection between the evolution of the atmospheric composition and the burial of CM in the strata had been proposed and grounded based on the scientific knowledge of the time.
4 Geological phenomena influencing the composition of the atmosphere
By the end of the 18th century, L. Spallanzani, influenced by A.L. Lavoisier's theory of matter8, formulated an original idea on the origin of volcanic phenomena (Spallanzani, 1796, ch. 21). L. Spallanzani suggested that the liberation of water or carbonic acid fixed in minerals sustains the most frequent and enduring gas emissions. In this process, if “everything is linked in this universe”, volcanoes would be, “according to H.B. de Saussure”, “like vents” that would “restitute these elements to the general circulation” (by J. Sénebier in the preface of L. Spallanzani's Voyages dans les deux siciles et dans quelques parties des appenins (Spallanzani, 1795)). This mention is evidence that the idea of a “general circulation” of elements, although not completely understood at that time (including the deep transfer to subduction zones), was clearly emerging by the end of the 18th century.
4.1 Volcanic gases are rich in CO2–S5
The “impossibility to collect the fluids” (Spallanzani, 1796) that escaped from the crater of the volcano Stromboli prevented L. Spallanzani and other naturalists from knowing their exact nature (Spallanzani, 1795). In the 1820s, J. B. Boussingault (1802–1887), a young French chemist from the Ecole des mineurs de St Etienne working as a mining engineer in the Andes, carried out sampling and analysis of volcanic exhalations. These measurements were precise and aimed to ascertain “the identity of these gases” (Boussingault, 1832b), identify the “chemical causes” at the origin of the volcanic phenomenon and verify some of the earlier hypotheses that were formulated by H. Davy (Boussingault, 1832b; Davy, 1808). J.B. Boussingault was also motivated to verify Laplace's ideas that volcanic phenomena were linked to the temperature rise inside the Earth (Boussingault, 1832b). He specifically focused on 6 volcanoes from the equator (Table 1) and reported his findings in the Recherches chimiques sur la nature des fluids élastiques qui se dégagent des volcans de l’équateur” (Boussingault, 1832b) as well as in the Considérations sur les eaux thermales de la cordière (Boussingault, 1832a), where he also reported the chemical analyses of thermal waters.
Résultats des analyses de gaz volcaniques issus de 6 volcans andins par J.B. Boussingault (Boussingault, 1832b).
| Volcanoes | Altitude (m) | Temperature of the gases (°C) | CO2 (%)a | Otherb |
| Tolima | 4300 | 50 | 0.14 | H2S, H20 |
| Azufral du Quindiu | 2500 | 19–20 | 95 | H2S |
| Puracé | 4359 | nc | 85 | H2S, H2O |
| Pasto | 4100 | 90 | 78 | |
| Tuquères (lac vert) | 3908 | 86 | ∼100 | H2S |
| Cumbal | 4761 | nc | ∼100 | H2S |
a Test with quicklime.
b Test with lead acetate for H2S, condensation on glass at 0 °C for water, and considered as non atmospheric air (measured using phosphorus eudiometer).
To sample the gases, J.B. Boussingault identified cracks where vapors escaped and positioned “graduated tube(s) filled with water”, which were completely replaced by the evolved gas. J.B. Boussingault measured the composition of the gas directly in the field by performing tests with caustic soda (for carbonic acid detection) and lead acetate mixed with acetic acid (for H2S detection), and he used a phosphorus eudiometer (for oxygen detection) in the laboratory (Boussingault, 1832b). The results of these investigations are presented in Table 1 and show that carbonic acid could account for over 90% by volume of the sampled gases. Combining the gas and thermal water analyses, he concluded that both the gas and water have “their source in the volcanic furnaces”, and that they are composed of “carbonic acid gas and some hydrosulfuric acid gas” (Boussingault, 1832a, p. 186) (Fig. 2, S5). The results of J.B. Boussingault were recognized throughout Europe as demonstrating the high content of carbonic acid in volcanic gases (Bischof, 1839; Ebelmen, 1847). J.B. Boussingault eventually proposed that the thermal destabilization of carbonates at depth, possibly favored by the presence of Si-rich minerals, could be the source of carbonic acid released by volcanoes:
« Ainsi, l’acide carbonique peut être considéré comme un produit de carbonate de chaux ou de soude. Dans le premier cas, la chaleur seule suffirait pour donner lieu au dégagement de l’acide; dans le second, indépendamment d’une haute température, il faudrait encore que le carbonate alcalin fût-il en contact avec une substance siliceuse ou alumineuse, par exemple avec la roche trachytique elle-même. »9 (Boussingault, 1832a).
4.2 Boussingault pioneering vision on the geological transfer of C – S5, S6
Similar to Adolphe Brongniard, J.B. Boussingault intended to summarize the different processes that would modify the composition of the atmosphere in his book Recherches sur la composition de l’atmosphère, published in 1834 (Boussingault, 1834). He formulated the idea of a global atmospheric regulation process that involved different counteracting processes (Fig. 2, S5). For J.B. Boussingault, among the “many causes tend[ing] to annually modify the composition of the atmosphere […] one of them that occur at the surface of the Earth is due to living beings. Organized beings […] are taking part of the carbon that is in the air in the form of carbonic acid. In plants, this assimilation occurs directly” (Boussingault, 1834). This statement was based on the experiments of the physiologists mentioned above, as J.B. Boussingault was aware of the “important works of Theodore de Saussure on carbonic acid of air” (Boussingault, 1834). As for the animals, “this is by eating plants that they assimilate this principle. […] This is the atmosphere that must be considered as the unique source where organic matter draws the carbon that enters in its composition” (Boussingault, 1834). Thus, having recognized the chemical action of plants on the atmosphere, and that atmospheric CO2 is the source of carbon for all living beings, he noted the antagonistic role of respiration and CM degradation with respect to plants action:
« Aussitôt que cesse l’action vitale, il se produit des phénomènes chimiques d’un ordre opposé à ceux qui ont contribué à l’assimilation ; la décomposition commence […] dont le résultat final est de restituer à l’atmosphère le carbone qui lui avait été enlevé. Si la quantité de matière vivante reste la même à la surface du globe, cette matière n’exerce en définitive aucune influence sur la proportion de l’acide carbonique atmosphérique »10 (Boussingault, 1834).
J.B. Boussingault is most likely one of the first scholars since the early conjectures of H. Davy (Davy, 1799a) to postulate a possible steady state between the activity of plants and respiration at the global scale, a hypothesis that neither L. Spallanzani nor A.L. Brongniard favored. He also described catastrophic events of coal burial that would cause a strong imbalance in this dynamic equilibrium over geologic time scales by storing large amounts of carbon: « Le seul cas où la matière organique pourra diminuer le carbone de l’atmosphère, serait celui où une catastrophe ferait disparaître subitement une grande masse d’êtres organisés. Telle a due être la cause qui a enfoui à diverses époques géologiques une partie de la végétation qui existait à la surface du sol. Le carbone qui forme la base des immenses dépôts de houille et de lignite a fait partie sans aucun doute de l’acide carbonique de l’atmosphère »11
Delving deeper into his attempt of a global synthesis and following A.L. Brongniard without subscribing to his views, J.B. Boussingault asked whether these antagonistic phenomena might have had an effect on the long-term evolution of the atmospheric composition:
« Toutefois, nous ne pouvons pas tirer de là la conséquence que l’air contint autrefois plus de cet acide qu’il n’en renferme à présent, parce qu’il existe sans doute différentes causes qui tendent à augmenter la proportion de l’acide carbonique dans l’atmosphère. »12 (Boussingault, 1834).
Indeed, he argued on the existence of an “energetic” antagonistic cause that would be “purely geological”, and would “depend on the intimate constitution of our globe” (Boussingault, 1834), and he described the volcanic exhalations of carbonic acid as that he had initially witnessed in the Andes:
« Il sort constamment de l’intérieur de la Terre des gaz qui se répandent continuellement dans l’atmosphère. Les volcans peuvent être considérés comme des soupiraux qui exhalent sans interruption aucune du gaz acide carbonique. »13 (Boussingault, 1834, p. 174).
Thus, J.B. Boussingault was not only well aware of the most recent findings on the role of plants on the atmospheric composition, but he also favored the biogenic origin of coal strata and recognized the role of CM burial and preservation on the composition of the atmosphere. Finally, he was the first to categorically identify the need to consider an additional geological cause: volcanic gases that tend to modify the carbonic acid content of the atmosphere.
Two syntheses of the recent findings on the role of plants and animals in the transfer of carbon at the global scale were published simultaneously in 1841 (Fig. 2, S6) by J.B. Dumas and J.B. Boussingault (Dumas and Boussingault, 1841) and the german chemist Justus Liebig (1803–1873) (Liebig, 1841). In their work, J.B. Dumas and J.B. Boussingault described the synthesis of biogenic material by the following reaction (the equation is derived by making use of the chemical nomenclature used by the authors following their own description of the reaction, from Dumas and Boussingault, 1841, pp. 25), 12 CO2 + 10 H2O -> C12(H2O)10 + 12 O2. However, despite tremendous progress in the understanding of the chemical regulation of atmospheric composition and of the transfers of carbon at the global scale, the complete links between “geochemical” processes and the transfer of carbon at the global scale were not yet understood or quantified.
5 The alteration of silicates and the geochemical carbon rotation
The aforementioned considerations showed that a global vision of the “circulation” of carbon on the Earth was emerging at the beginning of the 19th century. It was mainly derived from the discovery of the sources and sinks of atmospheric carbonic acid. Meanwhile, and most likely because of advances in the understanding of the role of life in the atmospheric composition, new ideas on the processes acting on long time scales started to emerge. Historically, two parallel approaches have included the role of atmospheric carbonic acid in the study of water-rock interactions on the surface of the Earth. The first approach involved the chemical analysis of mineral waters, and the second involved the study of the solid residues of weathering.
5.1 The dissolving power of carbonic acid: the aqueous and residual solid approach – S7, S8
The corrosive nature of acids had been known for many years. It had been explored by many scientists, and in particular by the French chemist Guillaume François Rouelle (1703–1770) in his Mémoire sur les sels acides published in 1752. He defined the notion of a base by stating that an acid must react with a base to form salts. Therefore, the idea that carbonic acid was able to react with the bases (the alkalis) present in rocks was already established at the beginning of the 19th century when John Dalton wrote “carbonic acid combines with alkalis, earths and metallic oxides and forms with them salts called carbonates” (Dalton, 1808, section 3). Further advances came from technical progress on the chemical analysis of waters, a field where the German chemist and geologist Gustav Bischof (1792–1870), professor at Bonn University and disciple of the father of neptunism Abraham G. Werner (1750–1817), exerted a crucial influence. He published his book entitled “Die vulkanischen Mineralquellen Deutschlands und Frankreichs” in 1826 in which, based on the analysis of mineral waters from diverse volcanic provinces in Germany and France and analyses reported by the famous Swedish chemist J.J. Berzelius, he concluded that carbonic acid, which is released to the atmosphere by the numerous “volcanic exhalations” of the investigated provinces, reacts chemically with the rocks to dissolve substances found in mineral waters and to form a residue depleted in the alkali elements:
“Die gewöhnliche Art der Zersetzung, die wir bei dem Feldspath bemerken, namentlich die des Feldspaths im Granit, scheint durch die unmittelbare atmosphärische Einwirkung veranlaßt zu sein.”14 (Bischof and Noggerath, 1826).
G. Bishof also understood that the acidic character of water, due to the dissolution of carbonic acid, was responsible for the progressive decomposition of feldspars, “so wird sie jene verbindungen nach und nach zersetzen, und mit den alkalien verint”15 (Bischof, 1826).
The famous German geologist Abraham G. Werner (1750–1817), one of the founders of the neptunist theory (Gohau, 2003; Rudwick, 1985), was G. Bishof's professor while at Bonn university. A.G. Werner developed an original theory on the origin of volcanoes due to the combustion of coal and pyrite layers at depth in the presence of water (Girardin, 1831). He also mentioned the role of carbonic acid in the formation of filons in 1791 (Werner, 1809, for the English translation). Whether A.G. Werner's ideas influenced G. Bischof may be suggested, but whatever his influences might have been, G. Bischof largely contributed to the discovery of chemical weathering as we describe it today, a process that releases most of the soluble elements from the rocks (Fig. 2, S7).
In addition to this approach based on the analysis of water, another approach was being developed that involved the study of kaolin. Indeed, at the end of the 18th and the beginning of the 19th century, kaolin was a rock of high economic value because it was used (by chinese in particular) for making porcelain. The first kaolin ore in Europe was found in Meissen, Germany. In France, Jean Etienne Guettard (1715–1786) discovered kaolin near Alençon in 1760, but its origin was still controversial at the beginning of the 19th century when the first detailed chemical analyses were performed (Adolf Ferdinand Gelhen (1775–1815) in Germany, Pierre Berthier (1782–1861) and Alexandre Brongniard (1770–1847) in France). It was clearly recognized that kaolin and granitic rocks coexisted in nature, but the processes governing the transformation of granite into kaolin was not yet well understood. At that time, the “galvanic” theory, initially proposed by A.F. Gelhen and supported by the French geologist Al. Brongniard (Brongniard and Malagutti, 1841), was popular. This theory attributed the alteration of granitic rocks to the action of electric currents in the crust. Challenging this “tendency to generalize unduly the application of galvanic forces” (Fournet, 1833), Joseph Fournet (1801–1869) gathered chemical analyses from Adolphe Ferdinand Gehlen (1775–1815) in Germany and Pierre Berthier (1782–1861) from the École des Mines de Paris, as well as his own chemical analyses of kaolin samples provided by Alexandre Brongniard (1770–1847) and published his results in 1833 (Fournet, 1833). According to J. Fournet, weathering was accompanied by a loss of bases such as potash, magnesia, lime and iron oxide, and kaolin could be defined as an hydrated alumino-silicate that was the natural end product of the systematic decomposition of the bedrock irrespective of the initial bedrock composition (Fournet, 1833). The mechanism he proposed involved dimorphism as a preliminary mechanical agent of disaggregation followed by the chemical action of water and carbonic acid. Indeed, echoing the studies of G. Bischof several years prior (Bischof, 1826, 1851), J. Fournet stated:
« si nous devons recourir à l’intermède d’un acide, nous verrons que le seul qui soit assez universellement répandu dans la nature auquel on puisse rapporter cet effet est l’acide carbonique »16 (Fournet, 1833, p. 250).
Although these studies highlighted the potential role of carbonic acid on silicate weathering, no clear link between silicate weathering and the chemical composition of the atmosphere was described by J. Fournet. However, he mentioned in his paper that the work could have some “possible geological consequences” (Fig. 2, S8) (Fournet, 1833), but this step was definitively achieved by Jacques Joseph Ebelmen (1814–1852) in the following decade. J.J. Ebelmen was a young and renowned chemist, adjunct professor in the docimasy laboratory of Pierre Berthier (quantitative ore analysis) at the École des Mines de Paris (from 1845 to 1852), and adjunct-administrator of the Manufacture de Porcelaine de Sèvres (from 1845 to 1852) at a time when outlines of the C transfers at the Earth scale was being described by J.B. Boussingault (Boussingault, 1834), J. Liebig (Liebig, 1841) and J.B. Dumas (Dumas and Boussingault, 1841). J.J. Ebelmen expanded upon some mineralogical processes affecting of the global carbon transfers.
5.2 From the art of docimasy to the role of chemical weathering on atmospheric composition – S9
J.J. Ebelmen wrote four main geological papers (Ebelmen, 1845, 1847, 1848, 1851), three on the decomposition of Ca-Mg silicate minerals and basaltic rocks by atmospheric agents, and the fourth on the decomposition of “stratified” (sedimentary) terranes. More than half of his first geology paper (Ebelmen, 1845) reported detailed analyses of rocks made under rigorous and thoroughly detailed analytical protocols, which were developed within the docimasy laboratory of the École de Mines. For example, he introduced the double normalization procedure to compare the unweathered bedrock with the weathered material (Fig. 3), a procedure widely used in modern geochemistry. This procedure is based on the observation that alumina is present in higher concentrations in the weathered parts of basalts and that it is thus immobile during rock decomposition. By normalizing all elemental concentrations in the weathered basalts by those in the unaltered bedrock, he classified the elements according to their mobile or immobile characteristics during water-rock interactions. Fig. 3 illustrates graphically the results that he obtained for a basaltic rocks from Linz (Germany) affected by spheroidal weathering. This diagram shows that lime, magnesia and iron oxide are released from the basalt together with silica, soda and potash. In addition, by analyzing the silicate minerals deprived of soda and potash (e.g., pyroxene or garnet minerals and weathered rind), he demonstrated that silica is mobilized and that this mobility does not depend on the presence of alkalis. In contrast to J. Fournet and G. Bischof, J.J. Ebelmen showed that soluble alkali-silicates do not exist and that alkalis are soluble. Along with his analyses of natural samples, he observed that the evaporation to dryness of mineral waters only lead to a mixing of carbonates and silica, which supported the hypothesis of G. Bischof and J. Fournet that the decomposition of silicate minerals is only possible by the action of carbonic acid present in significant proportions in soil waters.
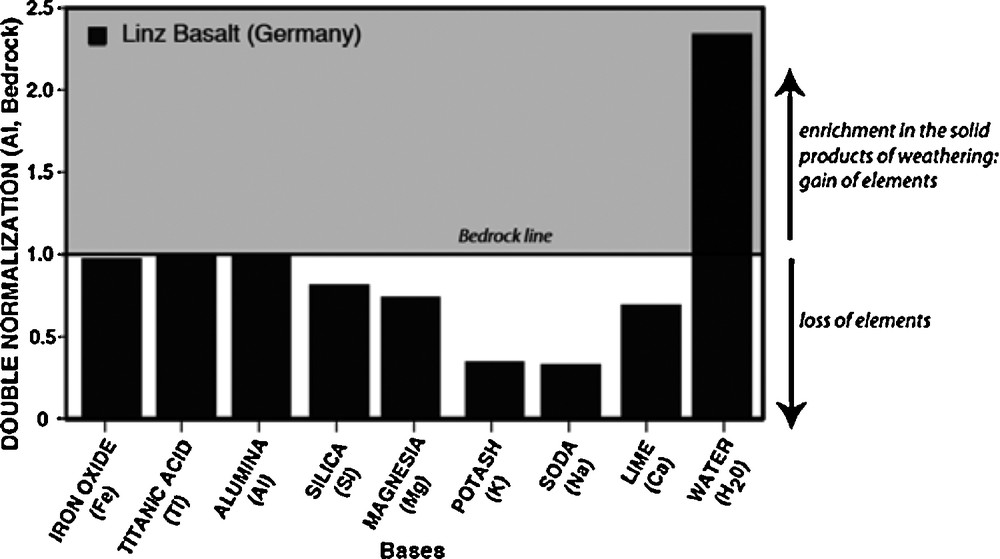
Graphic representation of Ebelmen's analyses of the Linz Basalt in Germany and its weathering rind. Bases are denoted according to Ebelmen's vocabulary; in parenthesis is the modern equivalent. Concentrations are first divided by the concentration of aluminum (Al), which is considered as an immobile element, and then divided by those in the bedrock. Values higher than 1 indicate enrichment in the weathered rind. Values lower than 1 indicate depletion of the same.
Représentation graphique des analyses d’Ebelmen réalisées sur le basalte de Linz en Allemagne (et ses auréoles d’altération). Les bases sont représentées conformément au vocabulaire d’Ebelmen. Un équivalent moderne est donné entre parenthèses. Les concentrations sont tout d’abord divisées par celle de l’aluminium (Al) considéré immobile, et ensuite divisées par celles de la roche non altérée. Les valeurs plus grandes que 1 signifient un enrichissement relatif de l’élément considéré, celles plus faibles que 1 correspondent à un appauvrissement relatif de l’élément considéré par rapport à l’aluminium. Masquer
Représentation graphique des analyses d’Ebelmen réalisées sur le basalte de Linz en Allemagne (et ses auréoles d’altération). Les bases sont représentées conformément au vocabulaire d’Ebelmen. Un équivalent moderne est donné entre parenthèses. Les concentrations sont tout d’abord divisées par celle ... Lire la suite
In contrast to the previous works of Al. Brongniard (Brongniard and Malagutti, 1841) and J. Fournet (Fournet, 1833) that J.J. Ebelmen refers to (Ebelmen, 1845), the aim of the French chemist was to interpret the chemical investigations made on kaolin and other materials in a broader perspective and demonstrate the “geological importance” (Ebelmen, 1845, 1847) of these natural phenomena of chemical weathering mentioned earlier by J. Fournet. By comparing the chemical composition of the igneous and so-called “stratified rocks” (sedimentary rocks), he concluded that each component present in the igneous rocks were also present in the sedimentary rocks, such as lime and magnesia in limestone and dolostone, respectively. However, he noticed that the sedimentary rocks contained carbonic acid as an additional component to igneous rocks. The author concluded that the only possible origin of stratified rocks at the surface of the Earth was the alteration of igneous rocks with geochemical sorting of the elements followed by the mechanical transport of the soluble products to the ocean (Ebelmen, 1845). He envisioned coupling this vast conveying process with the “alteration” of atmospheric air, namely, the consumption of carbonic gas and oxygen during chemical weathering. He writes:
« Les carbonates alcalins rencontreront, soit dans les eaux douces, soit dans la mer, des sels solubles à base de chaux et de magnesie, et produiront, en définitive, des carbonates de ces deux bases qui se décomposeront à leur tour, spontanément ou par l’intermédiaire des zoophytes et des mollusques. Les bases, qui tout à l’heure se trouvaient combinées à la silice dans les roches ignées ont précipité de l’atmosphère une proportion équivalente d’acide carbonique. »17 (Ebelmen, 1845).
This last sentence corresponds to what is known today as the forward pathway of the “Urey reaction” (Urey, 1952),
CaSiO3 + CO2 → CaCO3 + SiO2
Although J.J. Ebelmen did not translate it into a chemical equation (the chemical formula of CO2 and other compounds were already known by that time (Dalton, 1808)). In contrast to G. Bischof and J. Fournet, J.J. Ebelmen proposed that carbonic acid that was neutralized by chemical weathering was derived from the decomposition of biogenic material in soils.
5.3 Attempts toward a quantification of chemical weathering – S10
In 1847, J.J. Ebelmen's second paper aimed at quantifying the consumption of atmospheric carbonic acid during silicate weathering on a global scale. To reach this goal, he used the chemical notations introduced by J.J. Berzelius (Berzelius, 1819, 1835) and the accepted mineral stoechiometry of that time. Both the goal and the method used were clearly pioneering. J.J. Ebelmen calculated that 195 kg of carbonic acid was necessary to chemically weather 1 m3 of orthose (“12SiO2 Al2O3 KO”) assuming that all potassium would be released into solution. Considering an orthosic planet as the simplest hypothesis, he found that a 7 mm thick orthose layer covering the surface of the Earth would be sufficient to completely deplete the atmosphere from its carbonic acid content (using the known concentrations at that time). The same calculation revisited today with proper stoechiometry and atmospheric concentration would lead to an orthose layer only twice as thick, demonstrating the accuracy of his reasoning (Fig. 4).
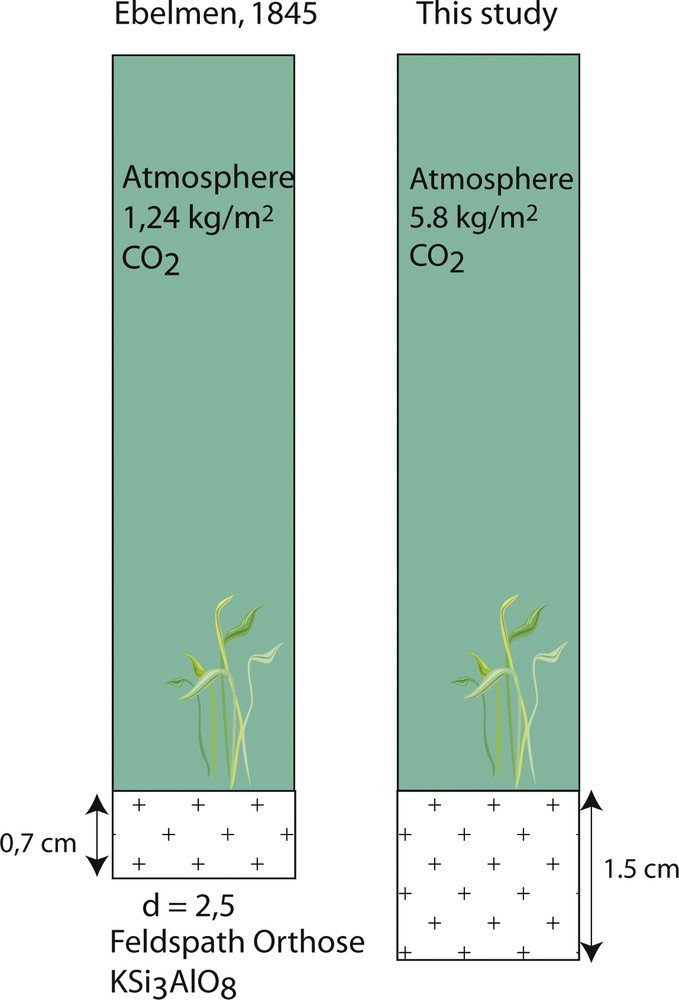
Thickness of an orthose layer exposed to the atmosphere necessary to consume the total amount of CO2 in the atmospheric column (1 m2). The values estimated by J.J. Ebelmen in 1845 are compared to those obtained using modern chemical formulas and CO2 content of the atmosphere and show that the order of magnitude is similar.
Représentation de l’épaisseur de la couche d’orthose exposée à l’atmosphère nécessaire pour consommer l’intégralité du CO2 atmosphérique (m2). Les calculs d’Ebelmen sont comparés aux résultats obtenus en utilisant les formules chimiques des minéraux et la concentration de CO2 atmosphérique moderne, et montrent que l’ordre de grandeur du résultat est similaire.
Under the same assumption of an orthosic planet, J.J. Ebelmen showed that the formation of a 500 m thick layer of kaolin – the mean thickness of siliciclastic sediments at the surface of the Earth, according to his senior colleague, the geologist Elie de Beaumont (1798–1874) – would necessitate 150 × 103 kg of carbonic acid per m2 of rocks, equivalent to 125 × 103 times the amount presumed to be present in the atmospheric air. Therefore, he argued that a counterbalancing process was necessary at the surface of the Earth to maintain the carbonic acid concentration of the atmosphere. Acknowledging the findings of J.B. Boussingault, which were discussed earlier (Boussingault, 1832a, 1832b, 1834), he proposed that the volcanic exhalations could constitute the source of the carbonic acid. The issue of the ultimate origin of the carbonic acid from volcanoes was not discussed in detail by J.J. Ebelmen; however, he ultimately concluded that:
« Quoi qu’il en soit de l’origine des gaz émis par les volcans, il est certain que c’est un phénomène considérable dont on conçoit que les effets puissant contrebalancer les résultats de l’absorption du même gaz dans la décomposition des roches. Ces faits font ressortir la liaison intime que tous les grands phénomènes de la nature ont les uns avec les autres »18.
He carefully added at the end of his paper that:
« rien n’établit que les causes variées qui tendent à modifier la composition de l’atmosphère aient en ce moment une résultante nulle »19
(Ebelmen, 1847, p. 653).
Thus, with great care, and echoing the thoughts of J.B. Boussingault (Boussingault, 1834), J.J. Ebelmen addressed the stability (homeostasis) of the atmospheric system and clearly established this question as being unsolved yet despite its foremost importance.
5.4 From atmospheric composition to the geological rotation of C – S10
In the 1820s, as illustrated by the works of Adolphe Brongniard and J.B. Boussingault, and until 1847, some geologists and early “geochemists” like J.J. Ebelmen were attempting to identify processes that were possibly counteracting one another and thus modifying the composition of the atmosphere (Fig. 5) and its equilibrium. However, to our knowledge, no clear mention of a global rotation of C was ever mentioned until the 1840s, with the initial synthetic works of Dumas and Boussingault (Dumas and Boussingault, 1841), J. Liebig (Liebig, 1841), and especially J.J. Ebelmen (Ebelmen, 1845, 1847). Furthermore, no attempt at the quantification of the problem had been made before that time since the early speculations of A.L. Lavoisier (Lavoisier, 1793). Indeed, these steps could not have been achieved without the foundation and wide diffusion of an atomistic theory of matter that was developed throughout the early 19th century. Accompanying the progressive adoption of A.L. Lavoisier's concepts, the combined works of the French chemist C. L. Berthollet (Berthollet, 1803), the English chemist John Dalton (1766–1844) (Dalton, 1808), and their Swedish colleague J.J. Berzelius (Berzelius, 1835) contributed heavily to these new developments. For example, in 1808, based on experiments he performed with his friend and physicist William Henry (1775–1836), J. Dalton showed that “carbonic acid appears to be a ternary compound, composed of one atom of charcoal, and two atoms of oxygen” (Dalton, 1808), to which J.J. Berzelius gave the chemical formulae “CO2” (e.g., Berzelius, 1819). After the landmark publication by J. Dalton (Dalton, 1808), the processes and laws governing the distribution of the elementary components of matter called “elements” or “atoms” between living or mineral bodies, their rule and pattern of combinations became a primary focus of many “scientists” such as J.J. Berzelius (Berzelius, 1835): “chemistry should provide us the knowledge of the elements and their combination”, irrespective of their presence in living or mineral bodies (Berzelius, 1819). As a consequence, J.J. Berzelius introduced and popularized the use of chemical formula (e.g., for minerals such as calcic carbonate
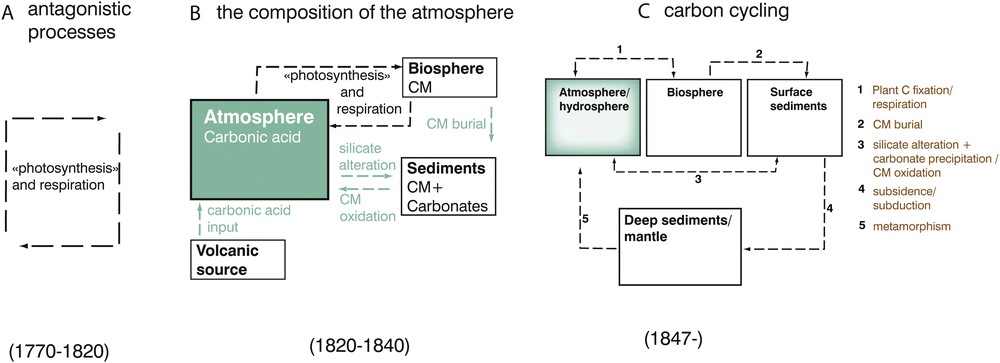
Three main conceptual steps leading from the idea of compensating processes to the “carbon rotation” and ultimately to the present day nomenclature of the “carbon cycle”. The details of each step are provided in the text.
Principales étapes conceptuelles ayant conduit de la découverte de processus « compensateurs » à la « rotation » du carbone à l’échelle globale, puis au « cycle du carbone » proprement dit.
« Quand Saussure eu démontré, par ses belles expériences, cette admirable loi de la nature en vertu de laquelle le carbone passe de l’atmosphère dans les végétaux pour être bientôt restitué à l’état d’acide carbonique, soit par leur décomposition, soit par les animaux qui s’en nourrissent, on cru que cette rotation du carbone assurait la permanence de la composition de l’air atmosphérique. On voit maintenant qu’il faut faire intervenir dans la solution de la question, des phénomènes d’un tout autre ordre, et que les éléments minéraux de la croûte terrestre concourent aussi, par des réactions inverses les unes des autres, à la production de cet équilibre »20 (Ebelmen, 1847, p. 652).
In the above quote, J.J. Ebelmen was the first to use the word “rotation” to refer to the transfer of carbon (not carbonic acid) as an element at the global scale. Although he attributes the origin of the idea of a rotation to N.T. de Saussure, we have shown that N.T. de Saussure never used this term explicitly, although he contributed heavily through his thorough experiments to the study of the counterbalancing effects of photosynthesis and respiration. The above quote concluded a 20-year long period that led Adolphe Brongniard, J.B. Boussingault and then J.J. Ebelmen to progressively extend the purely geological part of the global carbon “rotation”.
6 Discussion: from counteracting processes to the cycling of C at the Earth scale
6.1 Main steps leading to the carbon “rotation”
Based on this short historical account, we have shown that the foundation of the carbon rotation described between the years 1777 and 1847 can be represented by three main conceptual steps, as sketched in Fig. 5:
- • N.T. de Saussure, H. Davy and other chemists such as J.B. Dumas referred their work to A.L. Lavoisier's findings and interest in applying chemical principles to the study of plant physiology. The “counteracting” processes of respiration and carbon fixation by plants were established between the 1770s and the 1820s (Fig. 5a). This period approximately corresponds to the span between A.L. Lavoisier's works to Adolphe Brongniard's publications in the 1820s;
- • in the 1830s, the extension of these processes to the global scale (Figs. 2 and 5b) occurred. Adolphe Brongniard initiated a long period during which studies were focused on the composition of the Earth's atmosphere. The burial of CM in sediments was understood to be a major atmospheric carbonic acid sink (possibly triggered by catastrophic events of the Earth's history), since the work of Adolphe Bongniard (Brongniard, 1828). Soon after, between the 1830s and the 1840s, J.B. Boussingault (Boussingault, 1834) and J.J. Ebelmen explicitly recognized the interplay of different counteracting processes acting on the composition of the atmosphere, among which “purely geological causes” were identified. J.B. Boussingault recognized volcanic emissions of carbonic acid as one of these important “geologic” process at the scale of the Earth (Boussingault, 1834);
- • in 1847 (Figs. 2 and 5c), J.J. Ebelmen referred to the “admirable experiments by Saussure” as well as to the works of J. Fournet (and indirectly G. Bischof), on the role of carbonic acid on silicate weathering to suggest that the coupling between silicate weathering and carbonate precipitation in the ocean is another major “geological” carbonic acid sink. He described the rotation of the element carbon occurring on a global scale, and he made use of chemical formulas to quantify the impact of chemical weathering on atmospheric composition. The work of J.B. Dumas (Dumas and Boussingault, 1841) and J. Liebig (Liebig, 1841) in the same decade also indicates the increasing importance of questions related to the transfer of individual chemical elements (C, N, H) on a global scale.
These three steps also highlight the idea that the concept of a global carbon “rotation” involving chemical weathering and carbonate formation directly branched, in the mid 1840s, on studies developed throughout the 19th century that were dedicated to the processes acting on the atmospheric composition that included carbon fixation by plants, respiration and a constant supply of carbonic acid to the atmosphere by the volcanic emissions. In these early developments, living organisms played a central role, as they were also responsible for coal formation and even carbonate rock formation, as noted by J.J. Ebelmen (1845).
Our historical account also shows that the advent of the new “compositionalist” view of chemistry (the sociological details of this propagation remain a controversial issue, see (Allchin, 1997; Cavendish, 1766, 1784; Chang, 2010; Golinski, 1995; Hutton, 1795)), from the 1780s to the middle of the 19th century laid down the basis of the modern carbon cycle. A.L. Lavoisier's chemical findings sparked chemical inquisitions on animal (respiration) and plant physiology. His discovery of the compound nature of carbonic acid and his new theory of acidity that underpinned the understanding of the action of carbonic acid during rock weathering in works spanning from G. Bischof to J.J. Ebelmen's. Finally, his findings laid the grounds for future works by J. Dalton or J.J. Berzelius that led to the advent of the natural history of elements as proposed by J.J. Berzelius, which introduced the distribution of elements on Earth as an independent and fundamental question: “The history of inorganic substances of the globe is not less a part of natural history than is the history of organic substances” (Berzelius, 1819).
We will now show that prior to this construction, some forms of cyclic perception of matter already existed and were instrumental in giving birth to the later findings presented in this study. Furthermore, the carbon “rotation” described in 1847 was far from complete. In particular, inorganic processes were considered only for their action on the atmospheric composition, whereas the global cycling of carbonates was understood only after major progress was made in geodynamics later in the century, i.e., during the second half of the 19th century (theory of subsidence) and until the mid 20th century (plate tectonic and isotope geochemistry).
6.2 Hutton's cyclic conception of the Earth – S1
The concern with the Earth's system stability was a major change in the conception of carbon cycling at the turn of the 18th century. Indeed, before the advent of modern chemistry, some forms of carbon cycling had already been envisioned. James Hutton's theory of the Earth (Hutton, 1795) attributed to coal and life major roles in sustaining Earth's “machinery”, in which “all its different parts are adapted, in form, in quality, and in quantity, to a certain end […]” (Hutton, 1795 see also Allchin, 1997).
Based on the premise that the Earth is an “organized body”, the constitution of which must sustain life, James Hutton used the most recent discoveries of his time to build a cycling perception of the Earth, in which life, biogenic material and the geological transformations of this material played a fundamental role. These transformations served both as a means to create (accumulation of “solar principle” to form phlogiston) and release the heat that sustained the machine (coal combustion at depth, which he attributed to the action of heat on plant and animal remains (Hutton, 1795)), and an end of the whole “organized body” (Hutton, 1795). The fundamental point here is that the stability of the whole system is the mere premise of Hutton's conception, which could be considered a “proto-carbon cycle” where life played a major role. This can also be found in the homeostatic conceptions of H. Davy (Davy, 1799b) until some decades later, as previously shown, J.B. Boussingault and J.J. Ebelmen questioned this stability:
« La composition de notre atmosphère est-elle arrivée à un état permanent d’équilibre? Il faudra sans doute bien des siècles pour obtenir la solution de ce problème. Les moyens d’analyse que nous possédons sont maintenant assez précis pour que nous puissions léguer aux générations futures des éléments certains sur cette importante question. »21 (Ebelmen, 1845).
It is noteworthy that despite the ongoing apparent controversy between two contrasting world views – that is, between J. Hutton's cyclic conceptions of the Earth's dynamics and A.G. Werner's neptunism (for details refer to Gohau, 2003; Rudwick, 1985) – both the views contributed substantially and complementarily to the advent of the modern basis of the carbon cycle. The former heat-based model and cyclic concern were frequently referenced by Charles Lyell, especially in his conception of soil renovation on the Earth's surface, and pervaded the cyclic conceptions of 19th century scientists. The cyclic vision defended by J. Hutton, which was far more elaborate than a reframing of the old middle age's “fascination with cyclicity” (Koyre, 1971), continued to be used, although from a radically different point of view – for example, by physiologists and J.B. Boussingault. In contrast, A.G. Werner transmitted to a generation of geologists and mineralogists his interest in aqueous processes, especially their role in dissolution and mineral formation. As we have shown, G. Bischof and J.J. Ebelmen identified key aqueous processes, the dissolving power of carbonic acid on silicates, that were added to the processes acting on the atmospheric composition at the global scale as developed by physiologists and geologists (Ebelmen, 1847). A famous “controversy” between two geologists eventually bore fruit and served as a conceptual foundation of the modern carbon cycle.
6.3 Towards a modern carbon cycle: geodynamic meets geochemistry and petrology
6.3.1 Geodynamics of the crust: Vertical transfers of C in the crust – S11
Throughout the 19th century, more-detailed knowledge of the structure and dynamics of the Earth progressively added a clear connectivity between the surface and the “subterranean reservoirs” and offered a novel physical basis for the cycling of elements at the global scale (Fig. 2, S11). The main hypotheses with respect to the source of carbonic acid released by volcanism were the following: First, carbonic acid from deep reservoirs, possibly in a liquid form at great depths and under high pressure as it was first proposed by the French geologist Elie de Beaumont; the second possibility was the destabilization of carbonates at depth as suggested by H.B. de Saussure. Another option, which linked volcanic carbonic acid to the oxidation of “organic” compounds at depth, had been favored by A.G. Werner but was progressively abandoned during the 19th century. Prior works have recognized the British chemist and geologist Sir James Hall (1761–1832) as the first to investigate in Edinburgh the stability of carbonates under high temperature and pressure, using an experimental approach (reference in, Wyllie, 1998). This petrological knowledge was paralleled by the emergence of the theory of sediment subsidence, which was partly developed by the American geologist James Hall (1811–1898) and the English physicist John Frederick William Herschell (1792–1871) in his letter on phenomena connected with the internal temperature of the Earth to Charles Lyell in 1837, and published in 1838 (Herschell, 1838). This theory was further elaborated and renamed the “geosynclinal theory” 50 years later by the American geologist James Dwight Dana (1813–1895). It described the effect of internal heat and pressure on the subaerial material, a process that was called metamorphism. A contributor to this geodynamic view of the Earth, Thomas Sterry Hunt (1826–1892), summarized its development in his chemical essays (Hunt, 1875), and used this knowledge to link “carbonic acid evolved from the Earth interior to the decomposition of carbonates [previously generated by subaerial reaction] at great depths and high temperatures by reactions with siliceous matter”, referring to J.J. Ebelmen's hypothesis (Hunt, 1880); however he dismissed the deep molten reservoir hypothesis described by Elie de Beaumont. T.S. Hunt successfully applied the subsidence theory to the question of the fate of carbon as an element, a novel approach (Hunt, 1880), that complemented the understanding of the carbon rotation at that time. Although the detail of the subsidence theory was continuously developed during the second half of the 19th century, the idea of a connection between material formed by “subaerial” processes followed by their burial and decomposition at depth with the action of heat was a major step towards the first formulation of a “carbon cycle”, including the geochemical, petrologic and geodynamical aspects understood today. Within this new geodynamic framework, Viktor Moritz Goldschmidt (1888–1947), one of the founder of modern geochemistry, and Norman L. Bowen (1887–1956) described the thermodynamic and petrologic basis for the progressive destabilization of carbonates formed at the surface during incipient “metamorphism”, when they are buried at depth and release CO2 (Bowen, 1940; Goldschmidt, 1912).
6.3.2 New tools in geochemistry and petrology: the carbon cycle – S11, S12
Another development that led to the first formulation of the “carbon cycle” was the establishment of the science of thermodynamics in conjunction with industrial activities in Europe and North America developed throughout the 19th century (e.g., Sadi Carnot (1896–1832), Rudolf Clausius (1822–1888), Josiah W. Gibbs (1839–1903). Thermodynamics’ applications encompassed many domains described in this study, ranging from the understanding of plant C fixation pathways (which would be known as the “photosynthesis” by the end of the 19th after the works of the American botanist Charles Reid Barnes (1859–1910)), to the fate of carbonates at depth in the early 20th century (e.g., Bowen, 1940; Goldschmidt, 1912; Harker and Tuttle, 1956). Second, at the end of the 19th century, Svante Arrhenius (1859–1927) proposed linking the temperature at the surface of the Earth to the concentration of CO2 in the atmosphere (Arrhenius, 1896), drawing on the works of Joseph Fourier (1768–1830) and John Tyndall (1820–1893) (Bard, 2004). The carbon rotation became directly linked to the Earth's climate and thereby provided additional justification for its further development. In parallel, the birth of geochemistry as a scientific discipline applied to the carbon rotation, following the preliminary works of J. Dalton, J.J. Berzelius and J. Liebig, was definitively achieved by the two seminal works of M.I. Vernadsky (Vernadsky, 1924) and V.M. Goldschmidt (Goldschmidt, 1937). M.I. Vernadsky introduced the term “carbon cycle” in his book la géochimie (Vernadsky, 1924), first published in French, and quantified the main fluxes of the global carbon cycle (C fluxes exchanged by the biosphere, ocean, carbonate and coal formation, volcanic input). He clearly introduced the modern conception of geochemical reservoirs and the dynamic equilibrium of element cycles (Fig. 2, S11). Most recent developments, such as the model of plate tectonics (Fig. 2, S12) and the process of oceanic crust carbonatation (Alt and Teagle, 1999), demonstrate that the diversity of natural processes affecting the global carbon cycle at the geological timescale is far from being fully understood (Fig. 6).
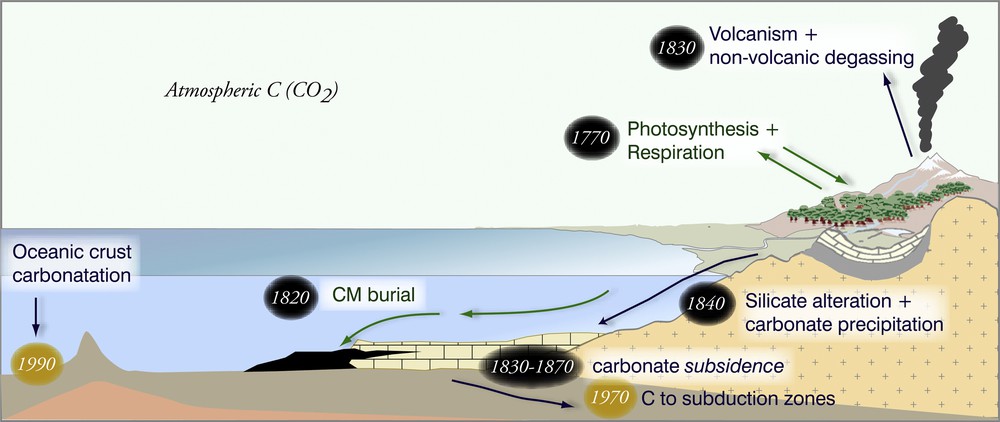
Key processes of the modern carbon cycle with the approximate dates of their identification. Processes corresponding to the so-called biochemical (Albrecht and Ourisson, 1971) or “organic” (Berner, 2003; Berner et al., 1983) sub-cycle are in green and those corresponding to the so-called “geochemical” (Albrecht and Ourisson, 1971) or “carbonate–silicate” sub-cycle are in blue. Recently discovered processes are indicated with a marker. The figure is modified after Gaillardet and Galy (2008).
Les processus principaux du cycle du carbone moderne avec indication des périodes approximatives de leur identification sont représentées. Les processus relevant du sous-cycle « biochimique » (Albrecht et Ourisson, 1971) ou « organique » (Berner, 2003 ; Berner et al., 1983) sont en vert. Les processus du cycle appelé aujourd’hui « géochimique » (Albrecht et Ourisson, 1971) ou du sous-cycle « carbonate-silicate » sont en bleu. Les découvertes plus récentes sont en jaune. La figure est modifiée à partir de Gaillardet et Galy (2008).
7 Conclusion
By the end of the 18th century, the chemical revolution had pervaded the study of the Earth and marked the advent of a chemistry-based investigation of the Earth and life processes. The description of the carbon cycle was preceded by major conceptual shifts (Fig. 5) and scientific breakthroughs (Fig. 2). It started as a process-oriented investigation of the counterbalancing effect of photosynthesis and respiration in laboratory experiments. Carbonic acid was recognized as being composed of C and O in the 1770s by A.L. Lavoisier and carbon acquired his current name by 1789 (de Morvaux et al., 1789). Questions regarding the stability of the atmospheric composition further drove these investigations: by 1809, the elementary pathways of carbon fixation in both terrestrial and aquatic plants was quantitatively demonstrated by N.T. de Saussure. In the early 1820s, Adolphe Brongniard proposed the first synthesis of processes acting on the carbonic acid content of the atmosphere (Fig. 5b) but did not clearly mention a carbon circulation. He showed that C burial of plant and animal remains served as a sink of carbonic acid from the atmosphere and directly acted on the atmospheres's long-term composition. In the 1830s, J.B. Boussingault described the continuous and “purely geologic” process of carbonic acid release by volcanoes as a potential control of atmospheric composition. By this time, carbonic acid had been discovered as a ternary compound by the works of J. Dalton (Dalton, 1808). Although its current chemical formula, CO2, existed since the works of J.J. Berzelius (Berzelius, 1819), the formula was not used in this context until the works of J.J. Ebelmen, who included the alteration of silicates– another “purely geological” process–and the carbonate precipitation to the processes acting on the CO2 content of the atmosphere, making use of the prior works of geologists such as G. Bishof or J. Fournet. In his 1845 paper (Ebelmen, 1847), J.J. Ebelmen clearly used the term carbon rotation for an idea that can be traced back to A.L. Lavoisier (Lavoisier, 1792, 1793). However the stability of the atmospheric composition had been the major concern sustaining these research. With J.J. Ebelmen's and subsequent works, the geochemical fate of an individual chemical element became itself a scientific question worthy of interest. This development was further fuelled by new models of the dynamic Earth, especially those affecting sedimentation in the oceans. In particular, the theory of subsidence (later called “geosynclinal” theory) first proposed in the 1830s allowed unambiguous attribution of volcanic CO2 to carbonate destabilization buried at depth following their biological formation in the ocean. V.M. Goldschmidt and M.I. Vernadsky introduced the term carbon cycle (Vernadsky, 1924) and founded modern geochemistry.The advent of the plate tectonic model between the 1930s and 1970s was a major event in the most recent developments of the Earth's carbon cycle concept (Berner, 2006; Berner et al., 1983; Garrels and Lerman, 1981; Garrels et al., 1972; Holland, 1978; Lasaga et al., 1985; Hayes and Waldbauer, 2006; Javoy et al., 1982), which now includes interconnections between the surface of the Earth and its deepest reservoirs (Hayes and Waldbauer, 2006; Javoy et al., 1982). The impact of this fundamental shift on the development of the carbon cycle (Fig. 2, S12) merits further dedicated studies in the coming years. Overall, the progressive construction of the C cycle is a remarkable illustration of the major role played by multidisciplinarity, technical developments, collaborations and controversies in the gradual development of scientific concepts and/or paradigms.
Acknowledgments
We thank Robert Berner for suggesting that the authors meet and for stimulating discussions on J.J. Ebelmen and the history of the carbon cycle. We are indebted to Jacques Touret for a very constructive review. The article benefited from advices of Gilles Fumey, Frédéric Perrier, Edouard Kaminski, Oleg Pokrovsky, Jacques Schott, Pascal Richet and Bernard Dupré. We finally wish to thank D. Allchin, R. Hazen, I. Martinez, M. Ader and R. Hilton for helpfull discussions during the elaboration of this work. Financial support for this work was IPGP program of Potamology and the Sloan Foundation.
1 If we burn pure coal under a bell jar filled with vital air and closed with mercury, a portion of the vital air is converted to fixed air or carbonic acid. If we absorb the carbonic acid with caustic alkali in liquor the residual air is still the vital air, in which we can burn a novel quantity of coal. By repeating these experiments, we can convert the entire amount of vital air into carbonic acid without any residue.
2 In reference to Van Helmont who coined the term (Lavoisier, 1789).
3 To redefine the basis of science, especially its nomenclature – “any physical science is composed of three things: the series of facts that constitute the science, the ideas that refer to them and the words that enunciate them.” (Lavoisier, 1789) and coauthored the result of this fundamental work (de Morvaux et al., 1789).
4 To charbon [the] ‘principle’ of coal substances we adapt the simplified expression of carbon, that will indicate the pure principle, essential of coal, and that has the great merit of specifying it with a single word.
5 If a plant substance, plus oxygen, forms carbonic acid and water, then as a corollary, by removing oxygen from carbonic acid and water, a plant combination should be retrievable.
6 Although art (the “art” of doing experiments) does not allow the means to perform this marvel, it is not unlikely, and analogy leads us to believe it, that it is the path that nature takes in the formation of plants.
7 Modern experiments do not permit doubt that charbon is one of the principle of bodies and that it is part in a number of combinations in the three kingdoms […] The charbon, accordingly, must be considered as a simple combustible body, or at least that chemistry has not succeeded yet to decompose, that forms fixed air or carbonic acid by combustion […]. We conceive after that the immense quantity of charbon locked in the bowels of the earth since marbles, chalk or calcareous rocks contain around three-tenth, and sometimes even a third of their weight of fixed air, and that this latter is composed of charbon for 28/100th of its weight […]. These consequences are far from all the ideas that we had until now; but they are not a less necessary consequence of the experiments that we have just reported. […] We will not follow here the changes of forms underwent by charbon when it passes from the mineral kingdom, to the plant kingdom and the animal kingdom. We would fall into chemical discussions unrelated to the object of this article.
8 A.L. Lavoisier's ideas that the state (aeriform, liquid or solid) of any material matter depends only on the quantity of heat provided and ambient pressure (Lavoisier, 1792) appeared to Spallanzani “very probable” (Spallanzani, 1796).
9 Carbonic acid might be considered as the product of carbonate of lime or of soda. In the first case, heat alone would be sufficient to disengage the carbonic acid; in the second, independent of high temperature, one would also require that the alkaline carbonate be in contact with a siliceous or aluminous substance, for example to trachytic rock itself.”
10 Once vital action is stopped, there occurs chemical phenomena of an opposite order than those that contributed to its assimilation; the decomposition begins […], whose the final result is to restitute to the atmosphere the carbon that was subtracted from it. If the amount of living matter stays the same at the surface of the earth, this matter does not ultimately exert any influence on the carbonic acid composition of the atmosphere.
11 The only case where organic matter will be able to diminish the carbon from the atmosphere would be if a catastrophic event leading to the death and burial of a large mass of organized beings. Such may be the case when large parts of the vegetation that existed at diverse geologic epochs were buried in sedimentary rocks. Carbon that is a part of the immense deposits of coal and lignite no doubt contributed to atmospheric carbonic acid.
12 Nevertheless, we cannot conclude that the air from the past contained more of this acid than it contains now, because there doubtlessly exist different sources that augment the proportion of carbonic acid in the atmosphere.”
13 Gases are emanating constantly from the interior of the earth into the atmosphere. Volcanoes can be considered vents releasing without interruption carbonic acid.
14 The usual mode of decomposition, which we observe for feldspar particularly in granites, appears to be due to direct exposure to the atmosphere.
15 It will break down those connections gradually and unify them with the alkalis.
16 Should we require an acid, we will see that the only one that is sufficiently universally spread in nature to which we may attribute this effect is carbonic acid.
17 Alkaline carbonates will meet, either in fresh water or in the sea, to form soluble salts made out of lime or magnesia, and they will produce carbonates of these two bases that will decompose at their turn, spontaneously or by the intermediate of zoophytes or mollusk. The bases, which once were combined with silica in igneous rocks, have precipitated from the atmosphere an equivalent portion of carbonic acid.
18 Whatever the origin of gases emitted by volcanoes, it is certain that it is a powerful phenomenon able to counterbalance the effect of the absorption of the same gas by the decomposition of rocks. These facts highlight the intimate relationship that these grand natural phenomena share.
19 Nothing has established that the various factors that modify the composition of the atmosphere have a null result.
20 When Saussure had demonstrated through his nice experiments the admirable natural law by which carbon passes from the atmosphere into the plants soon to be returned to the state of carbonic acid, either by their decomposition, or by animals that feed on them, it was believed that this rotation of carbon guaranteed the permanence of the composition of atmospheric air. We now see that we must include into the solution of this question a phenomenon of another kind and that the mineral elements of the Earth's crust also take part, through inverse chemical reactions, in achieving this state of equilibrium.
21 Is the composition of our atmosphere in a state of permanent equilibrium? This question will undoubted require many centuries resolve. The methods of the analyses that we now have are sufficiently precise for us to be able to pass on to the next generations clues that will help answer these important questions.
Vous devez vous connecter pour continuer.
S'authentifier