

1 Introduction
Natural organic matter (OM) is widespread in terrestrial ecosystems and has been known for long to play a major role in the global carbon cycle (Berner, 2012; Prentice, 2001; Raich and Potter, 1995). It encompasses OM present in various environments such as soil, groundwaters, and rivers. The molecular characterization of OM in these pools is of prime importance for several environmental purposes. Owing to the size and the dynamics of the soil carbon pool, small variations in the ability of soil to act as a carbon sink or source would induce differences in atmospheric CO2 concentrations, hence the key role of the soil OM pool in the exchanges between vegetation and atmosphere (Tate et al., 2000). Such variations may occur as the result of climate changes, but also after direct human activity or intervention. The latter include changes in land use such as afforestation or differences in tillage or cropping (e.g., Banwart et al., 2012; Foley et al., 2005; Paul et al., 2002; West and Post, 2002). Natural OM comprises a large diversity of constituents with different reactivity and dynamics. However, a stable pool of OM, with turnover up to millennia, is commonly considered. Several mechanisms are put forward to account for OM stabilization in soil, such as preservation of intrinsically resistant constituents and interaction with mineral matrix or microbial biomass (Krull et al., 2003). OM in groundwaters and rivers, which encompasses dissolved and particulate OM, is a major substrate for microbial communities and it is involved in nutrient transportation (Marschner and Kalbitz, 2003; Pizzeghello et al., 2006; Qualls and Haines, 1992). Moreover, this OM may also constitute drinking water sources. As a result, in addition to being important for the knowledge of environmental processes, a better understanding of this pool is also essential for its influence on drinking water treatment and the formation of disinfection by-products, which are then released in the environment (Margat, 1994; Sharp et al., 2004).
Despite this high environmental importance, natural OM remains poorly characterized at the molecular level (Hedges et al., 2000). Indeed, such a characterization is challenging because of the diversity and heterogeneity of OM (Frazier et al., 2003). OM is commonly divided into two major pools, the lipids, which are extracted using organic solvents and the remaining insoluble fraction, made of geomacromolecules. Although generally less abundant than the insoluble OM, lipids bear important information on sources (thanks to specific biomarkers) and degradation stage of OM. They exhibit a very large diversity and they would merit a dedicated review. However, their chemical characterization can be rather easily achieved through the combination of chromatography, which allows separation of the constituents of a complex mixture, and mass spectrometry, which leads to the identification of the individual molecules. On the contrary, specific tools have to be implemented for the molecular analysis of the insoluble fraction, due to its macromolecular structure (Kögel-Knabner, 2000). It must be noted that useful fingerprints can be derived from bulk analyses such as elemental analysis, specific ultraviolet absorbance, various colorimetric assays affording protein or carbohydrate contents or 3-dimensional excitation-emission matrix fluorescence (e.g. Rosario-Ortiza et al., 2007). However, these approaches do not provide insight in the OM chemical structure at a molecular level. Such a precise characterization is a prerequisite for modelling environmental processes in which natural OM is involved. As an example, bulk OM analyses fail in taking into account complexation mechanisms or more generally differences in its reactivity. In the following, we will discuss the analytical tools that have been recently developed to achieve the molecular characterization of this insoluble OM. Although this review is focused on natural OM from continental environments, it is important to keep in mind that the same approach can be followed in the molecular study of any other geomacromolecules, including those from oceanic sediments or even to extraterrestrial materials. Another common feature to the macromolecular OM from many natural environments is its complexity along with its tight association with the mineral matrix, which in addition, is often predominant. As a result, sample pre-treatment and/or fractionation are often necessary prior to analysis. They will be briefly reviewed below before the description of the main analytical tools recently developed for the molecular analysis of geomacromolecules (Fig. 1). The latter include non-destructive analytical spectroscopic methods along with thermal and chemical degradations followed by mass spectrometry identifications.
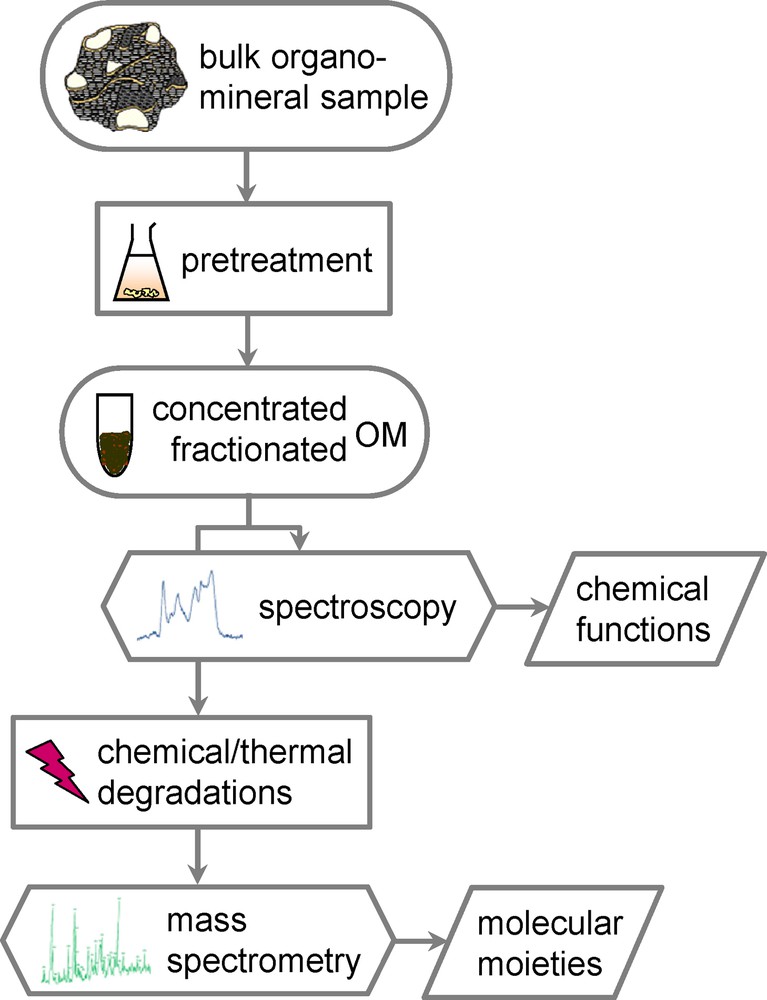
(Color online). Typical analytical flowchart for organic matter molecular analysis.
2 Sample pre-treatment
The organic carbon content in natural samples is often very low and OM can be concentrated by destruction of the associated mineral matrix through acid treatment using HCl and HF. Although this treatment is commonly performed in soil studies (Dai and Johnson, 1999), it may induce a loss of carbon, which may be associated with some specific organic constituents (Rumpel et al., 2006). As for river and groundwater samples, dissolved OM can be concentrated using reverse osmosis (Sun et al., 1995) but estuarine samples must be desalted prior to analyses as salts are concentrated along with OM upon reverse osmosis. This can be achieved through ultrafiltration, solid-phase extraction or electrodialysis (Dittmar et al., 2008; Koprivnjak et al., 2009; Liška, 2000; Simjouw et al., 2005). The main challenge during these pre-treatments is to recover representative OM without any alteration of its properties.
So as to go deeper in the chemical composition of the OM, it is often useful to perform a physical separation prior to analyses (Christensen, 1992). Such fractionation also aimed at isolating homogeneous fractions in terms of chemical composition or turnover (Golchin et al., 1997; Marzaioli et al., 2010; Moni et al., 2012; von Lützow et al., 2007). Several types of physical fractionation are carried out on soil samples. They are mainly based on particle size and density differences (Christensen, 1992) but the extent of aggregation must also be taken into account and aggregate fractionation was suggested as a first step prior to other physical fractionation (Six et al., 2002). However, none of these procedures can afford fractions with homogeneous turnover times (von Lützow et al., 2007). Due to the strong bond they form with OM, Fe oxides are often suggested to play a role in OM stabilization and a fractionation based on magnetic susceptibilities at different field strengths has been put forward (Shang and Tiessen, 1997). Water samples are commonly fractionated through the use of nonionic macroporous resin columns (Aiken et al., 1992), but some alterations of the OM may be associated with this fractionation step (Mace et al., 2001). Tangential ultrafiltration is also used to produce fractions of different molecular size and especially to isolate high molecular weight dissolved OM, which is considered as the most reactive pool of dissolved OM (Guo et al., 2009). However, some smaller or larger compounds than expected may contribute to the size fractions and some loss of carbon and nitrogen may take place upon fractionation (Kiikkilä et al., 2012).
For long, chemical extractions have been performed, resulting in the definition of fulvic and humic acids and humin (Stevenson, 1994); however, it is now more and more accepted that these treatments can alter molecular structures and result in operational rather than functional fractions (Baldock and Nelson, 2000; Kleber and Johnson, 2010). Chemical fractionation using oxidative reagents was also carried out to separate chemically resistant soil OM (Zimmermann et al., 2007) and was sometimes associated with HF treatment to derive mineral-associated OM (Mikutta et al., 2006; Sleutel et al., 2009).
Whatever the pre-treatment or the physical or chemical fractionation, the resulting OM has to be characterized using the methods described in the following.
3 Spectroscopic methods
The great advantage of the spectroscopic methods in the analysis of natural OM is that they are not destructive. They provide insight in the nature and relative abundance of the chemical functions involved in the OM such as carboxylic acids or aromatic moieties but are more limited to derive information at the molecular level as discussed below.
The most commonly used spectroscopic tools to analyse the natural OM have been for long Fourier-transform infrared (FTIR) and nuclear magnetic resonance (NMR, mainly 13C). However, FTIR was shown to release only little information on the chemical composition and numerous questions arose about observability of carbons in NMR. Advances in these techniques are thus reported below.
The most remarkable advance in FTIR spectroscopy lies on the replacement of the conventional light source of a spectromicroscope with synchrotron radiation, which results in a drastic increase in brightness and signal-to-noise ratio as highlighted by Lehmann and Solomon (2010). This improvement was applied to the study of the spatial distribution of organic carbon in soil microaggregates and suggested a preferential involvement of aliphatic molecules in organomineral interactions within microaggregates (Lehmann et al., 2007).
NMR analysis of natural OM is mainly achieved through 13C NMR using the so-called cross polarization and magic-angle spinning (CP-MAS). The CP sequence aims at enhancing the signal, thanks to magnetization transfer from the 1H reservoir to the 13C one, with respect to the single pulse (SP) sequence in which the 13C are directly observed. MAS is used to reduce line broadening. The released information is generally focused on the changes in relative abundances of the different types of C such as O-alkyl-C with respect to alkyl-C (e.g. Helfrich et al., 2006; Otto and Simpson, 2007; Fig. 2). It must be noted that integrations are commonly performed in defined chemical shift ranges but due to signal overlap, spectral decomposition appears more reliable. Moreover, additional insight can be gained from CP-MAS 13C NMR by varying the contact time, which is the key parameter in the 1H-13C magnetization transfer. When performed on size-separated fractions from a soil humic acid, this approach revealed heterogeneity both in molecular size and in supramolecular organization (Conte et al., 2006).
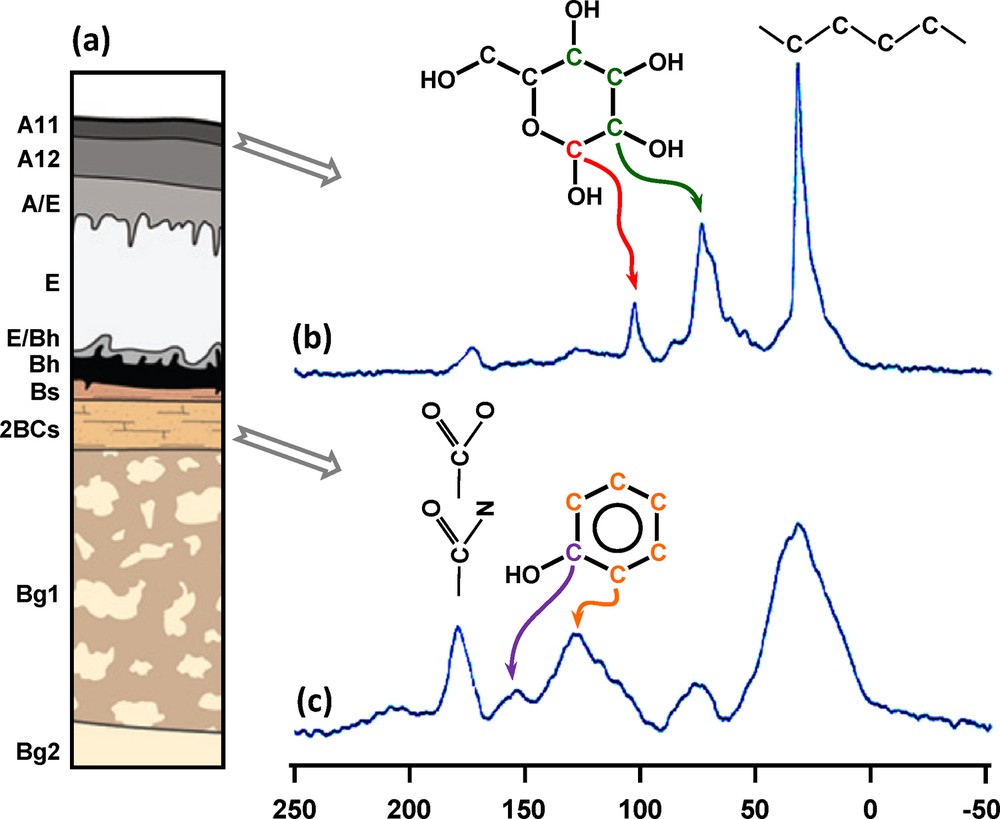
(Color online). Application of the cross polarization magic-angle spinning 13C nuclear magnetic resonance to the characterization of soil organic matter (OM) along a podzol profile (a). (b) OM in the A11 surficial horizon mostly comprises aliphatic and carbohydrate carbons, whereas (c) an increase in aromatic and carboxylic carbons is observed in the 2BCs deeper horizon.
Example taken from Bardy et al., 2008.
The main drawback in NMR is the difficulty in deriving:
- • information at the molecular level;
- • quantitative data.
So as to improve the information on the molecular composition of natural OM through solid-state 13C NMR, a molecular mixing model has been developed (Baldock et al., 2004). The basic principle of such an approach is that OM can be considered as a mixture of common classes of biomolecules, each being characterized by a representative chemical structure, distribution of 13C NMR signal intensity and elemental composition. Five components were defined to account for OM in soil and marine sediments, namely carbohydrate, lignin, protein, lipid and charcoal, with reference materials defined for each class of biomolecule. This approach was shown to be efficient in revealing differences between the marine and terrestrial studied systems. However, it appeared debatable due to the limitation related to the use of references. Nevertheless, a similar approach was used with a three end-member mixing model to follow the evolution of these compounds along a depth profile in oceanic OM (Sannigrahi et al., 2005). A rather good agreement was observed with NMR data and molecular information derived from chemical degradation.
A more direct insight in the chemical composition of natural OM can be obtained through the use of a combination of advanced solid-state NMR techniques (Mao et al., 2012). This includes spectral editing sequences such as dipolar dephasing or inversion recovery pulse sequences which allow distinguishing different types of carbons which resonate at the same chemical shift (such as protonated and non-protonated carbons). Two-dimensional techniques, such as 1H-13C heteronuclear correlation, can also be used in the same way. It led to decipher the major structural characteristics of two samples of surface-seawater dissolved organic matter.
Another approach involves the recent development of generalized two-dimensional (2D) correlation spectroscopy, which allows correlation between different types of spectra such as NMR and FTIR (Abdulla et al., 2013). When applied to high molecular weight dissolved OM along a salinity transect, this resulted in revealing single functional groups through deconvolution of complex overlapping signals. In a similar way, HF-treated soils were investigated using 2D correlation spectroscopy between 13C CP-MAS NMR on the one hand and near and mid infrared on the other hand allowing a better interpretation of the latter (Forouzangohar et al., 2013).
There are several reasons for which CP-MAS NMR spectra are not quantitative. Among them, interactions with paramagnetic components shorten relaxation times resulting in a selective loss of signal intensity. This can be partly circumvented thanks to the HF treatment but, as mentioned above, this treatment may induce some carbon loss, which may be selective (Rumpel et al., 2006). SP sequence, in which the nucleus is directly observed, is commonly used as a test for assessing to which extent CP NMR is quantitative. However in this sequence, one has to be aware that repetition delay between pulses must be long enough to avoid saturation effects, especially when some crystalline moieties are present in the studied material (Knicker, 2011a). All together, the most reliable method to derive a quantitative composition of natural OM through NMR is to acquire a series of CP–MAS spectra with variable contact time. The absolute intensity for each type of carbon can be derived from a diagram of intensity vs. contact time leading to the actual relative abundances of the different types of carbon.
As stressed above, 13C is the main nucleus involved in NMR studies of OM. However, it must be noted that 1H NMR (liquid state) is also performed especially for dissolved OM, including in the aforementioned 2D correlation spectroscopy approach. Of special interest is the use of 1H high-resolution magic-angle spinning NMR, in which the solid sample is swelled thanks to deuterated solvent and spectra are then acquired in the liquid state. This was performed to address the clay-organic interactions in model mixtures adsorbed onto montmorillonite and suggested that aliphatic components preferentially sorbed onto the clay surface (Simpson et al., 2006).
Another nucleus, which is increasingly used, is 15N. Due to the even lower natural abundance of the 15N isotope and its low gyromagnetic ratio (responsible for a low “NMR sensitivity”) when compared with 13C, 15N NMR is performed using the CP–MAS sequence except when 15N-labelled materials are concerned. Again, it is debated whether all nitrogen atoms are detected in the CP-MAS spectra, especially when condensed aromatic structures occur such as in chars (Knicker, 2011b). 15N NMR was also used in combination with another spectroscopic technique, namely X-ray photoelectron spectroscopy to identify the main N-bearing functions in riverine OM fractions (Templier et al., 2012). This study also pointed out that differences in response upon pyrolysis should be related to the chemical nature of the nitrogen moieties, and more specifically the occurrence of pyrrole units.
Although also an important nutrient, phosphorus has received little attention and 31P NMR chemical characterization is mainly performed in solution state after chemical extractions (e.g. Hamdan et al., 2012) but solid-state 31P NMR was also shown to be relevant to study the dynamics of soil P (Conte et al., 2008). Similarly, 27Al NMR was rarely used although it can provide information on Al environment in organo-Al complexes. In a study aiming at understanding Al dynamics during the podzolization of laterites in the upper Amazon Basin, the organo-Al complexes could be quantified by NMR and they were shown to accumulate in specific soil horizons (Bardy et al., 2007).
As mentioned above, X-ray spectroscopic techniques are also used to give additional constraints on the chemical composition of the OM. Among them, X-ray absorption near edge structure (XANES) spectroscopy is increasingly used despite limited access to synchrotron facility. It provides information on the nature of the chemical functions in the OM and is therefore comparable to NMR. Such a comparison was performed on several environmental matrices and black carbon reference materials (Heymann et al., 2011). Despite the advantages of XANES (high sensitivity, detection of all C), difficulties are still to overcome to derive quantitative data and to be able to distinguish black carbon from potential interferences such as coal. After the pioneering use of nitrogen K-edge XANES to characterise geomacromolecules (Vairavamurthy and Wang, 2002), this approach was recently used to reveal molecular changes in N-bearing functions upon soil burning (Kiersch et al., 2012). The potential of C and N XANES spectroscopy to analyse interactions of organic pollutants with soil OM was further demonstrated by following the evolution of the spectra after various types of alteration in laboratory experiments (Ahmed et al., 2012). In addition to carbon and nitrogen, sulphur K-edge XANES has been largely used for OM characterization in a wide range of environments, including soil (e.g., Prietzel et al., 2011; Solomon et al., 2003) where the accuracy of spectral decomposition was tested (Manceau and Nagy, 2012). Phosphorus K-edge XANES was shown to be suitable to identify inorganic and organic P species in natural environments, as well as 31P NMR (Kizewski et al., 2011).
Recently, C K-edge XANES was combined with scanning transmission X-ray microscopy (STXM) thus allowing simultaneous high-resolution imaging and spectroscopic characterization. According to the small-scale spatial heterogeneity of soil, this approach appears especially promising. It was therefore efficiently used to probe the chemical heterogeneity of organic matter in soil colloids (Schumacher et al., 2005) or microaggregates (Lehmann et al., 2007; Wan et al., 2007). It must be noted that these in situ techniques provide a direct analysis of organomineral interaction whereas previous approaches required separation of different organomineral classes prior to OM analysis through NMR (Helfrich et al., 2006) or pyrolysis (de Junet et al., 2013).
As described above, the main limitation of the spectroscopic analyses is their ability to give insight in the detailed OM molecular structure. The latter can be assessed through mass spectrometry analysis of products released from chemical and thermal degradations of geomacromolecules, as described below.
4 Chemical degradations
As mentioned in Section 2, some chemical treatments are performed to isolate chemically resistant OM prior to its analysis. In this section, we will consider the chemical degradations that are performed to access to the molecular structure of geomacromolecules through the composition of their degradation products, the latter being assumed to represent their building blocks (Fig. 3). As a result, the following chemical degradations aim at releasing low molecular weight molecules from the geomacromolecules. They are sometimes considered as depolymerisation as the released moieties can be likened to monomers. They mostly encompass oxidations and hydrolyses.
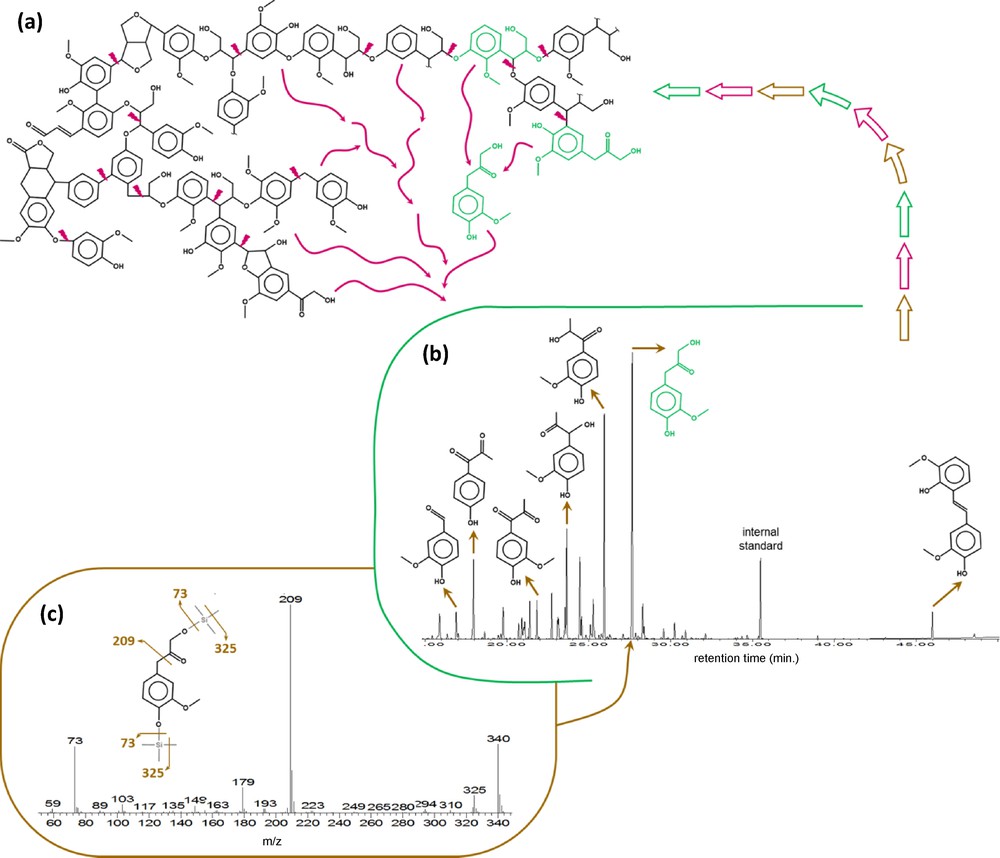
(Color online). Principle of characterization of a macromolecule through chemical degradation: a: microwave–assisted hydrolysis of Picea abies lignin releases building blocks from the macromolecule; b: separated by gas chromatography; c: identified by mass spectrometry (here as trimethylsilyl derivatives).
Example taken from Allard and Derenne, 2011.
Cupric oxide (CuO) oxidation has been used for more than 20 years to derive information about the oxidation state of lignin (from the relative abundance of some specific carboxylic acids to the aldehyde counterpart), its degradation stage and the nature of source plants in soils and sediments (Goni and Hedges, 1990). A vegetation index based on lignin phenol distribution was thus introduced by Tareq et al. (2004) to reveal vegetation changes along a peat core. It must be noted that CuO oxidation additionally yields hydroxyalkanoic acids derived from cutin and/or suberin, which are aliphatic biopolyesters occurring in the external parts of leaves, bark and roots (Goni and Hedges, 1990). This method was therefore recently used to follow the nature and abundance of both lignin and cutin/suberin moieties in various density fractions from a forest soil (Sollins et al., 2006). It has also been compared with transmethylation and with saponification (i.e. base hydrolysis) and the latter appeared as the most efficient way to analyse cutin in soils (Mendez-Millan et al., 2010a). It allowed revealing the selective preservation of root biomass with respect to shoots in a maize-cropped soil (Mendez-Millan et al., 2010b).
Oxidations have also been extensively used for black carbon quantification in soils and sediments based on the resistance of this material to oxidation (Hammes et al., 2007). However, most of these approaches such as chemo-thermal oxidation (Gustafsson et al., 2001) do not provide any molecular information on the black carbon structure. In contrast, in the so-called BPCA (benzenepolycarboxylic acids) method, the distribution of the acids formed after hot nitric acid oxidation gives insight in the condensation degree of the black carbon (Brodowski et al., 2005).
Acid hydrolyses are commonly used to release monomers such as sugars and amino acids. The main concern with these hydrolyses is their low yield and therefore to which extent they are representative. Indeed, in a study of a core profile of oceanic OM, total hydrolysable neutral sugars and amino acids only account for 9% and 28% of C in dissolved and particulate OM, respectively (Sannigrahi et al., 2005). This suggests either that molecularly uncharacterized OM is encapsulated in a matrix, which is not accessible to acids or that it is altered and thus not recognizable through molecular analyses. Interestingly, the use of methanesulfonic acid allowed increasing the hydrolysis yield by 46% with respect to classical HCl in soils and quantification through ion chromatography with pulsed amperometric detection showed that amino acids and aminosugars accounted for almost all N (Martens and Loeffelmann, 2003).
So as to derive a more complete view of soil OM chemical composition, a combination of the aforementioned methods is increasingly used, including sequential base and acid hydrolysis, which selectively cleaves ester and glycosidic bonds, respectively (Otto and Simpson, 2007). Acid and base (along with in situ methylation) microwave assisted hydrolysis were recently shown to be especially efficient in the characterization of the hydrophilic constituents of soil OM and thus constitute a promising approach for dissolved OM analysis (Allard and Derenne, 2011, 2013). It appears as a complementary tool to thermal degradations.
5 Thermal degradations
Pyrolysis has been for long a major tool in the characterization of the natural OM. Indeed, it allows the release of building blocks from the macromolecular OM. Moreover, when compared to chemical degradations, the cleavages are much less specific as a bond is cleaved as soon as enough energy has been brought by the heating. Several types of pyrolysis are used in OM analysis. They differ by their temperatures, the nature of the analytical system coupled to pyrolysis, the presence or not of a reagent and the open vs. closed device. Indeed, pyrolysis is usually performed at temperatures that are high enough (≥ 400 °C) to cleave covalent bonds in the macromolecule. However, sub-pyrolysis temperatures (250–350 °C) are also used to release products trapped within the macromolecular network without involvement of covalent bond. Such thermodesorbed products can therefore be distinguished from products actually resulting from the cleavage of the macromolecule by performing a two-step pyrolysis with a first heating at sub-pyrolysis temperature (Quénéa et al., 2006). Pyrolysis is commonly used in combination with gas chromatography and mass spectrometry (Py–GC–MS), which allows an easy identification of the pyrolysis products but pyrolysis can also be directly coupled to mass spectrometry (Py–MS) yielding a single mass spectrum for the total pyrolysis products. Pyrolysis can also be performed in the presence of a reagent, inducing in situ modification of the formed products. Two types of reactions are commonly performed, methylation or hydrogenation. Most of the analytical pyrolysis devices are open systems in which the released products are swept out of the heating system as soon as they are produced. However, closed systems usually operating at sub-pyrolysis temperatures were shown to be able to bring additional molecular information on OM composition (Berwick et al., 2010; Templier et al., 2005).
As pyrolysis breaks down the macromolecular network into small pieces, this raises the issue of how representative the pyrolysis products actually are. Indeed, the released compounds should account for all the constituents of the macromolecule (quantitative issue) and their chemical structure should be easily linked to that of the pristine moieties of the macromolecule (qualitative issue).
As a result, among the developments undergone by pyrolysis, a major aim was to increase the yield of pyrolysis products and, above all, to avoid any selective loss. As aforementioned, Py–GC–MS provides easier identification of the pyrolysis products thanks to their separation onto the GC column, but it is limited to GC-amenable compounds. High molecular weight or highly polar pyrolysis products may therefore escape detection. Py–MS therefore appears as complementary to Py–GC–MS but the resulting spectrum shows a high level of complexity, thus only providing little information on the OM chemical structure (Huang et al., 1998). A new device, termed in column pyrolysis, was designed so as to minimize transfer losses at the pyrolyzer–analytical system interface and it was compared with classical pyrolysis systems (Parsi et al., 2007). So as to increase the amount of GC-amenable compounds, especially the most polar ones, pyrolysis in the presence of a methylation reagent such as tetramethylammonium hydroxide (TMAH) was proposed (Challinor, 1995). This method, also termed thermochemolysis, provokes in situ methylation, making the pyrolysis products easier to analyze through GC (Shadkami and Helleur, 2010). It clearly highlighted some constituents of the OM such as lignin in hydrophobic fraction of riverine dissolved OM (Templier et al., 2005) or suberin in refractory soil OM (Quénéa et al., 2005). Another improvement was to combine the direct coupling of pyrolysis with MS and the in situ methylation. Temperature resolved in-source Py(TMAH)–MS involves sample heating at a given rate in the mass spectrometer source. It was especially successful in revealing a series of very long chain C43 to C53 fatty acids likely originating from mycobacteria in a humus horizon (Huang et al., 1998). Thermochemolysis was also shown to be effective at sub-pyrolysis temperatures, such as 250–350 °C, and off-line thermochemolysis performed in sealed glass tubes at 250 °C allowed revealing amino acid derivatives in riverine dissolved OM (Templier et al., 2005). However, more generally the identification of N-containing moieties using pyrolysis remains challenging (Templier et al., 2012). Another way to circumvent the detection problem of polar pyrolysis products is to reduce them into their hydrocarbon skeleton. This can be achieved through catalysed pyrolysis under hydrogen flow (termed hydropyrolysis) which results in increased yields and simplified pyrochromatograms when compared to classical Py–GC–MS (Berwick et al., 2010). However, this simplification leads to some loss of information on the building blocks of the macromolecule. Similar defunctionalisation was shown to take place under microscale sealed vessel (MSSV) pyrolysis performed during rather long time (up to several days) at sub-pyrolytic temperatures (Berwick et al., 2010). Depending on pyrolysis conditions, secondary reactions may take place during the thermal treatment. They would result in pyrolysis products different from the moieties that occur in the macromolecule, hence additional complexity in the interpretation of the pyrolysis data. For example, it has been known for long that:
- • aromatisation occurs upon heating as revealed by the formation of alkylbenzenes from aliphatic chains;
- • cyclisation leads to furan derivatives from carbohydrates;
- • decarboxylation and/or dehydration of lignin monomers are commonly observed.
To ensure the correlation between the pyrolysis products and the original structure, it is important to perform experiments on model compounds as recommended by Frazier et al. (2003) even though pure compounds may not fully reflect the nature of original biomolecules due to potential reactions they have undergone. Such an approach was recently followed on amino acids and dipeptides to derive new markers in Py(TMAH)-GC–MS (Gallois et al., 2007; Templier et al., 2013) and to assess the extent of conversion of amide-N into aromatic-N upon in-source Py–MS (Kruse et al., 2011) or MSSV pyrolysis (Berwick et al., 2007). Anyhow, the combination of various pyrolysis techniques appears powerful to provide a more complete evaluation of the natural OM molecular structure (Huang et al., 1998; Templier et al., 2005).
A major drawback in the pyrolysis techniques is the difficulty in deriving quantitative data. An attempt to assess pyrolysis yields was performed by using ion intensity in in-source Py–MS but large variations were noted depending on the pyrolysis conditions (Huang et al., 1998). However, this method was further developed to derive relative abundances of various pyrolysis product classes and thus to assess the evolution of the chemical composition of soil OM upon sequential chemical treatment (Sleutel et al., 2009). It revealed an enrichment of biologically labile compounds in soil after sodium hypochlorite treatment whereas the latter was supposed to isolate refractory material, suggesting some OM protection through mineral binding or encapsulation in macromolecular OM structures. The quantitation problem was also addressed upon thermochemolysis in the presence of TMAH in sealed ampoules at sub-pyrolysis temperatures (Frazier et al., 2003). Although this study revealed variations in reproducibility, it suggests the use of internal standards to be added after the TMAH reaction. It must be noted that relative abundances can be calculated from the ratios of the areas of the corresponding GC peaks in Py–GC–MS but the response factors have to be taken into account for comparison between compound classes. This raises an additional difficulty related to the commercial availability of the standard especially when in situ methylation is involved. White et al. (2007) proposed to overcome this difficulty by suggesting an “abundance index” based on comparison of summed relative abundances for sets of compounds belonging to given classes such as furfurals, cyclopentenones or polyaromatic hydrocarbons. This revealed significant variation in the sources of ultrafiltered dissolved OM along a salinity transect in the Mississippi River plume (Guo et al., 2009). Another approach to derive relative abundance of pyrolysis products was based on the intensity of two characteristic mass spectrometry fragments. It revealed enrichment of aliphatics in allophanic soil, without providing evidence for any specific mineral-organic binding (Buurman et al., 2007).
6 Mass spectrometry
Although mass spectrometry has been previously mentioned due to its large use as detection system when coupled with chromatographic separation (as in GC–MS) or pyrolysis, this section will be devoted to direct mass spectrometric systems. Because of the high complexity of natural OM, direct mass spectrometry requires high mass accuracy and resolution. Electrospray ionization coupled with Fourier-transform ion cyclotron resonance mass spectrometry (ESI FT–ICR MS) recently emerged as a powerful tool to analyse dissolved OM (Sleighter and Hatcher, 2007). It appeared especially efficient in revealing compounds with heteroelements (nitrogen and sulphur, e.g., Longnecke and Kujawinski, 2011), although spanning a restricted molecular weight range (m/z 300–1000). This approach was recently applied to study porewater OM samples collected along depth profiles in a fen bog complex (Tfaily et al., 2013). It revealed differences in OM reactivity between bog and fen that were shown to be consistent with data from UV absorbance, fluorescence spectroscopy and 1H NMR. The main limitation of FT–ICR MS is the solubility of the samples. To circumvent this problem, Zhong et al. (2011) compared the pyridine extracts from a set of geopolymers with their parent material, through the use of a combination of advanced NMR techniques, so as to determine whether the extracts are chemically representative of the parent material. In that case, FT–ICR MS analysis of the extracts can be used to represent the insoluble geopolymers at a molecular level.
The importance of in situ techniques able to analyse the OM at the nanoscale was aforementioned, due to the high spatial heterogeneity of natural OM samples. In this respect, elemental and isotopic imaging conducted via secondary ion mass spectrometry (SIMS) is a promising emerging technique. It exhibits a high potential in biogeochemistry and soil ecology as shown in proof-of-concept based on analysis of ex situ labelled materials within single microaggregate (Herrmann et al., 2007; Mueller et al., 2013) or of artificial soils (Heister et al., 2012). Combining NanoSIMS with other in situ nanoscale techniques such as STXM was proposed as a further step in the elucidation of environmental processes (Behrens et al., 2012). This approach, along with its coupling to XANES, made possible to analyse labelled OM located at the interface with minerals (Remusat et al., 2012). However, one must keep in mind that this technique so far only provides elemental and isotopic information on the OM.
7 Conclusion
Whereas it is widely recognized that natural OM plays a major role in the environmental processes, the chemical structure of natural OM is still poorly known at the molecular level. However, a wealth of analytical developments is being observed showing the importance to decipher this structure for organic geochemists. These developments include improvements of existing techniques but also new approaches. They concern both spectroscopic tools, which provide a general view of the geomacromolecules and degradations, which aim at releasing their low molecular weight building blocks. However some analytical problems still exist. One of the main challenges is to obtain representative quantitative data on the composition of the natural OM. To circumvent the limitations of a given method, the combination of several complementary approaches is increasingly applied, thus giving additional constraints on the derived data (Fig. 4). Although most of the techniques aim at elucidating carbon-containing structures, the importance of other elements in the characterization of the OM such as N, S or P must be noted and efforts should be put on their specific analysis. Besides the ongoing evolution of analyses of OM at a bulk scale, the emergence of nanoscale techniques must be emphasized. Their development to provide molecular information through coupling with molecular mass spectrometry is to encourage. However, a main challenge remains to reconcile data from nanoscale techniques with bulk analyses. This would provide an integrated understanding of the role of OM and its fate on Earth.
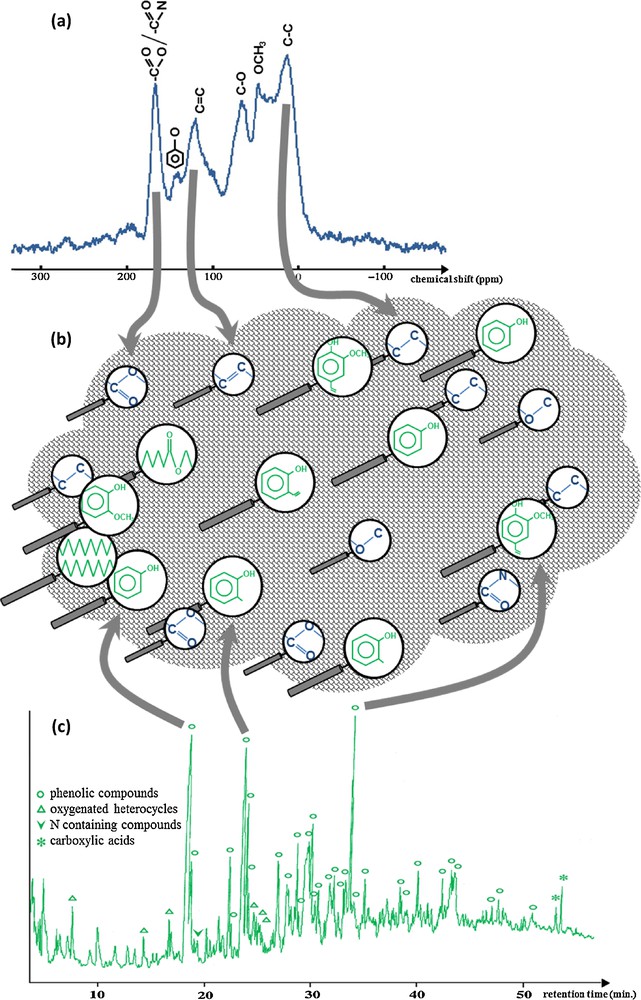
(Color online). Illustration of the different level of information derived from spectroscopic studies such as nuclear magnetic resonance (nature of the chemical functions) and degradations such as pyrolysis (molecular building blocks).
Example taken from Templier et al., 2005.
Acknowledgements
Joëlle Templier and Katell Quénéa (METIS, Paris) are gratefully thanked for their thorough review of a previous version of the manuscript.