

1 Introduction
Although average selenium concentrations in the continental crust are relatively low (0.05 ppm), diffuse geogenic pollutions are widely observed (Coleman and Delevaux, 1957; Yang et al., 1989; Presser et al., 1994; Seby et al., 1998). Selenium is in fact an important micronutrient with a narrow range between its toxic and deficiency concentration (Barron et al., 2009; Levander and Burk, 2006). Se intake for animals and humans is mainly provided by meats, cereals and, to a lesser extent, by drinking water (Appleton et al., 2006). Human intake is indirectly related to the selenium content in natural waters and to the bioavailable fraction of selenium in soils and geological materials that can be easily transferred to water and biota.
The geographical distribution and availability of Se in soils and natural waters is largely uneven and linked to the variable concentrations in rocks, as already recently emphasized by Schmitt et al. (2012) for other elements such as Mg, Ca, Li, and B, and for their isotopic signature. Limestone, sandstone, mafic and ultramafic rocks, intrusive and extrusive acid rocks often contain low Se concentrations (Coleman and Delevaux, 1957). In contrast, coals, shales, argillaceous sediments and phosphate rocks could be enriched with selenium (Coleman and Delevaux, 1957; Fernández-Martínez and Charlet, 2009; Vesper et al., 2008).
Selenium can thus be highly concentrated in black organic-rich shales and seleniferous black shales that are the geological parent material of the widespread seleniferous soils in the United States and in Europe (De Temmerman et al., 2014).
In natural waters, selenium concentrations are generally lower than 1 ppb (Ralston et al., 2008), but may significantly increase, influenced by anthropic or geogenic sources to reach toxic levels (Charlet et al., 2007; Fernández-Martínez and Charlet, 2009). The main aqueous selenium chemical speciation corresponds to the most labile and bioavailable forms: the oxyanions selenites (SeIV) and selenates (SeVI) (Ralston et al., 2008). Selenium speciation strongly depends on pH and Eh (Masscheleyn et al., 1990). Hence, selenium has a complex behavior and a large variety of selenium compounds may be found in the environment (Fernández-Martínez and Charlet, 2009; Masscheleyn et al., 1990; Ralston et al., 2008; Seby et al., 1998).
In France, the selenium concentration limit for safe drinking water has been set at 10 ppb, but in several areas this limit is exceeded (Chabart et al., 2006; Gourcy and Winckel, 2010; Karnay, 1999; Vernous et al., 1998). In the East and the West of the Paris Basin, geogenic sources of Se were identified and the enrichment of water by Se is considered as a result of oxidation processes after the drilling of wells (Gourcy and Winckel, 2010; Gourcy et al., 2011). In the French Department of Vienne, selenium anomalies (up to 40 ppb) were observed in different water distribution units and were attributed to the presence of continental selenium-rich facies formed by gravels and silty clay (Barron et al., 2009; Karnay, 1999).
This work presents a study that aims to identify and characterize the potential lithological source of selenium in the well-characterized limestone aquifer of the Hydrogeological Experimental Site of Poitiers (HESP), France.
2 Settings
The Hydrogeological Experimental Site of Poitiers (HESP) is a field research facility whose primary objective is to support the development of characterization methods and modelling approaches for groundwater flow and solute transport in heterogeneous carbonate aquifers, as a basis for the protection and management of groundwater resources. Hydrogeological and hydrogeochemical investigations focus on a confined limestone aquifer, which has been extensively characterized over an area of approximately 10 ha.
The HESP is located in a geological area called “Poitou Threshold” (Gabilly and Cariou, 2007), which makes the transition between two large Mesozoic–Cenozoic sedimentary basins, i.e. the Paris Basin to the northeast and the Aquitaine Basin to the southwest (Fig. 1). The Poitou Threshold consists of Jurassic carbonate rocks lying on a Hercynian crystalline basement. On the HESP area, the sedimentary cover begins with around 20-m-thick Aalenian dolomitic limestone and dolomite. These facies are overcome by the Bajocian limestone formations (50–60 m). Bathonian and Callovian rocks were completely eroded southward Poitiers, and the Cretaceous sedimentary formations are completely eroded in the region.
Location of the HESP in France.
Karst cavities partially filled with dark deposits are also present, and evidences based on the directional analysis of cave maps (Bodin and Razack, 1997) support the occurrence of karstification after the Cenozoic Pyrenean and Alpine tectonic phases.
Two limestone aquifers of regional extent occur in the Jurassic carbonate series:
- • the Lower- and Middle-Lias aquifer;
- • the Dogger aquifer.
These two aquifers are separated by the Toarcian aquitard consisting of low-permeability marls. The thickness of the Lower- and Middle-Lias aquifer is about 25 m, whereas the Toarcian aquitard and the Dogger aquifer are around 10 and 100 m thick, respectively. Previous works have demonstrated that these two aquifers are well isolated from each other by the Toarcian marls (Bodin et al., 2012; Chatelier et al., 2011). Furthermore, the coupling between flowmeter logs, borehole imaging logs and fluid temperature/conductivity logs indicates that the groundwater flow in the Dogger aquifer is strongly constrained within a three-dimensionally interconnected network and the low storativity values indicate that the Dogger aquifer is currently locally confined.
Water sampling campaigns between 2007 and 2012 revealed significant differences of aqueous total selenium content between wells located only a few tens meters from each other, from amounts lower to 5 ppb (detection limit) to more than 25 ppb. The HESP thus consists of an interesting area for the understanding of the Se behavior into a well-characterized aquifer.
3 Materials and methods
3.1 Rock sampling
A first set of samples was extracted from the C4 borehole drilled in 2004 (Table 1). These samples being stored at ambient room conditions, a partial alteration of these samples – mainly of the originally organic matter – has presumably occurred, limiting the investigations on the C4 samples to mineralogical and geochemical studies. The samples from Quaternary and Tertiary ages present specific anomalies (available as Supplementary Material, see Figs. 1 and 2, and Table 1) that will not be discussed in the present paper.
Concentrations of Se, SiO2, Al2O3, CaO, FeO, total sulfur, organic carbon and uranium in the samples from the C4 and the C5 wells.
| Age | Sample | Depth (m) | Description | Se (ppm) | SiO2 (%) | Al2O3 (%) | CaO (%) | FeO (%) | S (%) | Corg (%) | U (ppm) |
| Quaternary/Tertiary | C4 Qu1 | 9.97 | Conglomerate + shale + sandstone | 0.18 | 51.61 | 10.01 | 0.70 | < 0.05 | < 0.01 | < 0.01 | 6.75 |
| C4 Qu2 | 15.98 | Conglomerate + shale + sandstone | 0.05 | 66.74 | 12.57 | 0.87 | < 0.05 | < 0.01 | < 0.01 | 4.39 | |
| C4 Qu3 | 18.11 | Conglomerate + shale + sandstone | 1.72 | 53.77 | 15.16 | 0.52 | < 0.05 | < 0.01 | < 0.01 | 6.83 | |
| Bathonian | C4 Bt1 | 20.49 | Limestone | 0.07 | 0.40 | 0.07 | 55.84 | < 0.05 | 0.03 | 0.03 | 2.78 |
| C4 Bt2 | 26.24 | Silicified limestone | 0.28 | 2.23 | 0.20 | 54.73 | 0.07 | 0.02 | 0.03 | 1.46 | |
| C4 Bt3 | 31.56 | Silicified limestone | 0.03 | 1.19 | 0.30 | 55.25 | 0.07 | 0.01 | 0.02 | 3.79 | |
| C4 Bt4 | 35.67 | Silicified limestone | 0.05 | 0.78 | 0.13 | 55.91 | 0.05 | 0.03 | 0.04 | 1.72 | |
| C4 Bt5 | 40.57 | Silicified limestone | 0.21 | 0.71 | 0.09 | 56.00 | < 0.05 | 0.02 | 0.04 | 2.33 | |
| C4 Bt6 | 46.51 | Limestone | 0.07 | 0.17 | 0.15 | 56.10 | < 0.05 | 0.03 | 0.05 | 1.40 | |
| Bajocian | C4 Bj1 | 50.88 | Limestone | 0.11 | 0.06 | 0.08 | 56.25 | 0.03 | 0.03 | 0.04 | 1.55 |
| C4 Bj2 | 55.76 | Limestone | 28.00 | 0.07 | 0.13 | 55.61 | 0.14 | 0.07 | 0.06 | 3.11 | |
| C4 Bj3 | 57.81 | Argillaceous/Marly limestone | 153.00 | 59.99 | 16.09 | 6.42 | 0.53 | 0.34 | 0.91 | 27.28 | |
| C4 Bj4 | 61.68 | Limestone | 0.19 | 0.58 | 0.35 | 54.71 | 0.10 | 0.02 | 0.03 | 1.54 | |
| C4 Bj5 | 67.71 | Limestone | 0.14 | 0.59 | 0.34 | 54.85 | 0.11 | 0.23 | 0.07 | 7.07 | |
| C5 Bj1 | 67.86 | Argillaceous | 1.70 | 15.57 | 12.00 | 34.18 | 0.8 | 3.02 | 2.94 | 406.28 | |
| C5 Bj2 | 69.36 | Argillaceous | 2.10 | 39.49 | 29.23 | 1.22 | 1.5 | 5.98 | 7.51 | 912.60 | |
| C4 Bj6 | 72.14 | Argillaceous/Marly limestone | 6.40 | 1.38 | 0.65 | 53.96 | 0.09 | 0.04 | 0.06 | 0.53 | |
| C4 Bj7 | 74.88 | Argillaceous/Marly limestone | 8.10 | 33.02 | 24.23 | 14.44 | 0.46 | 0.13 | 0.57 | 25.94 | |
| C4 Bj8 | 82.23 | Argillaceous/Marly limestone | 10.50 | 0.83 | 0.42 | 54.70 | 0.10 | 0.04 | 0.04 | 0.58 | |
| C4 Bj9 | 86.04 | Limestone | 0.11 | 2.41 | 0.63 | 52.58 | 0.20 | 0.36 | 0.09 | 24.73 | |
| C4 Bj10 | 91.50 | Argillaceous/Marly limestone | 6.10 | 25.10 | 9.37 | 15.90 | 1.86 | 13.17 | 0.73 | 12.58 | |
| C4 Bj12 | 92.58 | Limestone | 3.80 | 13.85 | 7.81 | 32.02 | 1.42 | 3.72 | 1.08 | 18.53 | |
| C4 Bj13 | 93.29 | Argillaceous | 495.00 | 34.26 | 21.86 | 4.17 | 2.52 | 7.26 | 1.22 | 36.09 | |
| C4 Bj14 | 94.81 | Argillaceous | 0.05 | 0.83 | 0.48 | 54.37 | 0.15 | 0.02 | 0.04 | 1.16 | |
| C4 Bj15 | 96.38 | Limestone | 0.14 | 4.12 | 1.07 | 51.56 | 0.20 | 0.03 | 0.04 | 2.96 | |
| C4 Bj16 | 102.13 | Argillaceous/Marly limestone | 0.12 | 1.91 | 0.54 | 35.89 | 0.61 | 0.36 | 0.10 | 3.25 | |
| Aalenian | C4 Aa1 | 105.50 | Limestone | 0.04 | 2.35 | 0.55 | 52.45 | 0.22 | 0.17 | 0.07 | 2.82 |
| C4 Aa2 | 110.72 | Limestone | 0.17 | 2.86 | 0.60 | 43.59 | 0.42 | 0.65 | 0.12 | 3.00 | |
| C4 Aa3 | 115.67 | Limestone | 0.09 | 6.96 | 1.02 | 30.45 | 0.54 | 0.53 | 0.13 | 1.40 | |
| C4 Aa4 | 119.02 | Cherty limestone | 0.12 | 12.64 | 2.38 | 40.54 | 0.31 | 1.11 | 0.27 | 2.53 |
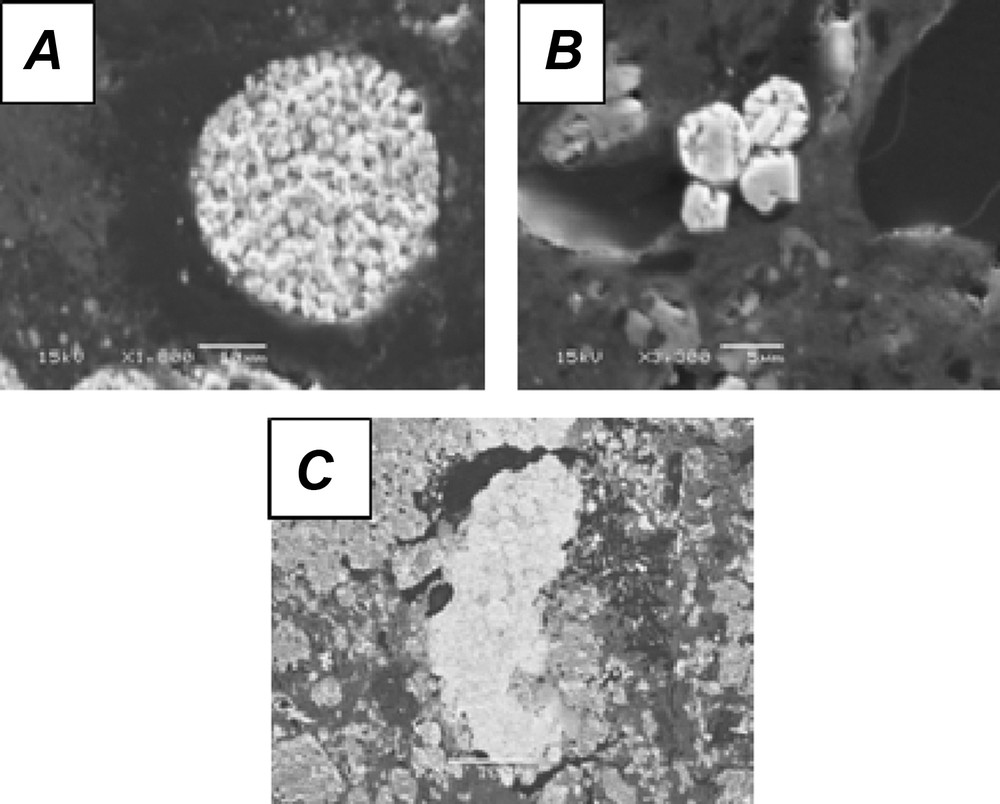
Main identified pyrite morphologies (secondary electron images). A. Framboïdal pyrite. B. Euhedral pyrite. C. Elongated pyrite.
During the drilling of the C5 well (2012), fresh samples were collected as cylinders of 10 cm diameter per 15 cm length and stored under anaerobic atmosphere (argon) at low temperature (–18 °C). Extensive studies were performed on two C5 argillaceous samples from different depths.
3.2 Mineralogy
Powders were obtained by grinding in an agate mortar to get the infra 50 μm fraction, while oriented preparations were obtained from suspensions in osmosed water under ultrasonic treatment. The mineralogical study of the samples was performed following the standard powder procedures outlined by Brindley and Brown (1980) and Moore and Reynolds (1997) for X-ray diffraction (XRD) analysis. The oriented preparation are analyzed after being air dried and after vapor salvation with ethylene glycol at 60 °C for 12 hours and heat for 4 hours at 350 °C and 550 °C. The diffractogram patterns were acquired with a Philips Panalytical X’pert Pro apparatus using copper Cu Kα radiation (λCu Kα = 1.5418 μm), a Soller slit of 0,4 rad, an antiscattered slit of 1/4°2θ, an Ni filter and an Xccelerator detector. The analytical conditions were: 40 kV, 40 mA, steps of 0,017° 2θ and 0.033° 2θ and investigated angular ranges from 2 to 65° 2θ and from 2 to 35° 2θ for powders and oriented preparations, respectively. Phase identification was made with X’pert HighScore software using JCPDS Pdf2 mineralogical database. The semi-quantifications were realized after deconvolution and integration of the main peak area of each phase using Fytyk software.
3.3 Elemental analysis
Elemental analysis was performed on powered samples representing all the geological facies of the C4 well and the two fresh clayey powdered samples (C5Bj1 and C5Bj2) of the C5 well. The samples were analyzed at the SARM laboratory (CRPG, CNRS, Nancy, France). Elemental compositions were obtained after LiBO2 fusion and acid dissolution. Major elements were quantified by ICP–AES Thermo Fischer Cap 6500 and trace elements by ICP-MS Agilent 7700XI, following the methods described by Carignan et al. (2001). Total organic carbon (after treatment with concentrated HCl to remove inorganic carbon) and total sulfur were determined using a carbon–sulfur elemental analyzer LECO SC 144DR (CRPG, CNRS, Nancy, France).
The number of data (inferior or equal to 30 per element) is insufficient to realize a multivariable analysis and statistical studies were only realized using Pearson correlations coefficients calculated using “R3.0.2” software between the normalized values of the elemental contents.
3.4 Petrography
To prepare standard-type thin sections (26 mm × 46 mm), the frozen samples were cut off by dry sawing before impregnation at room temperature under atmospheric pressure with a two-component glue composed of a polymerizing agent (Araldite) and 20% acetone. The thin sections were first observed using an optical polarized light microscope to investigate the rock structure and the different sedimentary figures. After carbon coating, the samples were then observed using a scanning electron microscope JEOL 5600-LV [Low Vacuum] (IC2MP, Poitiers) equipped with a secondary electron detector Everhart–Thornley and a backscattered electron detector Centaurus (KE Development ic, Cambridge) with a Bruker energy dispersive X-ray spectrometer for scanning electron microanalyses (SEMA). Data processing was provided by chemical analysis software QUANTAX. The petrographic investigation focuses on the identification, location and description of significant minerals morphologies and tends to define the distribution of selenium into the matrix.
3.5 Organic matter analysis
A molecular characterization of the organic matter was performed by thermochemolysis coupled with a GC–MS on the C5 samples. The experimental method described by Grasset and Amblès (1998) was applied to powdered samples (2.5 g) placed in a ceramic boat with an excess of a 50% (w/w) methanol solution of tetramethylammonium hydroxide (TMAH). The sample was left stand for 1 h to allow the TMAH solution to fill all the pores. Then the boat was placed in a Pyrex tube and heated at 400 °C for 30 min in a tubular furnace. Thermochemolysis products were then swept by nitrogen N2 (flow rate: 100 mL/min) to a trap containing dichloromethane. After heating, the pyrolysate was transferred quantitatively into a round-bottom flask, vacuum-dried, weighed, and re-dissolved in dichloromethane and stored at 0 °C until GC–MS analysis.
Gas Chromatography–Mass Spectrometry analysis was achieved with a Trace GCThermo Finnigan (split injector, 250 °C; FID, 300 °C) with a fused silica capillary column (Supelco Equity 5%, 30 m in length, 0.25 mm i.d., 0.25 m in film thickness), and helium as the carrier gas. The oven was initially kept at 60 °C for 1 min, and then heated at a rate of 5 °C·min−1 to 300 °C and maintained at that temperature for 15 min. The column was coupled with a Finnigan Trace MS quadrupole mass spectrometer (ionization energy 70 eV, mass range m/z 45–600, cycle time 1 s). The various organic products were finally identified on the basis of their GC retention times, their mass spectra (comparison with standards), and the literature data.
3.6 Palynological study
The C5 samples were analyzed by Geobiostratdata, consulting company using the following conventional treatment method of argillaceous sediments for palynological studies: acid digestion successively by HCl–HF–HCl, density gradient in ZnCl2, concentration by sieving at 10 μm and holding between slide and cover-slip of 50 μL of the treatment residue diluted in glycerol. The obtained slides were examined at a magnification of ×250 using a Zeiss light microscope.
4 Results and discussion
4.1 Mineralogical investigations
According to the lithology described above (see Settings), the most common minerals are carbonates (Table 2). Dolomite is associated with calcite at highly variable levels within Aalenian levels and at the bottom of the Lower Bajocian. Calcite is the only carbonate found in the Bathonian and, as expected, the contents of calcite drop sharply into levels corresponding to the karst infillings and in some sandy clay levels.
Mineralogy of the carbonated and argillaceous samples.
| Well | Age | Sample | Sm (%) | C (%) | I (%) | K (%) | Arg (%) | Gy (%) | Jar (%) | An (%) | Q (%) | F (%) | P (%) | Ca (%) | Do (%) | Go (%) | Py (%) |
| C4 | Quaternary/ | C4Qu1 | 3.1 | 0 | 0 | 0 | 11.6 | 0 | 0 | 4.4 | 82.8 | 1.2 | 0 | 0 | 0 | 0 | 0 |
| Tertiary | C4Qu2 | 58.9 | 0 | 1 | 40.1 | 50.4 | 0 | 0 | 3.3 | 46.3 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Bathonian | C4Bt1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.6 | 0 | 0 | 99.4 | 0 | 0 | 0 | |
| C4Bt2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 84 | 0 | 0 | 15.7 | 0 | 0.4 | 0 | ||
| C4Bt4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1.2 | 0 | 0 | 98.8 | 0 | 0 | 0 | ||
| Bajocian | C4Bj1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | |
| C4Bj3* | 20.6 | 0 | 0 | 79.4 | 52.2 | 0 | 0 | 6 | 27.6 | 1.2 | 0 | 0 | 0 | 13 | 0 | ||
| C4Bj4 | 28.1 | 2 | 43.6 | 26.4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | ||
| C4Bj5 | 21.1 | 1.2 | 40.8 | 36.9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0 | 0 | 0 | ||
| C4Bj7* | 34 | 0 | 0.6 | 65.4 | 51.5 | 0 | 0 | 8 | 2 | 2 | 0 | 36.5 | 0 | 0 | 0 | ||
| C4Bj9 | 0 | 1.3 | 62.4 | 36.3 | 2.7 | 0 | 0 | 0 | 1.5 | 0 | 0 | 94.4 | 0.6 | 0 | 0.8 | ||
| C4Bj10* | 49.6 | 0 | 4.8 | 42.7 | 15.7 | 12.4 | 3.3 | 0 | 13.3 | 1.7 | 0 | 43.7 | 0 | 5.6 | 4.3 | ||
| C4Bj11 | 0 | 0 | 47.6 | 52.4 | 2.1 | 0 | 0 | 0 | 1.6 | 0 | 0 | 95.5 | 0.7 | 0 | 0.1 | ||
| C4Bj12* | 52.8 | 0 | 17.7 | 29.5 | 4.5 | 3.2 | 1.5 | 0 | 0.7 | 0 | 0 | 89.2 | 0 | 0 | 0.9 | ||
| C4Bj13* | 51.6 | 1.1 | 20.5 | 26.8 | 16.3 | 6.1 | 0.8 | 0 | 2.4 | 0 | 0 | 72.5 | 0 | 0 | 2 | ||
| C4Bj14 | 78 | 0 | 7.4 | 14.5 | 1.4 | 0 | 0 | 0 | 0.7 | 0 | 0 | 96.2 | 0 | 1.6 | 0.1 | ||
| C4Bj16 | 22.6 | 4.3 | 36.7 | 36.4 | 1.2 | 0 | 0 | 0 | 1.4 | 0 | 0.3 | 13.3 | 83.5 | 0 | 0.3 | ||
| Aalenian | C4Aa2 | 30.3 | 2.1 | 43.7 | 24 | 0.7 | 0.5 | 0 | 0 | 5.6 | 0.1 | 0.3 | 0 | 92.2 | 0 | 0.6 | |
| C4Aa4 | 0 | 0 | 56.1 | 43.9 | 0.7 | 0 | 0 | 0 | 43.5 | 0 | 0 | 29.7 | 25.7 | 0 | 0.4 | ||
| C5 | Bajocian | C5Bj1* | 0 | 0 | 0 | 100 | 19.5 | 0 | 0 | 2.9 | 0 | 0 | 0 | 76.6 | 0 | 0 | 1 |
| C5Bj2* | 8 | 0 | 0 | 92 | 79 | 0 | 0 | 14.8 | 3.7 | 0 | 0 | 0 | 0 | 0 | 2.5 |
Within the carbonated levels, clays are only present in low quantities (0.7–2.4%) in some samples of Bajocian and Aalenian ages. Clay quantities are variable within Bajocian argillaceous infillings (4.5–79%). In carbonated levels, illite is the most abundant clay mineral except two Bajocian fillings, where smectite and kaolinite are dominant. Kaolinite is dominant in Upper and Middle Bajocian infillings, while smectite is the most abundant clay mineral in Lower Bajocian infillings.
The detrital assemblage is mainly composed of quartz. In levels where this mineral is most expressed, it is usually associated with microcline, except in the Aalenian and the Lower Bajocian bottom, where an albite-type plagioclase is present. In the Bajocian argillaceous levels, quartz may be associated primarily with anatase and small amounts of other heavy minerals, such as zircon and rutile, identified by XRD and SEM-EDX.
The Lower and Middle Bajocian terranes are further characterized by the presence of iron-rich minerals, with pyrite (FeS2) generally associated or not with gœthite, and sulfates, mainly small amounts of gypsum accompanied by jarosite. In the Lower Bajocian, pyrite and sulfates are found both in the host limestone and in the karst infillings. Sulfates are absent in karst infillings located in limestones of the Middle to the Upper Bajocian. Finally, gœthite is only well expressed in the sandy clay level.
The differences in clay type can demonstrate that the argillaceous infillings are not originated from a simple dissolution of the limestone, because the weathering of a parental rock does not produce the same original argillaceous assemblages (Velde and Meunier, 2008). The high quartz quantities in some argillaceous levels and the presence of other detrital minerals such as anatase, zircon and rutile also support a detrital origin of these argillaceous infillings.
4.2 Occurrence of selenium in the HESP
Se-enriched zones were attributed to samples with Se content above 1 ppm. Such samples are observed in some Bajocian levels (Table 1) along with a depletion of calcium and inorganic carbon (not shown), which corresponds to the carbonated aquifer. On the contrary, these samples present a considerable enrichment in silicon and aluminum, consistent with the presence of clays as the most abundant phases of these levels. The exceptions – samples C4Bj2, C4Bj6 and C4Bj8 – correspond to the border samples between the limestone host rock and the identified selenium-enriched argillaceous levels. The argillaceous levels are therefore suspected to be the main carrier matrix of selenium in the Dogger's aquifer.
The argillaceous levels rich in selenium are consistent with the three karstified levels described by Audouin et al. (2008) at approximately 50, 80 and 110 m, suggesting that clayey matrices fulfill the main karst cavities. The argillaceous levels were identified as black clays with high values in the natural gamma log (NG > 1000 API American Petroleum Institute unit) (Keys, 1990) observed in several wells (Audouin et al., 2008). The high natural gamma logs were interpreted by the presence of high uranium content in these clays that reach 200 ppm, as calculated by the conversion of the API levels to ppm. Elemental quantifications (Table 1) show an important enrichment with uranium, but also an important heterogeneity of uranium concentrations in the argillaceous levels.
Some Se-rich argillaceous levels are also enriched with many other elements including Fe (II), sulfur and/or organic carbon. As the association of selenium with organic matter and pyrite was described by many authors (Kulp and Pratt, 2004; Martens and Suarez, 1997; Matamoros-Veloza et al., 2011), the enrichment of the argillaceous levels in selenium would therefore be easily explained by the presence of pyrite and organic matter in the argillaceous matrix. But other samples are rich in selenium and not enriched in iron, sulfur or organic carbon. The correlations are therefore more complex than a “simple” pyritic and/or an organic association, and should be consolidated by a statistical study.
4.3 Correlation analysis and selenium distribution
The present correlation analysis (Table 3) is consistent, for major elements such as Ca, Si or Al, with the lithological description and the mineralogical studies (Table 2). For instance, within the “limestone” samples, negative correlations between Ca and Mg (–0.95) or reduced iron FeII (–0.95) are coherent with the presence of dolomite and ferrous carbonates that substituted calcium carbonate in the Aalenian levels and at the bottom of the Lower Bathonian.
Semi-matrices of correlation of the argillaceous samples and of the limestone samples.
| Al | As | C inorg | C org | Ca | Cs | Cu | F | Fe II | Mg | Mn | P | Rb | REE | S | Se | Si | Sr | U | V | |
| Al | 1.00 | 0.40b | –0.88 b | 0.86 b | –0.59b | 0.98 b | 0.33b | 0.75 b | 0.46b | 0.30b | –0.25b | 0.32b | 1.00 b | 0.60b | 0.81 b | –0.21b | 0.96 b | 0.38b | 0.03b | 0.51b |
| As | –0.18a | 1.00 | –0.22b | 0.48b | –0.52b | 0.38b | 0.68b | 0.50b | 0.57b | 0.43b | –0.09b | 0.14b | 0.38b | 0.32b | 0.62b | –0.09b | 0.38b | –0.11b | –0.01b | 0.46b |
| C inorg | –0.57a | 0.39a | 1.00 | –0.77 b | 0.28b | –0.89 b | –0.22b | –0.47b | –0.12b | 0.03b | 0.31b | –0.32b | –0.90 b | –0.47b | –0.70 b | 0.21b | –0.87 b | –0.42b | –0.09b | –0.25b |
| C org | –0.16a | 0.77 a | 0.19a | 1.00 | –0.69b | 0.82 b | 0.41b | 0.66b | 0.57b | 0.46b | –0.31b | 0.00b | 0.85 b | 0.39b | 0.97 b | –0.14b | 0.91 b | 0.19b | 0.10b | 0.19b |
| Ca | –0.67a | 0.25a | 0.96 a | –0.04a | 1.00 | –0.54b | –0.35b | –0.87 b | –0.95 b | –0.95 b | 0.11b | –0.10b | –0.55b | –0.37b | –0.73 b | 0.23b | –0.68b | 0.11b | 0.05b | –0.39b |
| Cs | 0.20a | 0.79 a | –0.17a | 0.86 a | –0.37a | 1.00 | 0.27b | 0.77 b | 0.44b | 0.25b | –0.21b | 0.44b | 0.99 b | 0.60b | 0.76 b | –0.21b | 0.93 b | 0.38b | 0.04b | 0.56b |
| Cu | 0.08a | 0.85 a | 0.22a | 0.36a | 0.17a | 0.60a | 1.00 | 0.25b | 0.34b | 0.26b | –0.02b | –0.09b | 0.28b | 0.21b | 0.52b | –0.25b | 0.33b | –0.07b | –0.12b | 0.13b |
| F | 0.20a | 0.88 a | –0.05a | 0.81 a | –0.23 | 0.98 a | 0.75 a | 1.00 | 0.84 b | 0.74 b | 0.00b | 0.49b | 0.74 b | 0.57b | 0.67b | –0.25b | 0.76 b | 0.08b | –0.05b | 0.70 b |
| Fe II | –0.17a | 0.85 a | –0.04a | 0.64a | –0.09a | 0.81 a | 0.78 a | 0.85 a | 1.00 | 0.93 b | 0.06b | 0.14b | 0.43b | 0.31b | 0.65b | –0.17b | 0.52b | –0.23b | 0.04b | 0.47b |
| Mg | 0.03a | 0.97 a | 0.29a | 0.84 a | 0.09a | 0.88 a | 0.79 a | 0.94 a | 0.78 a | 1.00 | –0.05b | –0.01b | 0.25b | 0.21b | 0.53b | –0.19b | 0.42b | –0.25b | –0.11b | 0.26b |
| Mn | –0.65a | 0.28a | 0.98 a | 0.03a | 1.00 a | –0.33a | 0.16a | –0.19a | –0.10a | 0.13a | 1.00 | 0.35b | –0.25b | –0.33b | –0.27b | 0.20b | –0.29b | –0.61b | 0.23b | 0.28b |
| P | 0.27a | 0.83 a | –0.17a | 0.67a | –0.31a | 0.94 a | 0.81 a | 0.98 a | 0.88 a | 0.88 a | –0.29a | 1.00 | 0.36b | 0.40b | 0.01b | 0.04b | 0.25b | –0.12b | 0.07b | 0.63b |
| Rb | 0.02a | 0.68a | –0.15a | 0.94 a | –0.35a | 0.94 a | 0.34a | 0.86 a | 0.71 a | 0.77 a | –0.30a | 0.77 a | 1.00 | 0.59b | 0.79 b | –0.21b | 0.96 b | 0.39b | 0.07b | 0.52b |
| REE | 0.82 a | –0.42a | –0.36a | –0.65a | –0.32a | –0.30a | 0.03a | –0.23a | –0.43a | –0.32a | –0.34a | –0.11a | –0.54a | 1.00 | 0.39b | –0.29b | 0.49b | 0.42b | 0.19b | 0.53b |
| S | –0.45a | 0.42a | –0.13a | 0.12a | 0.02a | 0.30a | 0.51a | 0.33a | 0.78 a | 0.24a | –0.05a | 0.44a | 0.23a | –0.39a | 1.00 | –0.21b | 0.86 b | 0.09b | 0.17b | 0.24b |
| Se | 0.48a | 0.50a | –0.49a | 0.70 a | –0.68a | 0.92 a | 0.36a | 0.84 a | 0.60a | 0.66a | –0.64a | 0.83 a | 0.87 a | –0.07a | 0.10a | 1.00 | –0.23b | –0.49b | –0.10b | –0.29b |
| Si | 0.44a | –0.64a | –0.73 a | –0.08a | –0.77 a | –0.05a | –0.73 a | –0.24a | –0.43a | –0.46a | –0.75 a | –0.25a | 0.12a | 0.17a | –0.44a | 0.31a | 1.00 | 0.27b | 0.01b | 0.37b |
| Sr | –0.54a | 0.41a | 1.00 a | 0.14a | 0.96 a | –0.18a | 0.29a | –0.04a | –0.02a | 0.29a | 0.98 a | –0.15a | –0.19a | –0.30a | –0.09a | –0.50a | –0.78a | 1.00 | –0.21b | 0.16b |
| U | 0.79 a | 0.19a | –0.44a | 0.45a | –0.67a | 0.64a | 0.12a | 0.57a | 0.11a | 0.43a | –0.60a | 0.54a | 0.57a | 0.31a | –0.43a | 0.84 a | 0.49a | –0.45a | 1.00 | 0.15b |
| V | 0.62a | 0.57a | -0.40a | 0.59a | -0.59a | 0.89 a | 0.56a | 0.88 a | 0.60a | 0.72 a | -0.55a | 0.89 a | 0.74 a | 0.15a | 0.07a | 0.95 a | 0.12a | -0.38a | 0.85 a | 1.00 |
a Argillaceous samples.
b Limestone samples.
Within the limestone aquifer, no element is significantly correlated with selenium, probably disseminated between different structural components (Table 3, top). However, selenium concentrations are relatively low (Table 1) in the limestone host rock samples, and high positive anomalies are only observed in the “argillaceous” and “border” samples.
Literature data generally reported selenium affinities for pyritic materials, and more generally for metallic sulfide minerals (Fernández-Martínez and Charlet, 2009; Perkins and Foster, 2004; Wang et al., 2010), but also for mature organic matrices like coals (Coleman et al., 1993; Finkelman et al., 1999) and for phosphates (Fernández-Martínez and Charlet, 2009). When focusing on “argillaceous” levels (Table 3, bottom), selenium is well correlated with P (0.83), moderately with Corg (0.70) and FeII (0.60), which could be consistent with a distribution between the pyritic, apatite and organic materials present in the matrix. Selenium is also well correlated with U (0.84) and surprisingly highly correlated with vanadium (0.95), alkali metals (Cs [0.92] and Rb [0.87]) and fluorine (0.84). If a chemical affinity is highly unlikely, the abundance of selenium and of these elements is clearly concomitant. In parallel, it is interesting to note that similar high correlations are observed between these elements and organic carbon (Corg–Rb: 0.94; Corg–Cs: 0.86; Corg–F: 0.81; Corg–V: 0.59) within the argillaceous levels. Association between V and organic matter was reported for coals (Huggins and Huffman, 2004) and soils (Połedniok and Buhl, 2003). Rb and Cs have similar chemical properties and are besides strongly correlated (0.94). The association between Cs and organic matter has already been described in soils (Mabit and Bernard, 1998; Takenaka et al., 1998; Tegen and Dörr, 1996) and organic groundwater colloids (Caron et al., 2008). The affinity of these two alkaline elements with organic matter may indirectly explain their affinity for selenium, through the presence of ternary complexes for instance (Gustafsson and Johnsson, 1994).
Other elements present high correlations with the organic carbon and could be used to better define the nature of organic matter. Within the “limestone” aquifer, organic carbon is well correlated with Si (0.91) and Al (0.86), and especially with sulfur (0.97), which presumably corresponds to a marine-influenced diagenesis environment, where high concentrations of sulfate and iron salts in marine water leads to the formation of pyrite and organic sulfur (Wang et al., 2008; Wang et al., 2010). These correlations are not observed in the “argillaceous” filling materials (Corg–Si:–0.08; Corg–Al:–0.16; Corg–S: 0.12).
As for the organic C, most of the correlations within the “limestone” aquifer are distinguishable from the “argillaceous” samples, which demonstrates that the filling materials have an external origin and are not simply originated from the dissolution of the host rock. This tendency is particularly observed for iron, which is weakly correlated with S (0.65), As (0.57) and Cu (0.34), and strongly correlated with Mg (0.93), due to its carbonated nature in the “limestone” samples, whereas high correlations with S (0.78), Cu (0.78) and As (0.85) are observed in the “argillaceous” levels, probably due to an incorporation of these elements in pyrite during its early stage of formation (Huerta-Diaz and Morse, 1992; Morse and Arakaki, 1993) or to their presence as sulfide micro-inclusions (Deditius et al., 2010; Large et al., 2009).
The correlation analysis thus confirmed that the argillaceous infillings are detrital sediments from terrestrial external origin: they are not originated from the limestone local rocks alteration. Within the argillaceous sediments, selenium should be mainly associated with the organic fraction.
4.4 Extensive characterization of the karst infillings
4.4.1 Petrographic investigations
The black clayey samples present an alternation of inhomogeneous depositions observed as non-parallel crossed laminations more or less rich in carbonates or clays. Carbonate minerals are present from submillimetric to millimetric grains either as fossil fragments or as grains (< 0.1 mm) that probably belong to the matrix background. A part of the carbonates would thus have precipitated in situ.
Clays appear as homogeneous millimetric light areas separated by dark brown millimetric laminations. Elemental analysis shows a chemical composition similar to those of pure kaolinite in homogeneous domains and mixtures of kaolinite and smectite minerals, but also quartz, iron oxides, and calcite in dark zones. Within the laminated argillaceous deposits, organic matter appears as black or dark brown patches.
Three main morphologies of pyrite (Fig. 2) are observed, suggesting different ways of their formation (Raiswell, 1982). Framboidal pyrite, isolated or in clusters, is the main type encountered. These crystals have a composition similar to the theoretical formula of pyrite (FeS2), without detectable substitution of sulfur by selenium. The formation of framboidal pyrite is interpreted as a combination of biotic and inorganic processes (Wilkin, 1995). Large framboidal pyrite – from 4 to 50 μm – is generally formed in pore water of anoxic sediments during diagenesis, while smaller framboidal pyrite (< 6 μm) is formed in anoxic water and then buried in the sediments (Wilkin and Barnes, 1997). As most of the pyritic crystals observed in the samples from the C5 well have large diameters, a formation in the porosity after deposition is assumed. These crystals were then probably preserved after an early diagenetic stage and a rock-forming process (Pierret et al., 2000; Wilkin et al., 1996).
Less commonly observed are euhedral pyrite crystals that could be directly formed by nucleation and growth in supersaturated solution (Giblin and Howarth, 1984) or are transformed from FeS precursor precipitated from an initial supersaturated solution (Schoonen and Barnes, 1991). Elongated framboidal pyrites were only identified in relatively carbonated-rich levels and consist of spherical pyrite crystals cemented with uranium phosphate abundantly present in the spaces between the spheres. The association between pyrite and uranium was observed in uranium ores (Cunningham et al., 1998; England et al., 2001; Simpson and Bowles, 1977). No uranium ore was described in the Poitou threshold, suggesting a transportation of these materials probably from the Massif Central or the Armorican Massif (Fig. 1).
Abundance of titanium oxides, zircons, and quartz emphasizes the detrital origin of these deposits. Most rarely, calcium phosphates were identified; their presence may be of biological origin or result from igneous rocks erosion. Organic matter was also observed disseminated between the carbonated and the argillaceous areas or as dark brown filling cavities of homogenous color, suggesting a dissociation of organic and mineral matters in some areas.
Organic matter areas, argillaceous zones rich in organic matter, apatite and pyrite are all potential candidates for selenium-bearing species. Elemental mapping of these specific hosts in the studied samples was thus performed to better understand the distribution of selenium. The concentration of selenium in the samples being lower than the limit of detection of the X-ray spectrometer used in the present study, only localized positive anomalies are expected to be observable (Se concentrations higher than 0.1%). The observations of the thin sections tend to show no areas with high concentration of selenium, suggesting no specific association with a particular component of the geological matrix, but rather a diffuse distribution of selenium within the individual components.
4.4.2 Characterization of the “argillaceous” organic matter
All the analyzed argillaceous samples from the C5 well present similar chromatographic responses after thermochemolysis. The chromatogram of the thermochemolysis products derived from the C5Bj2 sample is depicted in Fig. 3. No n-alkane/n-alkene series were identified, phenols, alkylphenanthrenes and alkylanthracenes being the major products. The relative intensity of the chromatographic peaks (unresolved complex mixture – UCM) between 20 and 40 min. Fig. 3 suggests a high degree of condensation of the aromatic structures. Characteristic ions were used to examine the alkylnaphthalene and alkylphenanthrene isomers (Fig. 3).
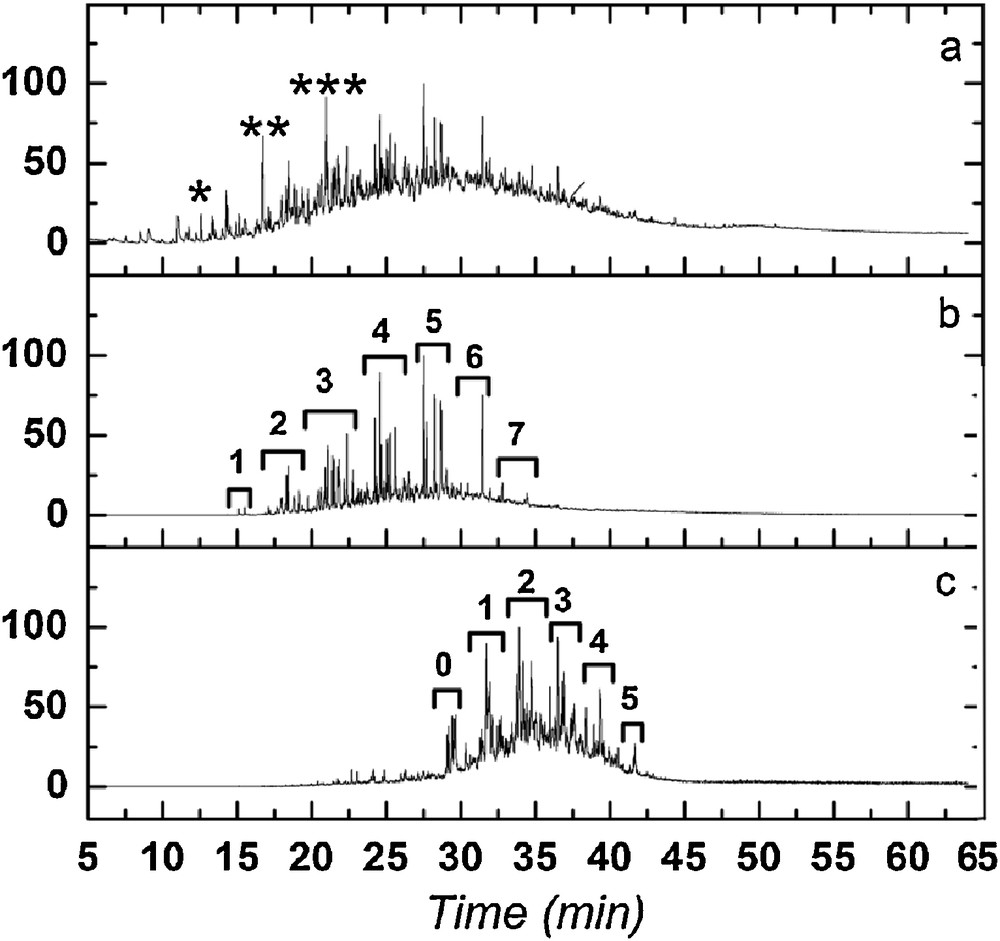
Chromatogram from thermochemolysed black shales (C5Bj2). a: total Ion current Chromatogram (TIC) (*: dimethylphenol [as methoxy], **:trimethylphenol, ***: tetramethylphenol); b: extracted Ion Chromatogram (XIC) of alkylnaphtalenes ([m/z] = 142, 156, 170, 184, 198, 212 and 226]); c: extracted Ion Chromatogram (XIC) of alkykphenanthrenes ([m/z] = 192, 206, 220, 234, 248 and 262). Numbers in Fig. 3b and c indicate the number of methyl-substituted carbons.
The identified compounds could have a recent anthropogenic origin from industrial activities (Purcaro et al., 2013), but the dominant economic activities – mainly agriculture – in the local area around the HES are not coherent with this interpretation. Moreover, soils and sediments polluted by hydrocarbons should present a more complex and diversified mixture of organic hydrocarbons (Biache et al., 2014; Wakeham et al., 2003; Wilcke et al., 2014; Witt, 1995).
The phenolic nature of the degradation products agrees with the relatively low rank of the raw materials (Hartgers et al., 1994). Phenolic compounds in the degradation products of organic sediments are usually associated with diagenetically altered lignin. It is however difficult to relate in the studied samples phenols with the structure of lignin. It should be taken into account that the organic matter present in the C5Bj2 sample cannot be unequivocally established as arising solely from woody tissues, because other sources for phenolic compounds such as tannins (Christiansen et al., 1995), which may at least in part explain the presence of the phenol isomers found here, cannot be discarded.
The distribution of phenols depends on several factors such as the initial composition of the raw organic material and the physicochemical conditions of the depositional environment. However, the maturation of the organic matter during the transformation from soft brown coal to sub-bituminous coal involves the elimination of methoxy and phenolic groups and the formation of polycyclic aromatics such as phenanthrenes and anthracenes (Philp et al., 1982), and high proportions of alkylnaphtalenes and alkylphenanthrenes were found to be dominant in mature coal (Armstroff et al., 2006).
1,2,5-Trimethylnaphtalene and 1,2,5,6-tetraramethylnaphtalene were found to be abundant in shales and coal samples (Püttmann and Villar, 1987), and 1,7-dimethylphenanthrene could derive from pimaric acid, common to plants and pine resins (Wakeham et al., 1979). In lignite coals, the presence of alkylnaphtalene was also attributed to the degradation of terpenoid and alkylnaphtalene probably derived from abiandic and sadarocopimaric acids, abundant in resins and pine forests (Fazeelat and Asif, 2005). We can thus confirm the high degree of maturation of the analyzed organic matter and affirm that the identified phenanthrenes and naphthalenes are derived from coniferous plants.
4.4.3 Palynological investigations
Samples C5Bj1 and C5Bj2 contain enough and sufficiently well-preserved palynomorphs to be characterized. Palynological assemblages are dominated by pollens of Gymnosperms (Pinaceae, Cupressaceae, Taxodiaceae). Angiosperms are rare, thus eliminating a Neogene or Paleogene origin. Spores – mainly Pteridophytes – are abundant and frequently separated from their perispore, suggesting an input by aquatic run-off. The presence of Cicatricosisporites tends also to locate both samples in a Mesozoic period (Kremp and Kawasaki, 1972).
The abundance of Cupressaceae and Cyperaceae combined with the presence of Alnus in C5Bj1 (Leopold et al., 2012) demonstrates that the deposition of C5Bj1 is more recent than that of C5Bj2. The presence of Classopollis in C5Bj2 is conclusive. Classopollis is described from the Jurassic to the Lower Cretaceous included (Pocock and Jansonius, 1961). In the Vendée region, near to HESP site, its extinction was reported at the end of the Santonian (Azéma et al., 1981). From the Turonian to the Santonian, Classopollis was abundant in Vendée, and Normapolles was present with a high frequency. The comparison with pollen assemblages from the Vendée region allows us to date the sample C5Bj2, in which Normapolles is absent, from the Cenomanian, when Pinaceae pollens were abundant in Vendée (Azéma et al., 1981).
Finally, palynological assemblages suggest an Upper Cretaceous age for both samples, probably Cenomanian for C5Bj2 and Santonian to Maastrichtian for C5Bj1.
5 Conclusion: origin and history of the filling materials
This study aims to characterize the geogenic source of selenium in the HESP to further understand Se dynamics in a well-characterized Jurassic limestone aquifer. Selenium appears mainly concentrated in argillaceous sediments that fulfill some karstic cavities developed within the Bajocian host rock paleokarst.
These filling materials mainly concentrated light REE, Y, heavy elements such as Th, U and Zr, but also Se, iron, sulfur and organic carbon. A wide variability of enrichments suggests a wide variability of sources for the different elements, and thus a continental origin (Dinis et al., 2016).
Correlations between elemental analysis data allow us to distinguish many geochemical differences between “argillaceous” and “limestone” samples, which supports an external input. Markers of transport were also clearly identified in the argillaceous matrix and some of them cannot be originated from the dissolution of the limestone host rocks. The argillaceous composition proved also heterogeneous: while in the “C4 well” deep argillaceous samples are enriched in smectite, in the “C5 well” and in the “C4 well” more superficial samples, kaolinite is dominant. This indicates climate differences at the time of the formation of these minerals: kaolinite being formed in hot and moist climates, and smectite in dry climates (Eberl et al., 1984).
Organic matter is assumed to be originated from vascular plants that have been transported from the surface into karst cavities. Palynological assemblages date the C5 samples from the Upper Cretaceous, which means before the Cenozoic Pyrenean and Alpine tectonic phases, as assumed by Bodin and Razack (1997) through the directional analysis of the cave map.
Furthermore, selenium is not concentrated in a specific component of the argillaceous matrix and seems rather diffuse within the individual components. Nevertheless, reduced conditions favored after deposition the formation of pyrite crystals and the reduction of organic matter. The induced pyritic and organic materials are heterogeneous, limiting the interpretation of the correlation studies.
At last, correlative studies of total rock data demonstrated that within the argillaceous sediments, Se is highly correlated with elements (Rb, Cs, V and F) that are also correlated with organic carbon. An affinity between Se and organic matter is thus deduced. But the components extracted by thermochemolysis – GC – MS are very mature and make it difficult, at this step of the study, to deduce any chemical bonds between Se and the organic matter or the presence of organoselenide compounds. Concordances in the history of Se and the organic matter give rise to further studies to determine the nature of the Se compounds, to elucidate the nature of the affinity between selenium and the present organic matter, and to understand the mechanisms inducing the release of selenium into groundwater from this association.
Acknowledgments
The financial support of this study by the Poitou-Charentes Water Research Program (CPER #1) and by the French National Observatory System Service (NOS) H+ is gratefully acknowledged.