


 CC-BY 4.0
CC-BY 4.0
1. Introduction
La structure des liquides silicatés est d'une importance considérable dans les processus ignés et dans les applications industrielles (conditions des fours) car l'organisation à l'échelle atomique affecte fortement leurs propriétés dynamiques et physico-chimiques (par exemple, la densité, l'entropie, la répartition des éléments entre les phases cristallines et fondues, les propriétés de transport, y compris la viscosité et la diffusivité). Les verres sont depuis longtemps reconnus comme de bons analogues des liquides dont la structure peut être facilement révélée expérimentalement. Au contraire, les mesures à des températures élevées peuvent être difficiles et peuvent donner des informations structurales moins détaillées. Cependant, des changements importants de propriétés se produisent au-dessus de la température de transition vitreuse, Tg, tels qu'une augmentation de la capacité thermique et de la dilatation thermique, une diminution de la viscosité, etc. Ces observations impliquent que la structure du verre ne reflète que la structure du liquide surfondue figée à Tg avec, en plus, une forte dépendance due à l'histoire thermique, c'est-à-dire typiquement la vitesse de trempe [Allwardt et Stebbins 2004, Henderson et al. 2006]. Par conséquent, les verres peuvent présenter des différences structurales significatives par rapport aux liquides à haute température. Pour comprendre comment les données obtenues sur les verres peuvent être extrapolées aux produits fondus, il est pertinent de suivre l'évolution de la structure en fonction de la température, la rapidité avec laquelle les changements structuraux se produisent et la manière dont ils peuvent affecter les propriétés générales [Stebbins 2008].
Les liquides et les verres sont tous deux caractérisés par un manque d'ordre à grande échelle. Contrairement aux cristaux pour lesquels un modèle structural unique et précis peut être établi, la structure du verre/liquide est décrite statistiquement, en considérant des informations moyennes sur la structure s'étendant sur des échelles de portée courte et intermédiaire, appelée SRO et IRO (respectivement short range order et intermediate range order). Parmi les nombreux outils expérimentaux permettant d'élucider la complexité des verres/liquides silicatés, la diffraction des rayons X et des neutrons offre la possibilité de sonder à la fois les SRO et les IRO [Cormier 2019, Cormier et al. 2001, Wilding et Benmore 2006] et bénéficie de la disponibilité d'appareils opérationnels à haute température [Cuello et al. 2014, Skinner et al. 2012]. La diffraction donne accès à une fonction de distribution de paires (PDF, pair distribution function) dans l'espace réel qui représente la probabilité de trouver un atome à une distance r d'un atome pris à l'origine. En plus des données expérimentales, une description structurale fournie par des approches de modélisation est généralement nécessaire pour révéler de nombreux aspects structuraux. Ceci peut être facilement obtenu à partir de données de diffraction en utilisant des méthodes telles que le Monte Carlo inverse (RMC, reverse Monte Carlo) ou l'affinement structural avec potentiel empirique (EPSR, Empirical Potential Structure Refinement) [Majérus et al. 2004, Nienhuis et al. 2021]. Comparativement aux verres, les données structurales pour les liquides font défaut, ce qui exclut une évaluation de la comparaison verre/liquide et n'offre pas de validation expérimentale pour développer de nouveaux potentiels plus précis dans les simulations de dynamique moléculaire (DM) [Shih et al. 2021].
Le joint MgSiO3–CaSiO3 (CMS) est un système simple avec deux cations non-formateurs de réseaux (Mg, Ca), et ayant une région vitrifiable complète entre les deux membres terminaux, MgSiO3 et CaSiO3. Il contient trois minéraux d'intérêt pour la géochimie du manteau supérieur, l'enstatite (MgSiO3), le diopside (CaMgSi2O6) et la wollastonite (CaSiO3). L'enstatite et le diopside sont des minéraux pyroxéniques [Thompson et Downs 2004, 2003]. Leur structure est constituée de chaînes simples de tétraèdres SiO4 à sommets partagés. Ces chaînes sont reliées par leurs sommets à des sites M1 octaédriques réguliers qui forment une chaîne d'octaèdres à arêtes partagés. Parallèlement aux chaînes M1, il existe une deuxième chaîne octaédrique avec des sites M2 plus grands et plus déformés que les sites M1 permettant l'introduction de grands cations. Dans le diopside, les atomes de Ca occupent les grands sites M2 tandis que les atomes de Mg occupent les sites M1 standards. En raison de leur taille, les atomes de Ca ne peuvent pas être accueilli dans les sites M1, de sorte que CaSiO3 a une structure légèrement différente et est appelé un pyroxénoïde. Dans la wollastonite [Hesse 1984, Trojer 1968], la périodicité de la chaîne tétraédrique est modifiée (3 répétitions de SiO4 au lieu de 2 répétitions de SiO4 pour les pyroxènes) pour permettre au Ca d'accéder aux sites de type M1. Il en résulte une symétrie cristalline plus faible.
Composition (en %mol, ±0.05) et densité, d (en g⋅cm−3, ±0.002) pour les verres CaOx ⋅ MgO1−x ⋅ 2SiO2
| Nom | Composition nominale | SiO2 | CaO | MgO | NBO/Q∗ | dglass (g⋅cm−3) | dliquide (g⋅cm−3) | Tneutron (K) | TX-ray (K) |
|---|---|---|---|---|---|---|---|---|---|
| CMS00.50 (enstatite composition) | MgO–SiO2 | 50.38 | — | 49.62 | 1.97 | 2.693 | 2.502 (1923 K) | 1873 | 1973 |
| CMS12.38 | 0.5CaO–1.5MgO–2SiO2 | 49.60 | 14.08 | 36.32 | 2.03 | 2.789 | 2.547 (1773 K) | 1773 | |
| CMS25.25 (composé de diopside) | CaO–MgO–2SiO2 | 50.24 | 25.33 | 25.32 | 2.02 | 2.805 | 2.563 (1873 K) | 1673 | 1973 |
| CMS38.12 | 1.5CaO–0.5MgO–2SiO2 | 48.6 | 36.98 | 14.42 | 2.11 | 2.871 | 2.612 (1773) | 1773 | |
| CMS50.00 (composition wollastonite) | CaO–SiO2 | 50.42 | 49.58 | — | 1.97 | 2.891 | 2.615 (1923 K) | 1873 | 1973 |
*Nombre d'oxygène non-pontant (ONP) par tétraèdre de SiO4 (Q), calculé avec la formule ONP∕Q = (2O–4Q)/Q.
Le mélange Ca–Mg dans les verres/liquides ajoute de la complexité à la structure avec une évolution possible en fonction de la température. Comme les cations ont la même charge électronique nominale mais une taille cationique différente (rMg2+ = 0.71 Å, rCa2+ = 1.14 Å [Shannon 1976]), on s'attend à des préférences de site distinctes et un changement de coordinence locale ou une distorsion locale du site pourrait survenir avec la température. Le mélange Ca–Mg le long du joint MgSiO3–CaSiO3 produit une variation non linéaire de la viscosité, avec pour la composition de diopside une viscosité plus faible (à une température donnée) par rapport aux deux membres terminaux et un minimum qui est augmenté près de Tg [Neuville et Richet 1991]. Les auteurs ont montré que ce comportement peut être interprété par un excès d'entropie de configuration pour les liquides impliquant un comportement idéal de mélange de cations. Une grande partie de l'entropie excédentaire est conservée dans des compositions intermédiaires à Tg. L'analyse structurale des verres CMS soutient le mélange aléatoire des cations car ces composés sont décrits avec une distribution désordonnée des cations Ca et Mg [Allwardt et Stebbins 2004, Cormier et al. 2010]. Bien que les verres dans ce système aient été largement étudiées [Cormier et Cuello 2012, 2011, De Sousa Meneses et al. 2006, Gaskell et al. 1991, Kalampounias et al. 2009, Kohara et al. 2011, Kroeker and Stebbins 2000, Schneider et al. 2003, Shimoda et al. 2008, 2007, Taniguchi et al. 1997, 1995, Wilding et al. 2004, Yin et al. 1983, Zhang et al. 1997], les études expérimentales à l'état liquide sont limitées au MgSiO3 [Wilding et al. 2010, 2008] et CaSiO3 [Benmore et al. 2020, 2010, Skinner et al. 2012]. Il serait donc intéressant de poursuivre l'étude de la structure de la phase fondue des compositions intermédiaires, y compris la composition importante du diopside, pour laquelle les effets entropiques et structuraux sont maximisés.
Dans cet article, nous avons pour objectif d'explorer l'évolution structurale le long du joint MgSiO3–CaSiO3 en utilisant la diffraction des neutrons (DN) et la diffraction des rayons X (DRX) à haute température. Les données expérimentales ont été ajustées en utilisant l'approche EPSR pour révéler les différences structurales entre les verres et les liquides.
2. Expérimental
2.1. Préparation d'échantillon
Des verres le long du joint pyroxène Ca–Mg (CaxMg1−1xSi2O6 où x = 0, 0.25, 0.5, 0.75, et 1) ont été préparés à partir de poudres de SiO2, CaCO3 et MgO sous des formes commerciales très pures. La décomposition des carbonates a été obtenue par chauffage à 800 °C pendant une nuit. Les mélanges ont ensuite été fondus, à 100 °C au-dessus de leurs températures de fusion respectives, pendant 1 h dans un creuset Pt-10%Rh. Les verres ont été obtenus par trempe des verres ramollis, en plongeant le fond des creusets dans l'eau. Les échantillons ont été broyés dans un mortier d'agate et refondus deux fois pour améliorer l'homogénéité chimique du verre. Les verres sont notés CMSx.y où x = %mol CaO, y = %mol MgO et (100 − x − y) = %mol SiO2.
Des morceaux de chaque verre ont été intégrés dans une résine époxy, polis, recouverts de carbone et analysés chimiquement à l'aide d'un micro-analyseur à sonde électronique (CAMECA SX50) au Centre Camparis (Sorbonne Université, France), comme indiqué précédemment [Cormier et Cuello 2012]. Les compositions chimiques (tableau 1) sont obtenues en faisant la moyenne de dix points différents. Les densités de verre ont été mesurées à température ambiante en utilisant la poussée d'Archimède avec du toluène comme liquide d'immersion. Les densités des verres fondus ont été obtenues ou extrapolées à partir de données expérimentales [Courtial et Dingwell 1999, 1995]. Le tableau 1 indique les densités et les températures expérimentales utilisées dans les mesures de diffraction.
2.2. Diffraction des rayons X
Des mesures DRX à haute énergie ont été effectuées sur la ligne de faisceau ID11 à l'installation européenne de rayonnement synchrotron (ESRF) à l'aide d'un dispositif de lévitation aérodynamique et de chauffage laser [Drewitt et al. 2011, Hennet et al. 2011]. Le faisceau synchrotron de rayons X, d'une section transversale de 100 × 100 μm2 et d'une énergie de 100.456 keV, a été incident sur les gouttelettes liquides sphérique qui sont en lévitation au-dessus de la buse d'un lévitateur à l'aide d'un flux de gaz argon à travers une buse conique en aluminium. Deux faisceaux laser CO2 de 125 W ont été utilisés pour la chauffe, avec des températures mesurées à 1973 K (CMS00.50, CMS38.12, CMS50.00) ou 1773 K (CMS12.38, CMS25.25). Des diagrammes de diffraction bidimensionnels ont été enregistrés avec une caméra CCD FReLoN conçue par l'ESRF [Labiche et al. 2007]. Le faisceau diffusé a été positionné à l'angle du détecteur, ce qui permet une large gamme de vecteurs de diffusion total (Q) s'étendant jusqu'à Qmax = 23.6 Å−1. Les distances échantillon-détecteur, le centre du faisceau et l'inclinaison du détecteur ont été déterminés par comparaison entre les distances-d connus d'un calibrant Si. Les données de diffraction ont d'abord subi une correction de champ plat (flat-field) puis ont été corrigées pour tenir compte du bruit du courant de signaux 'noirs', des effets géométriques et de la polarisation du faisceau incident et réduites à des figures unidimensionnelles à l'aide du logiciel FIT2D. Les intensités diffractées mesurées ont été mises à l'échelle pour correspondre à la somme de l'effet Compton et de l'autodiffusion, qui a ensuite été soustraite pour obtenir le facteur de structure total FX(Q), en utilisant une procédure standard [Drewitt et al. 2013, 2011].
2.3. Diffraction des neutrons
Les mesures de DN ont été réalisées sur le diffractomètre D4C de l'Institut Laue-Langevin, à l'aide d'un dispositif de lévitation aérodynamique et de chauffage laser [Hennet et al. 2006]. Ces acquisitions à haute température ont été obtenues sur les échantillons liquides et le dispositif de lévitation vide à l'intérieur de la chambre de diffraction. Un étalon au vanadium a également été mesuré à température ambiante pour la normalisation absolue de l'intensité de diffusion. La longueur d'onde des neutrons est de 0.4979 Å−1, ce qui produit une gamme de Q utilisable jusqu'à Qmax = 23.6 Å−1. Le programme CORRECT a été utilisé pour corriger les données des effets d'atténuation, de bruit de fond, de diffusion multiple et d'inélasticité [Howe et al. 1996], donnant le facteur de structure totale des neutrons, FN(Q). Les données DN ambiantes sont celles précédemment publiées, également acquises sur le diffractomètre D4C [Cormier et al. 2010], à l'exception du verre CMS25.25 qui a été mesuré au cours de cette expérience.
3. Affinement structural avec potentiel empirique
Les modèles structuraux pour les verres et des liquides ont été dérivés des données de diffraction en utilisant l'EPSR [Soper 2005]. Cette méthode affine les positions atomiques dans un modèle de boîte et les potentiels interatomiques entre les paires atomiques pour obtenir progressivement une adéquation entre les données de diffraction simulées et mesurées. La procédure d'ajustement a été effectuée simultanément sur les données de diffraction des neutrons et des rayons X pour tous les verres, tandis que pour les liquides, seules les données de rayons X étaient disponibles pour CMS12.38 et CMS38.12. Les paramètres des potentiels utilisés pour la modélisation de l'EPSR ont été déterminés comme indiqué précédemment [Weigel et al. 2008] et figurent dans le tableau 2. Les spécifications de la boîte atomique cubique sont énumérées dans le tableau 3. Des configurations aléatoires initiales de 5000 atomes ont été générées et équilibrées à température ambiante (verre) ou à haute température (liquide) proche de l'expérimentale, en utilisant le potentiel de référence. Ensuite, les potentiels empiriques ont été affinés par variation de l'amplitude énergétique demandée (valeurs ereq) entre 0–50 kJ⋅ mol-1 afin de reproduire les jeux de données expérimentales. Une fois qu'un bon ajustement est obtenu, avec une énergie interne équilibrée, des données structurales statistiques ont été collectées en faisant la moyenne sur des ensembles de >1500 configurations.
Paramètres des potentiels de référence dans les simulations EPSR
| Élément | Charge électrique | 𝜀 (kJ⋅mol−1) | 𝜎 (Å) |
|---|---|---|---|
| Si | +2 | 0.175 | 1.06 |
| O | −1 | 0.1625 | 3.6 |
| Mg | +1 | 0.2 | 1.8 |
| Ca | +1 | 0.2 | 2.4 |
Longueurs de boîtes et densités des modèles EPSR
| Verre | Longueur de la boîte (Å) | Numéro atomique, (atomes Å−3) | R𝜒(neutron) | R𝜒(rayons X) | Température (K) |
|---|---|---|---|---|---|
| CMS00.50-verre | 39.56 | 0.08076 | 0.222 | 0.431 | 300 |
| CMS00.50-liquid | 40.51 | 0.07503 | 0.339 | 0.265 | 1923 |
| CMS12.38-verre | 39.61 | 0.08048 | 0.124 | 0.339 | 300 |
| CMS12.38-liquide | 40.82 | 0.07349 | — | 0.132 | 1773 |
| CMS25.25-verre | 40.05 | 0.07781 | 0.212 | 0.603 | 300 |
| CMS25.25-liquide | 41.24 | 0.07126 | 0.369 | 0.549 | 1723 |
| CMS38.12-verre | 40.19 | 0.07702 | — | 0.359 | 300 |
| CMS38.12-liquide | 41.48 | 0.07008 | 0.088 | 0.210 | 1773 |
| CMS50.00-verre | 40.56 | 0.07493 | 0.050 | 0.272 | 300 |
| CMS50.00-liquide | 41.94 | 0.06777 | 0.247 | 0.286 | 1923 |
R𝜒 sont des facteurs de fiabilité calculés dans l'espace réel sur la plage de 1⩽r⩽8 Å [Wright 1993]. Températures utilisées lors des simulations.
4. Résultats
4.1. Facteurs de structure
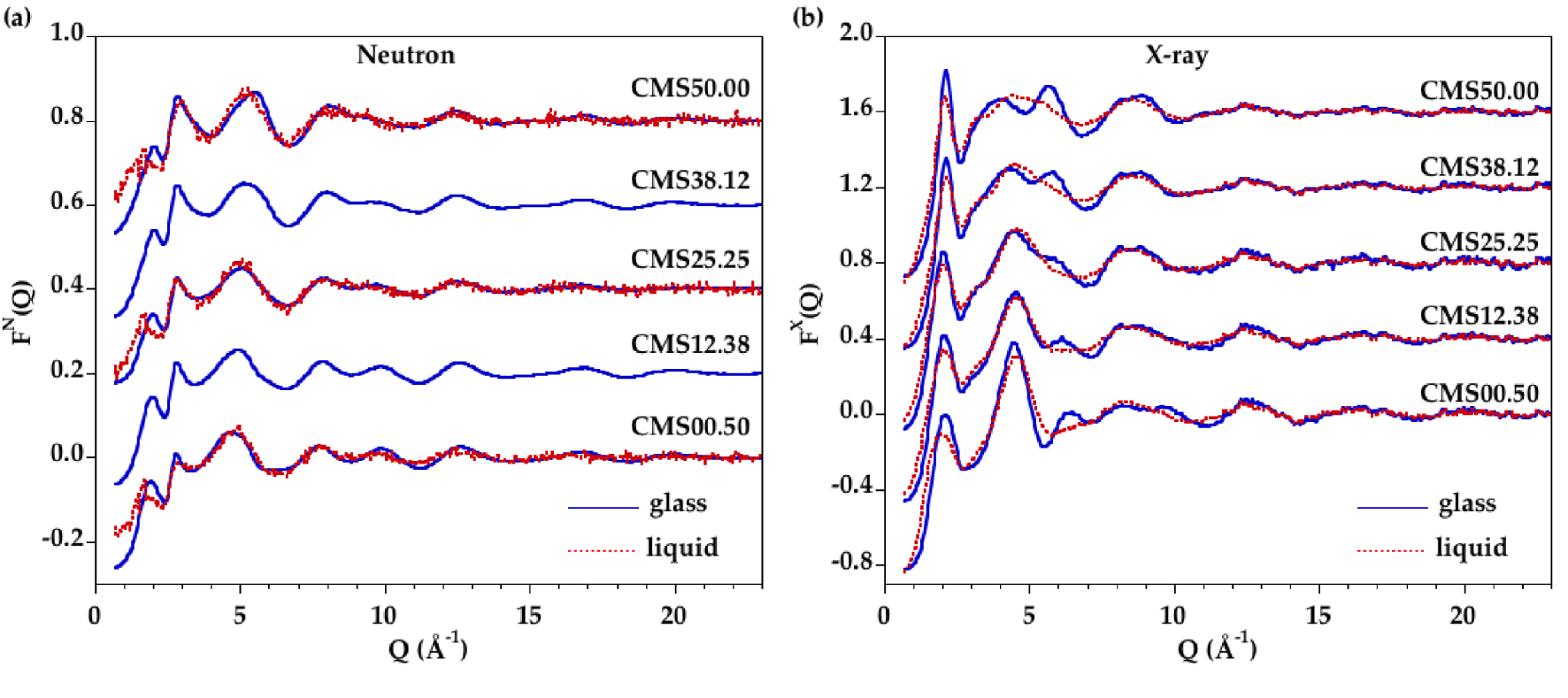
Comparaison des facteurs de structure mesurés pour les verres et liquides, en (a) la diffraction des neutrons et en (b) la diffraction des rayons X. La courbe bleue continue correspond au verre et la courbe rouge en pointillés correspond au liquide.
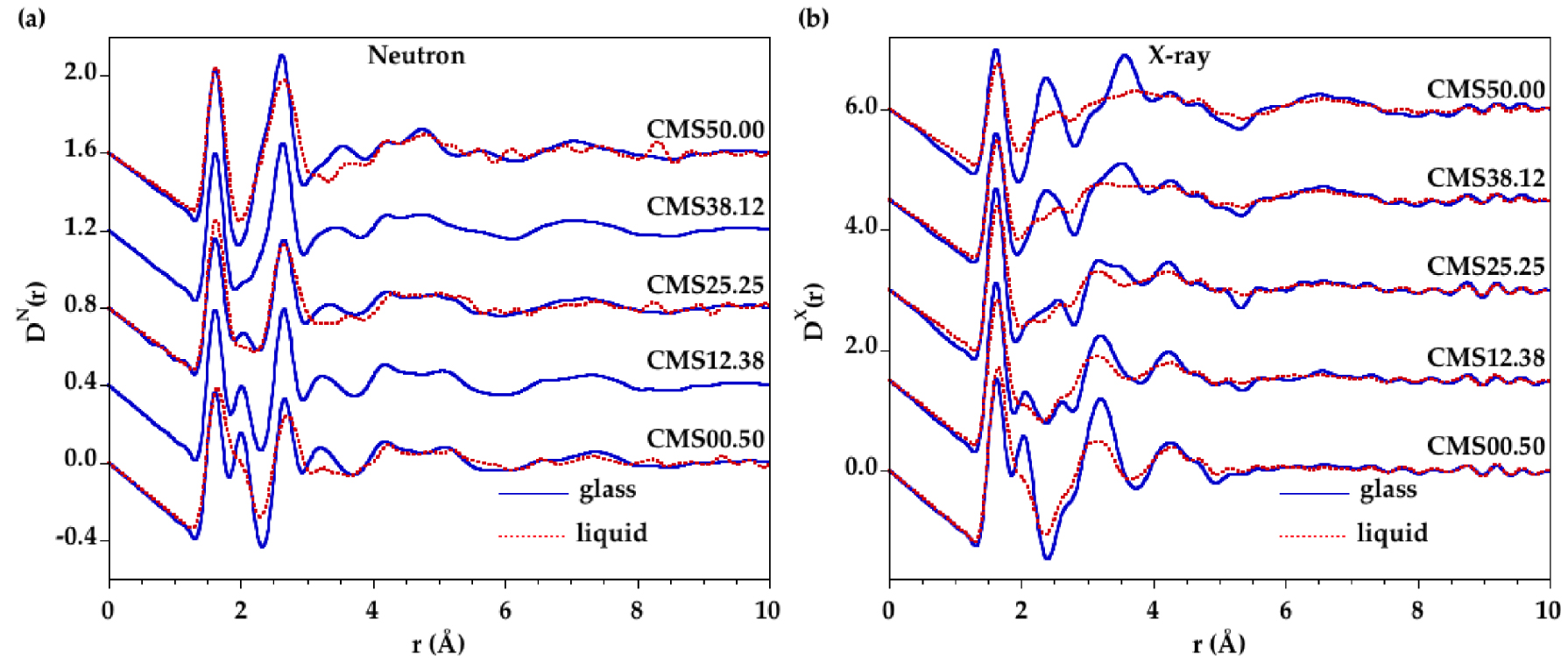
Comparaison des fonctions de distribution totale des paires pour les verres et les liquides, en (a) la diffraction des neutrons et en (b) la diffraction des rayons X. La courbe bleue continue correspond au verre et la courbe rouge en pointillés correspond au liquide. Les transformées de Fourier ont été obtenues à partir des facteurs de structure de la figure 1 sans fonction de modification et en utilisant l'intervalle 0.56 Å−1⩽Q⩽22 Å−1.
Les facteurs de structure expérimentaux des rayons X/neutrons pour les échantillons à l'état vitreux et liquide sont présentés à la figure 1. Quand MgO est remplacé par CaO dans les verres, la position du premier pic, Qp, dans les fonctions FN(Q) est décalée de 1.92 ± 0.01 Å−1 pour CMS00.50 à 2.05 ± 0.01 Å−1 pour CMS50.00, et l'intensité du pic Qp diminue légèrement. Ces positions sont reliées à des fluctuations de densité séparées par une distance d = 2π∕Qp, produisant des valeurs comprises entre 3.29 ± 0.02 Å pour CMS00.50 et 3.06 ± 0.02 Å pour CMS50.00. Lors de la fusion, le pic Qp est clairement décalé vers des valeurs de Q inférieures, à environ 1.67 Å−1, mais les faibles statistiques ne permettent pas de distinguer des variations avec la composition. Ce déplacement implique des distances de corrélation plus élevées dans les liquides que dans les verres. Cela peut être attribué soit à un effet de densité (densités plus faibles à l'état liquide), soit à des changements structuraux. Un pic similaire est observé dans les fonctions FX(Q) pour les verres et les liquides et, dans ce cas, on observe une forte dépendance chimique : l'intensité augmente à mesure que CaO se substitue à MgO. De plus, la position du pic passe de 2.04 ± 0.02 Å−1 à 2.12 ± 0.01 Å−1 dans les verres à teneur croissante en CaO et de 1.98 ± 0.02 Å−1 à 2.15 ± 0.02 Å−1 dans les liquides à teneur croissante en CaO. Ce pic est donc particulièrement sensible à l'organisation des cations.
Pour les ensembles de données sur les neutrons et les rayons X, on observe la présence de points isobestiques (points communs dans les diagrammes de diffraction) pour les séries verre et liquide (figure supplémentaire S1). Ces points indiquent que les fonctions FN(Q) ou FX(Q) pour des compositions intermédiaires peuvent être obtenues en utilisant une combinaison linéaire des fonctions pour les deux membres terminaux, c'est-à-dire les échantillons CMS00.50 et CMS50.00 [Cormier et al. 2010, Cormier and Cuello 2012].
Les fonctions FN(Q) varient principalement entre 4 et 12 Å−1 alors que les oscillations sont très similaires au-dessus de 12 Å−1. Cette dernière région est principalement dominée par le SRO associé au réseau silicaté. A l'état liquide, les différences entre échantillons sont observées sur une région Q limitée entre 3 et 7 Å−1. Les différences en fonction de la température reflètent essentiellement un degré accru de désordre structural. Au-dessus de 7 Å−1, les facteurs de structure ne sont pas modifiés de manière importante parce que le SRO reste très similaire dans les verres et les liquides. Pour de faibles valeurs de Q, l'élargissement des caractéristiques du signal est apparent à l'état liquide.
Les compositions de wollastonite CMS50.00 ont été précédemment étudiées en détail à l'état liquide en utilisant des techniques de diffraction des neutrons et des rayons X combinées à des simulations DM [Benmore et al. 2010, Skinner et al. 2012]. Nos données DN ont de meilleures statistiques, permettant une confiance accrue sur les valeurs de Q élevées. Le pic Qp a une intensité supérieure à celle de Skinner et al.[Skinner et al. 2012], ce qui est en meilleur accord avec les données à température ambiante. Les fonctions FX(Q) pour le verre et le liquide CMS50.00 sont similaires à celles mesurées par Benmore et al. [2010], indiquant une évaporation réduite de CaO pendant les mesures. CMS00.50 a également été précédemment étudié à l'état liquide par Wilding et al. [Wilding et al. 2010, 2008] et leurs données DRX sont similaires à celles de la présente étude.
4.2. Fonctions dans l'espace réel
La transformée de Fourier du facteur de structure total fournit la fonction correspondante dans l'espace réel (figure 2), appelée fonction de distribution totale de paires (PDF) ou D(r). Les trois premiers pics principaux de D(r) sont bien compris et correspondent aux corrélations Si–O (1.62 Å), Mg–O (2.00 Å) et Ca–O (2.40 Å). Chaque rayonnement a une sensibilité différente aux éléments : les données DRX sont dominées par les éléments de numéro atomique élevé (O < Mg < Si < Ca) tandis que les données DN sont dominées par les éléments ayant une longueur de diffusion de neutrons élevée, b, évoluant comme bSi = 4.153 fm < bCa = 4.76 fm < bMg = 5.37 fm < bO = 5.803 fm. Ces différentes sensibilités expliquent que la corrélation Ca–O se distingue clairement à 2.38 Å dans les fonctions DX(r) alors qu'elles apparaissent dans les fonctions DN(r) sous la forme d'un petit épaulement sur le côté des r bas du pic à 2.63 Å qui est dominé par les contributions O-O. La présence de points isobestiques peut également être observée dans les fonctions D(r) (figure supplémentaire S2).
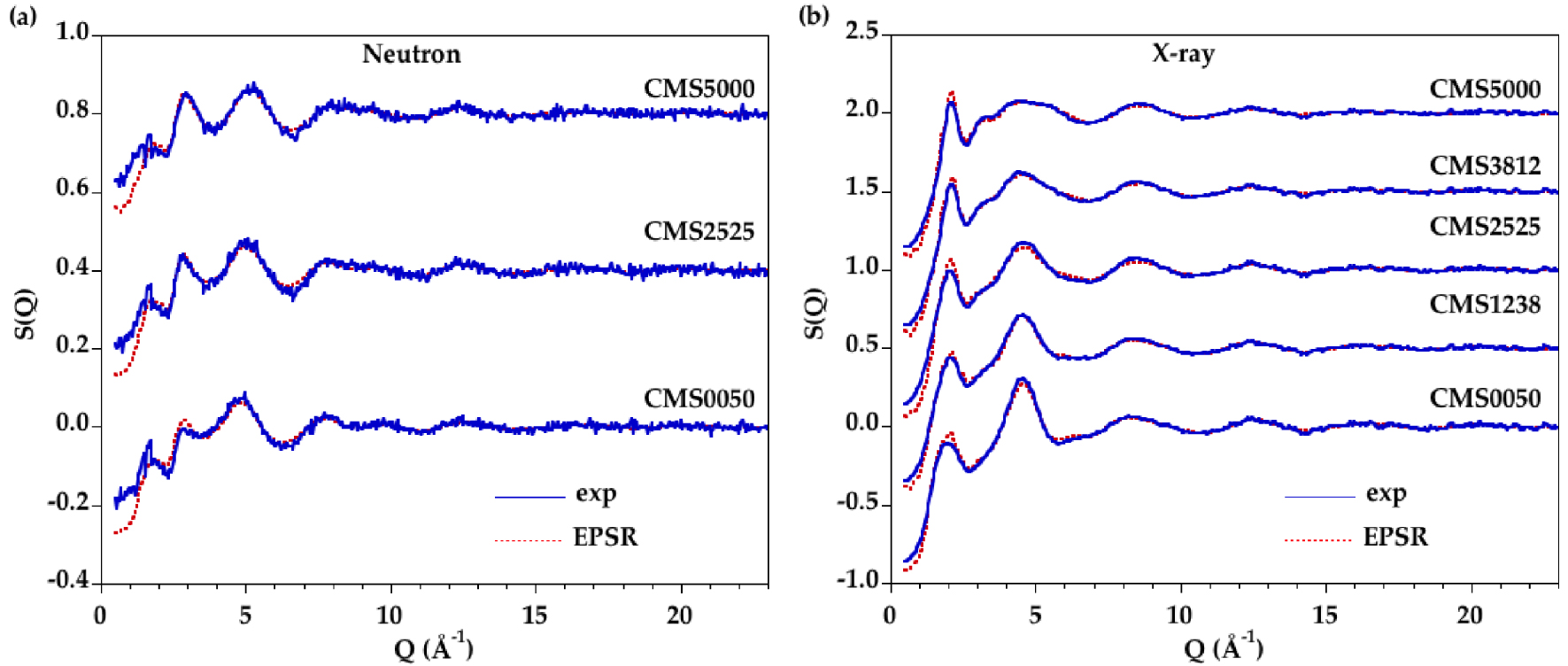
Facteurs de structure expérimentaux (courbe bleue) et modélisés (courbe rouge en pointillés), obtenus par diffraction de neutrons (a) et de rayons X (b) pour les verres CMS.
Les variations à moyenne échelle sont principalement associées au réseau silicaté dans les fonctions DN(r) et à des corrélations impliquant des cations dans les fonctions DX(r) Dans les données DRX, l'attribution du pic a précédemment indiqué que les contributions proches de 3.16 Å sont principalement dues aux corrélations Si-Mg et Mg–Mg et les contributions proches de 3.56 Å sont principalement dues aux corrélations Ca–Ca et Ca–Si [Cormier et Cuello 2012]. Les fonctions de tous les liquides ont des SRO semblables à celles des verres, avec une contribution distincte de Si–O qui est légèrement décalée de 1.62 Å à 1.63 Å en fonction de la température. Cependant, les pics de Mg–O et de Ca–O ne sont plus résolus, ce qui indique un désordre thermique important dû à de larges distributions des distances Mg-O et Ca-O. Dans les liquides, les pics de Mg–O et de Si-O se chevauchent et le pic de Ca–O est moins résolu dans les données DRX. Les pics structuraux dans les fonctions DX(r)) sont nettement moins résolus que dans les fonctions DN(r) à haute température et l'élargissement des pics dans les fonctions DX(r) augmente à mesure que la teneur en CaO augmente.
4.3. Modèles EPSR
Il est difficile d'obtenir des informations précises sur l'environnement local cation-oxygène à partir d'une analyse directe, par exemple à partir d'ajustements gaussiens, en raison du chevauchement important entre les différentes contributions, qui s'accentue encore lorsque la température augmente. La modélisation EPSR est une méthode d'ajustement alternative permettant l'extraction d'informations structurales détaillées à partir de configurations atomiques. On obtient une correspondance correcte en ajustant simultanément les données issues des diffractions de neutrons et de rayons X, comme on le voit à la figure 3 pour tous les verres et à la figure 4 pour tous les liquides. Les fonctions D(r) correspondantes apparaissent dans les figures supplémentaires S3 et S4. Les facteurs de fiabilité R𝜒 sont déterminés sur les fonctions D(r) suivant Wright [Wright 1993] et les valeurs sont reportées dans le tableau 3. Un bon niveau de correspondance entre les modèles EPSR et les données expérimentales est atteint avec des facteurs R𝜒 comparables ou meilleurs que ceux publiés dans les études précédentes [Gong et al. 2021, Guignard and Cormier 2008].
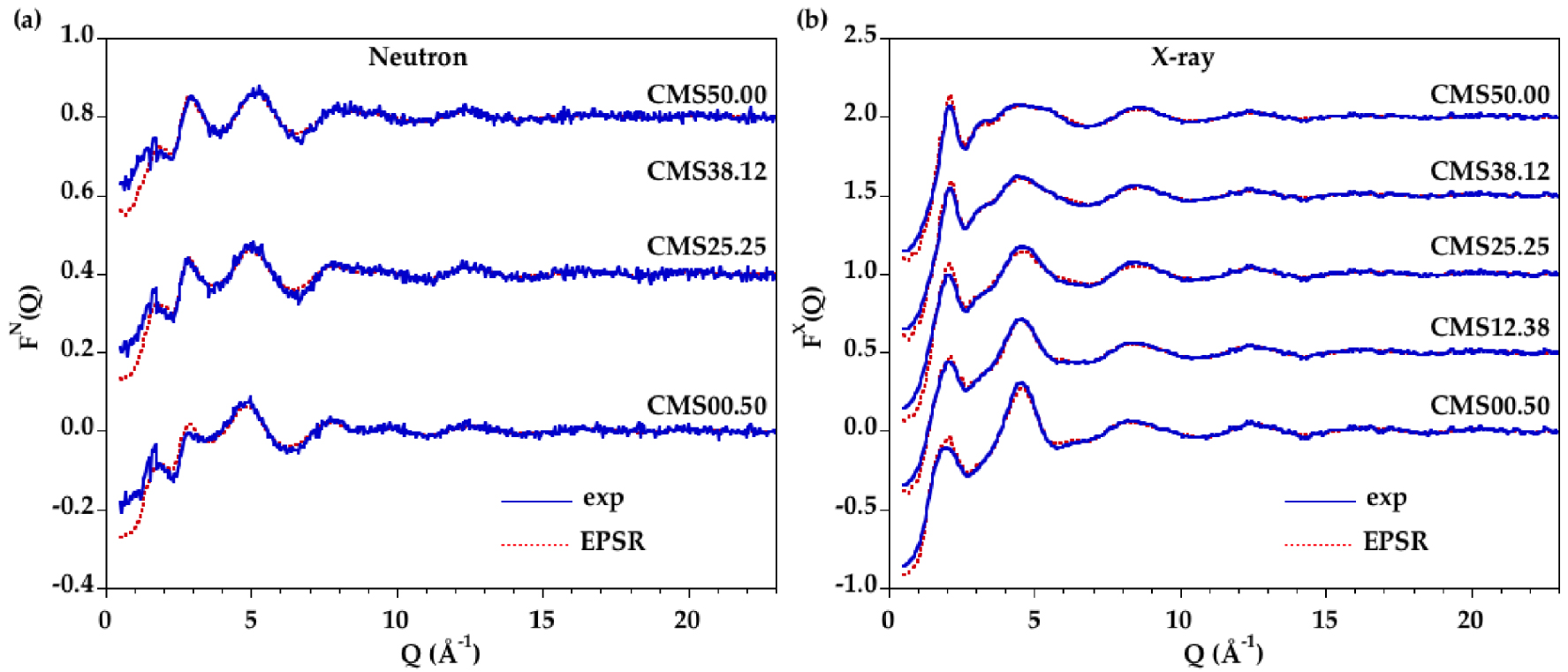
Facteurs de structure expérimentaux (courbe bleue) et modélisés (courbe rouge en pointillés), obtenus par diffraction de neutrons (a) et de rayons X (b) pour les liquides CMS.
Les fonctions de distribution partielle de paires (PPDF, partial pair distribution functions) dérivées de l'EPSR sont illustrées à la figure 5 uniquement pour CMS00.50, CMS25.25 et CMS50.00 pour plus de clarté (tous les PPDF pour chaque verre/liquides apparaissent dans la figure supplémentaire S5). Le premier pic des PPDF Mg–O et Ca–O présente un élargissement considérable à l'état liquide indiquant que le désordre de l'oxygène augmente autour de ces cations en fonction de la température.
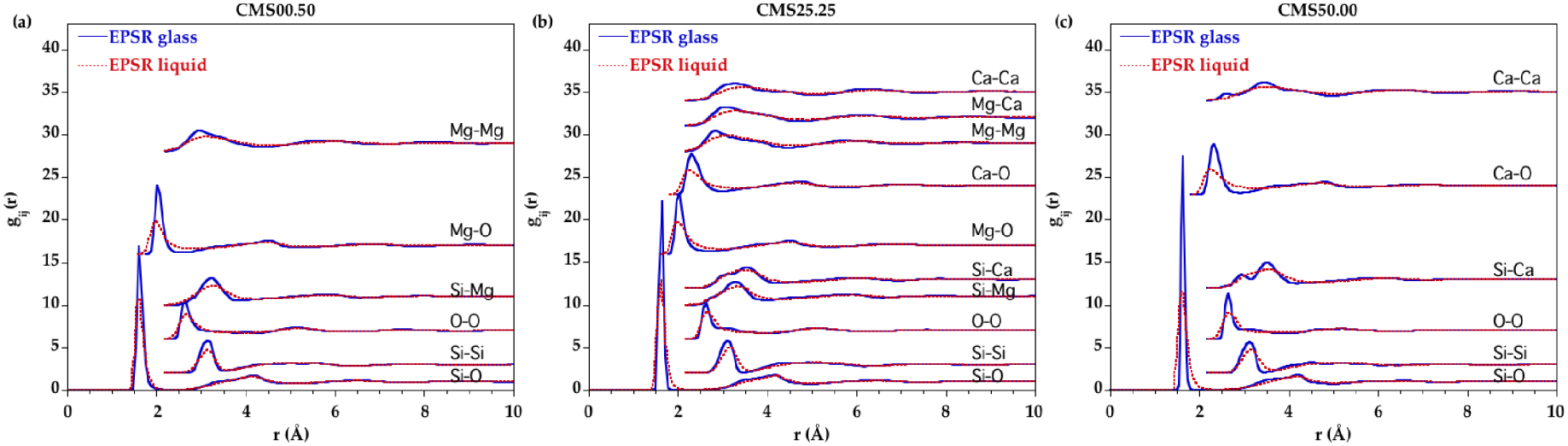
Comparaison des fonctions de distribution partielle EPSR pour le verre (courbe bleue) et le liquide (courbe rouge en pointillés) correspondant aux échantillons (a) CMS00.50, (b) CMS25.25 et (c) CMS50.00.
En utilisant le minimum après le premier pic principal dans les PPDF Si–O, Mg–O et Ca–O comme distance de coupure, le nombre de voisins O autour de Si, Mg et Ca a été déterminé. Les distances de coupure sont indiquées dans le tableau 4. Les nombres moyens de coordination (NC) sont indiqués à la figure 6. Le silicium est principalement quadricoordonné dans toutes les compositions pour tous les verres et liquides. De petites quantités de sites Si tricoordonnés et pentacoordonnés (<3%) sont observées et leur proportion ne change pas significativement avec la composition ou la température. Ceci est cohérent avec un réseau silicaté de tétraèdres partageants les sommets qui dominent la structure liquide à pression ambiante. Le NC(Mg–O) est de ∼4.5 dans les verres et augmente de ∼0.25 avec la température. De même, le NC(Ca–O) passe de ∼5.8 dans les verres à ∼6.3 dans les liquides. Cependant, le NC en fonction de r, NC(r) (figure 7), montre qu'il est difficile de définir une distance de coupure à l'état liquide puisqu'aucun plateau clair n'apparaît pour Mg et Ca, contrairement à Si. Par conséquent, la définition de la première couche de voisins de l'oxygène autour de Mg et Ca à l'état liquide est non ambiguë. Ceci est dû à la distribution continue des distances Mg–O et Ca–O révélée par l'absence de minimum atteignant zéro dans les PPDF Mg–O et Ca-O.
Distances de coupure utilisées dans le calcul des nombres de coordination moyens
| Si–O (Å) | Mg–O (Å) | Ca–O (Å) | |
|---|---|---|---|
| Verre | 2.10 | 2.60 | 2.95 |
| Liquide | 2.15 | 2.9 | 3.28 |
La distribution des sites pour Mg et Ca dans les verres et les liquides est fournie à la figure 8, en utilisant les distances de coupure du tableau 4. Les sites en coordinence x sont notés [x]Mg ou [x]Ca. Dans les verres, la distribution des sites indique une majorité de sites [4]Mg, avec certains sites [5]Mg, et une majorité de sites [6]Ca avec la coexistence de sites [5]Ca et [7]Ca. Les distributions des sites Ca et Mg montrent de grandes similitudes avec les modèles précédents obtenus en couplant DM et RMC (voir la figure supplémentaire S6) [Cormier et Cuello 2012, 2011].
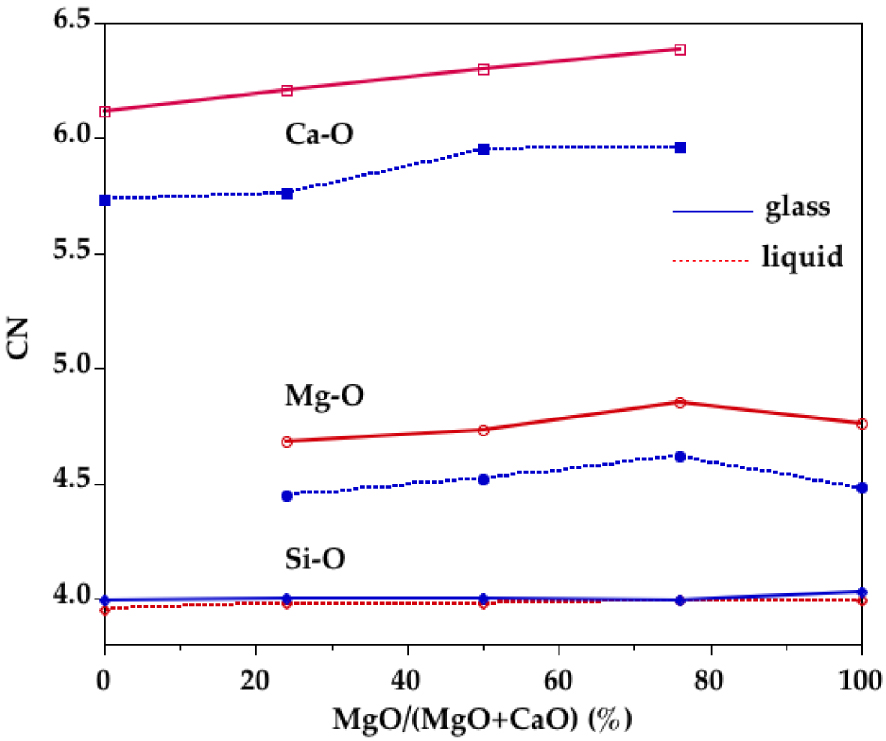
Dépendance de la composition des nombres de coordination Si–O, Mg–O et Ca–O déterminés à partir des modèles EPSR pour les verres (courbes bleues) et les liquides (courbes rouges en pointillés).
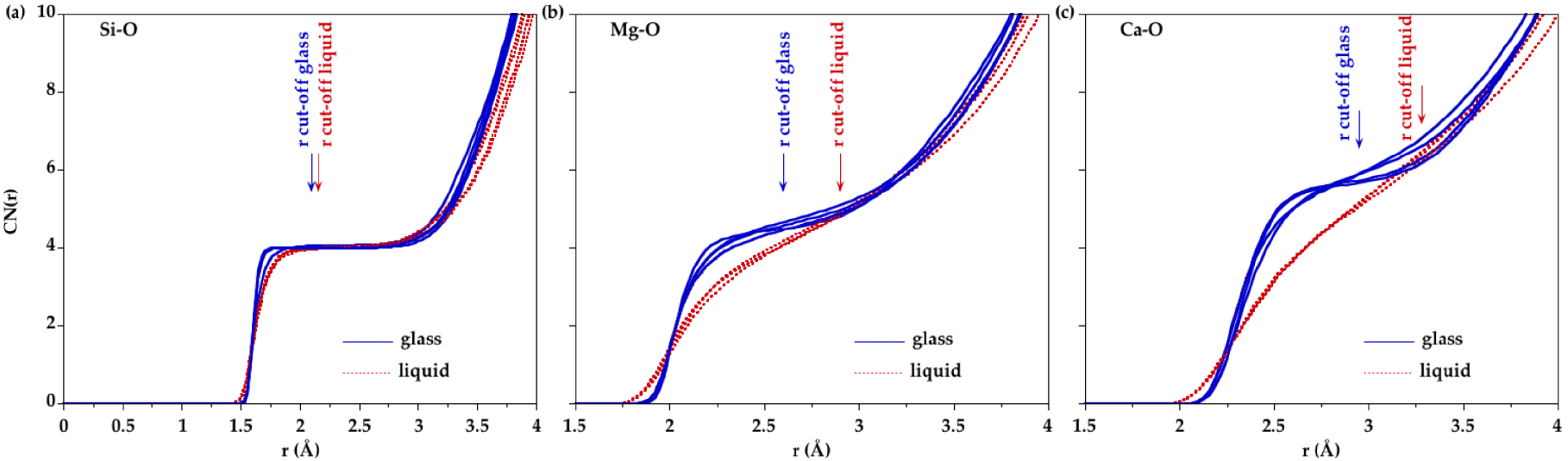
Evolution du nombre de coordination avec la distance de coupure pour les paires (a) Si–O, (b) Mg–O et (c) Ca–O tel que déterminé à partir des modèles EPSR.
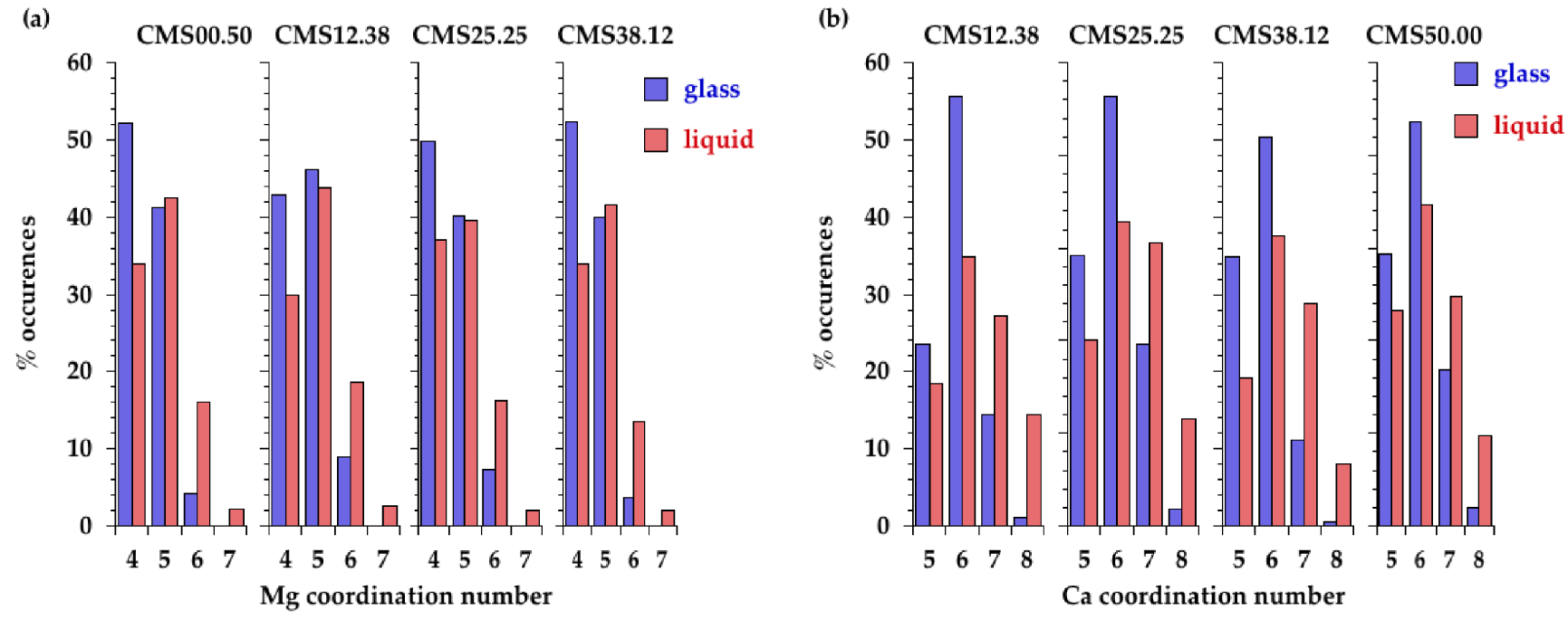
Distribution des polyèdres (a) Mg et (b) Ca extraits des modèles EPSR dans les différents verres et liquides.
Les distributions des oxygènes pontants (OP), des oxygènes non-pontants (ONP) et des oxygènes libres (OL) (OL ne sont liés à aucun atome de Si) sont représentées sur la figure 9a. La fraction ONP a une tendance décroissante avec l'augmentation de la teneur en MgO. La présence de O triplement lié est toujours inférieure à 0.9 % de la totalité des atomes d'oxygène dans les verres/liquides et la teneur n'est donc pas significative. A l'état liquide, la proportion de ONP augmente, ce qui est concomitant avec une diminution de OP et OL. La faible proportion de OP se traduit par une connectivité moyenne du réseau plus faible qui est quantifiée par le nombre moyen de n atomes de OP autour d'un atome de silicium (figure 9b). La connectivité moyenne du réseau montre des valeurs croissantes à mesure que la teneur en MgO augmente dans les verres et les liquides.
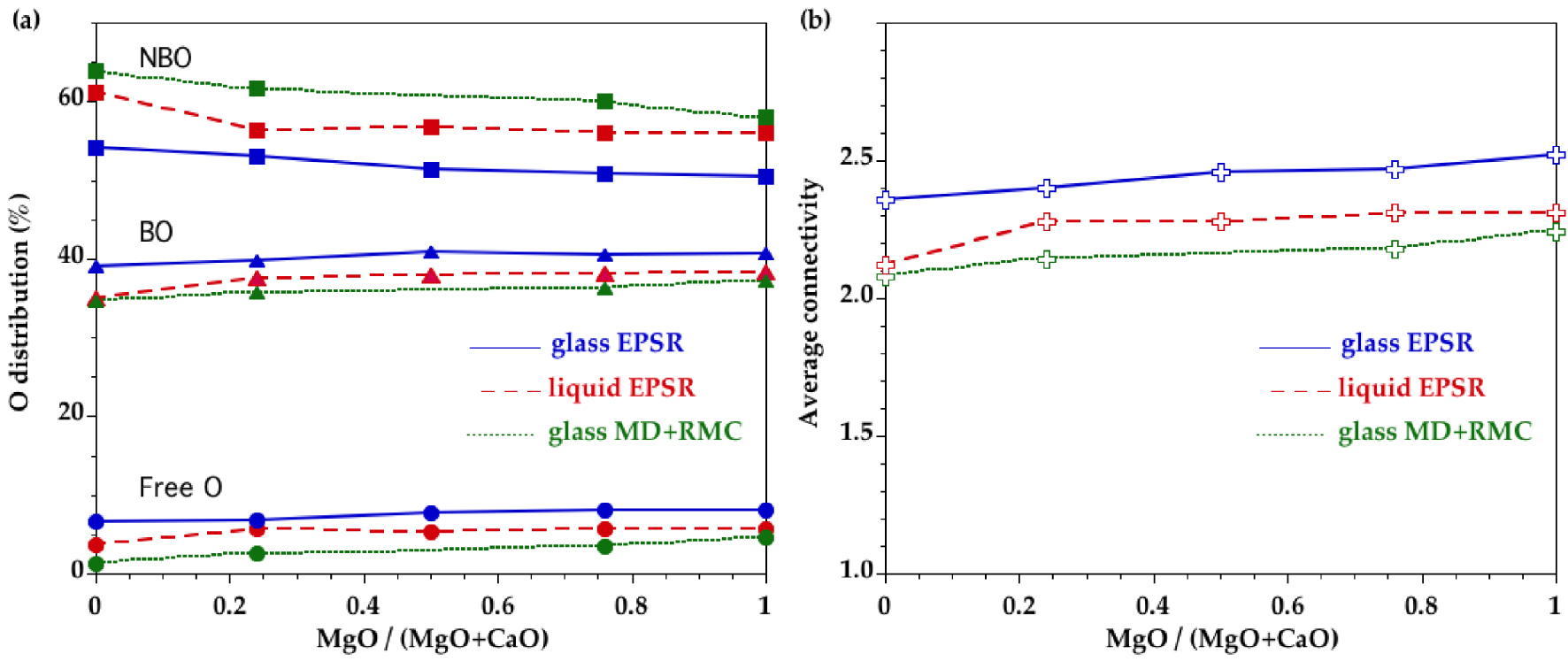
a) Distribution de l'oxygène entre oxygène non-pontant (ONP, carrés), oxygène pontant (OP, triangles) et oxygène libre (O libre, cercles), calculée à partir des modèles EPSR pour les verres CMS (courbes continues) et les liquides (courbes en pointillés longs). La comparaison est faite avec les modèles de verre précédents obtenus en combinant DM et RMC (courbes en pointillés courts) [Cormier et Cuello 2012]. Les tracés servent uniquement à guider l'œil. b) Connectivité moyenne, n, dérivée des modèles EPSR pour les verres CMS (courbe continue) et les liquides (courbe en pointillés longs). La comparaison est faite avec les modèles de verre précédents obtenus en combinant DM et RMC (courbe en pointillés courts) [Cormier et Cuello 2012]. Les tracés servent uniquement à guider l'œil.
5. Discussion
5.1. Comparaison entre les modèles atomistiques
Les modèles EPSR peuvent être comparés à nos résultats précédents obtenus en combinant des simulations DM avec la modélisation RMC [Cormier et Cuello 2012]. Dans CMS00.50, le NC moyen pour le Mg reste similaire :CNDM+RMC = 4.35 et CNEPSR = 4.48 (figure supplémentaire S6a,b). La distribution des différents sites de coordination produit une quantité presque égale de [4]Mg (52%) et [5]Mg (41%) dans l'EPSR tandis que [4]Mg était prédominant (68%) dans le modèle DM+RMC. Pour le Ca dans la CMS50.00, le NC moyen et la distribution du site ne sont pas significativement modifiés (figure supplémentaire S6c,d). Il apparaît donc que les deux méthodes d'ajustement des données produisent des résultats convergents, renforçant la validité de cette approche pour déterminer le SRO.
Les distributions de OP, ONP et OL montrent que les modèles EPSR contiennent plus de OP et d'O libre que les modèles DM+RMC, avec une légère dépendance de composition (figure 9a). La teneur plus élevée en OL explique une connectivité moyenne élevée dans les modèles EPSR (figure 9b) qui augmente avec la teneur en MgO comme dans les modèles DM+RMC. Le joint MgSiO3–CaSiO3 correspond à des compositions de métasilicate de sort que l'on s'attend à ce que n ait une valeur de 2. L'écart par rapport à cette valeur pour les verres est plus élevé pour EPSR que pour DM+RMC. Il est intéressant de noter que les modèles EPSR liquides ont des valeurs plus proches des modèles DM+RMC, probablement parce que les modèles DM+RMC conservent davantage les caractéristiques structurales de l'état à haute température en raison des vitesses de trempe rapides intrinsèques dans les simulations DM et de la relaxation insuffisante de la structure dans la modélisation RMC des verres.
La spéciation Qn (Qn est un tétraèdre SiO4 contenant n atomes de OP) montre plus de Q3 et de Q4 et moins de Q2, de Q1 et de Q0 dans les modèles EPSR par rapport aux modèles DM+RMC (figure 10). Les deux modèles présentent des écarts avec les proportions de Qn déterminées expérimentalement par RMN de 29Si pour les verres CMS00.50, CMS25.25 et CMS50.00 [Schneider et al. 2003, Sen et al. 2009, Zhang et al. 1997], car ces mesures ont déterminé des teneurs plus élevées en Q2 et en Q1 et des teneurs plus faibles en Q3 et en Q4 par rapport aux modèles. Une récente étude DM avec des potentiels avancés indique une spéciation Qn pour CMS00.50 et CMS50.00 très similaire à nos résultats avec une sous-estimation de Q2 et une surestimation de Q3 et de Q4 [Shih et al. 2021]. Ces résultats indiquent que des contraintes supplémentaires devraient être incorporées dans les modèles pour améliorer la description de certaines unités ou groupes structuraux spécifiques. En outre, une comparaison minutieuse entre les modèles expérimentaux et atomistiques devrait être fournie pour assurer une amélioration continue de la modélisation et des simulations de verres et de liquides.
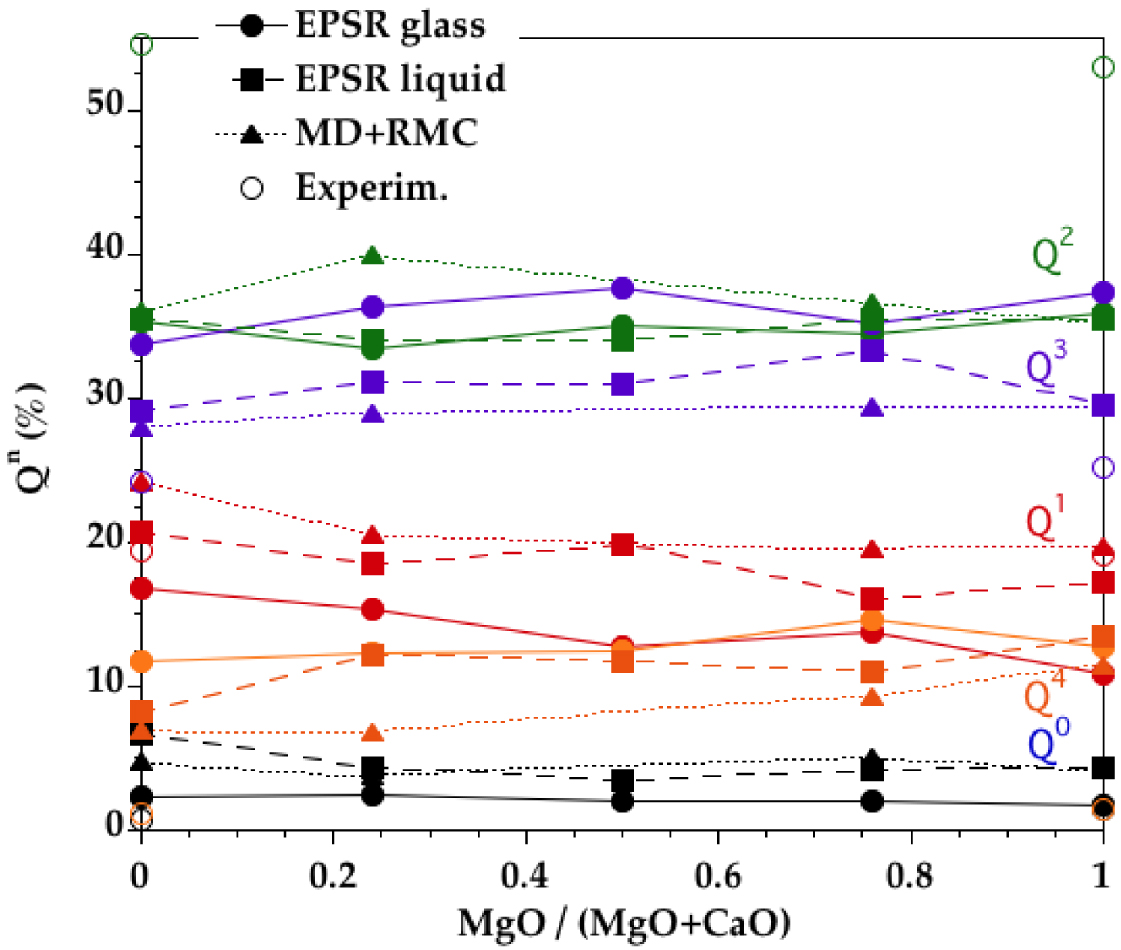
Distribution de Qn dérivée des modèles EPSR pour les verres CMS (courbes continues, cercles) et les liquides (courbes en pointillés longs, carrés). La comparaison est faite avec des modèles de verre précédents obtenus en combinant DM et RMC (courbes en pointillés courts, triangles) [Cormier et Cuello 2012] et point expérimental (symboles ouverts) [Schneider et al. 2003, Sen et al. 2009, Zhang et al. 1997]. Les tracés servent uniquement à guider l'œil.
5.2. Évolution de l'environnement de Si en fonction de la température
Pour la paire Si–O, le NC(r) montre un plateau clair (figure 7a) qui indique un environnement bien défini autour des atomes de Si correspondant aux tétraèdres de SiO4 dans les verres et les liquides, avec de très faibles proportions d'autres sites coordonnés. Les tétraèdres de SiO4 sont peu affectés par une augmentation de la température. La position du premier pic dans les fonctions totales D(r) n'est pas informative car elle est affectée par les contributions de Mg-O. Dans CMS50.00 (échantillon sans Mg), la position du pic Si–O est augmentée de + 0.01 Å avec la température. Les PPDF Si–O déterminés à partir des modèles EPSR ne montrent pas de changements significatifs en fonction de la température, à l'exception d'un élargissement cohérent avec un désordre thermique.
La structure le long du joint MgSiO3–CaSiO3 est fortement dépolymérisée avec une proportion élevée d'espèces Q2 et Q3, en accord avec les résultats expérimentaux précédents [De Sousa Meneses et al. 2006, Schneider et al. 2003, Zhang et al. 1997]. Cependant, le degré de connectivité affiché à la figure 9b est plus élevé que prévu compte tenu de la stoechiométrie. Les structures verre/liquide présentent une large gamme d'espèces Qn comparées à la structure des homologues cristallins qui est composée uniquement d'espèces Q2. Sur la base des observations sur les boîtes de configuration 3D, le réseau silicaté pourrait être décrit comme une structure de réseau en forme de chaîne de tétraèdres SiO4 partageant les sommets formant des chaînes brisées coexistant avec quelques anneaux. Ceci diffère de la topologie des pyroxènes et des pyroxénoïdes qui est uniquement composée de chaînes tétraédriques. De plus, des évolutions chimiques sont visibles sur la figure 9 : la substitution du Ca par le Mg conduit à une légère augmentation de la connectivité associée à plus de OP et de OL. Cependant, comme les cations Mg et Ca ont la même charge, le mélange Mg/Ca ne nécessite pas de redistribution significative des charges dans la structure, ce qui explique les petites modifications du réseau (figures 9 et 10). La différence de rayons cationiques est essentiellement compensée par une distribution des deux cations dans de multiples sites distincts (figures 7 et 8).
On observe une nette tendance à une teneur plus élevée en ONP et à des teneurs plus faibles en OP et en OL dans les liquides par rapport aux verres (figure 9a). Ces changements dans la spéciation de l'oxygène, associés à des espèces Q3 plus faibles et à des espèces Q1 plus élevées, indiquent que le réseau liquide est moins connecté. Les espèces OL correspondent à de l'oxygène non lié à des tétraèdres SiO4 et peuvent donc être une signature pour des régions enrichies en Mg et Ca. Inversement, la forte proportion de OL peut indiquer des régions de silicate hautement polymérisées appauvries en Mg et Ca. Au fur et à mesure que les teneurs en OL et en OP augmentent lors du refroidissement, la ségrégation entre les régions riches en silicate et les régions riches en (Mg, Ca) est plus importante dans les verres que dans les liquides.
5.3. Évolution de l'environnement de Mg en fonction de la température
Dans les verres, la distribution des sites de Mg indique non seulement une prévalence des sites de [4]Mg, mais aussi une grande quantité de sites de [5]Mg. La présence de polyèdres quadricoordonné et pentacoordonné est également bien établie pour d'autres cations divalents tels que Fe2+, Ni2+ ou Zn2+ [Cormier et al. 2021, Galoisy et Calas 1993, Jackson et al. 2005, Waychunas et al. 1988]. Dans les liquides, des sites à coordinence plus élevée sont présents, principalement [5]Mg et une petite quantité de [6]Mg, coexistant avec encore une grande quantité de [4]Mg (figure 8). Lors de l'augmentation de la température, il y a donc une redistribution des sites qui se reflète dans l'évolution du NC moyen (Mg–O) proche de 4.5 dans les verres et proche de 4.7-4.8 dans les liquides. Ces valeurs concordent avec des simulations de DM très récentes pour CMS00.50 [Shih et al. 2021]. L'environnement Mg–O est plus ordonné dans le verre CMS00.50 que dans d'autres verres, comme on le voit dans le PPDF de Mg–O qui présente un premier pic bien défini (figure 5a). Le NC(Mg–O) moyen dans CMS00.50 est le plus bas, en accord avec les résultats précédents [Cormier et Cuello 2012]. Autrement, il n'y a pas de dépendance claire avec la composition, que ce soit dans les verres ou dans les liquides, au vu de la dispersion des données. Un plateau est visible dans le NC(r) des verres (figure 7b), permettant de définir la première couche de coordination de l'oxygène autour des atomes de Mg, alors qu'il est plus difficile de déterminer les points d'inflexion corrects pour les liquides. La lecture de la position du premier pic dans les PPDF de Mg–O révèle un passage de 2.02 ± 0.02 Å dans les verres à 1.98 ± 0.02 Å dans les liquides (figure 5 et figure supplémentaire S7a). Bien que cette évolution de la position maximale du pic puisse indiquer une diminution de la coordinence, une longue traîne du côté des r élevés pour les liquides implique la contribution additionnelle de longues distances Mg–O qui expliquent l'augmentation du NC avec la température. Les études précédentes de DRX ou d'EXAFS ont généralement obtenu des NC bas pour Mg [Matsubara et al. 1988, Taniguchi et al. 1997, Yin et al. 1983] en raison des difficultés à déterminer correctement le NC lorsqu'il existe une distribution asymétrique des distances. En raison du désordre et de la distorsion du site, les contributions de r élevées ne sont régulièrement pas détectées dans les données EXAFS car cette méthode est plus sensible aux voisins les plus proches [Kroeker et Stebbins 2000]. Les approches de modélisation (EPSR ou RMC) assurent une détermination plus précise du NC car cette valeur n'est pas basée sur un ajustement gaussien.
Le raccourcissement des distances Mg–O dans les liquides est remarquable puisqu'une dilatation thermique est attendue. Ces courtes distances de Mg–O sont compensées par la présence de longues distances de Mg–O, ce qui entraîne la traîne du côté des r élevés et produit un environnement plus déformé dans le liquide que dans le verre. Un comportement différent peut être observé pour les métaux de transition divalents à l'état fondu. En effet, un raccourcissement des distances Ni–O dans un liquide Na1.95Ni0.05Si2O5 a été déduit par spectroscopie d'absorption des rayons X à haute température [Farges et al. 1994]. Ceci a été interprété comme une teneur croissante en [4]Ni pendant la transition du verre à la l'état liquide. Du Fe2+ quadricoordonné a également été trouvé dans les verres et des liquides de silicate alcalin sans effets de température [Waychunas et al. 1988]. Cependant, de telles études EXAFS peuvent être insensibles aux longues distances Ni–O ou Fe–O, en particulier à l'état liquide.
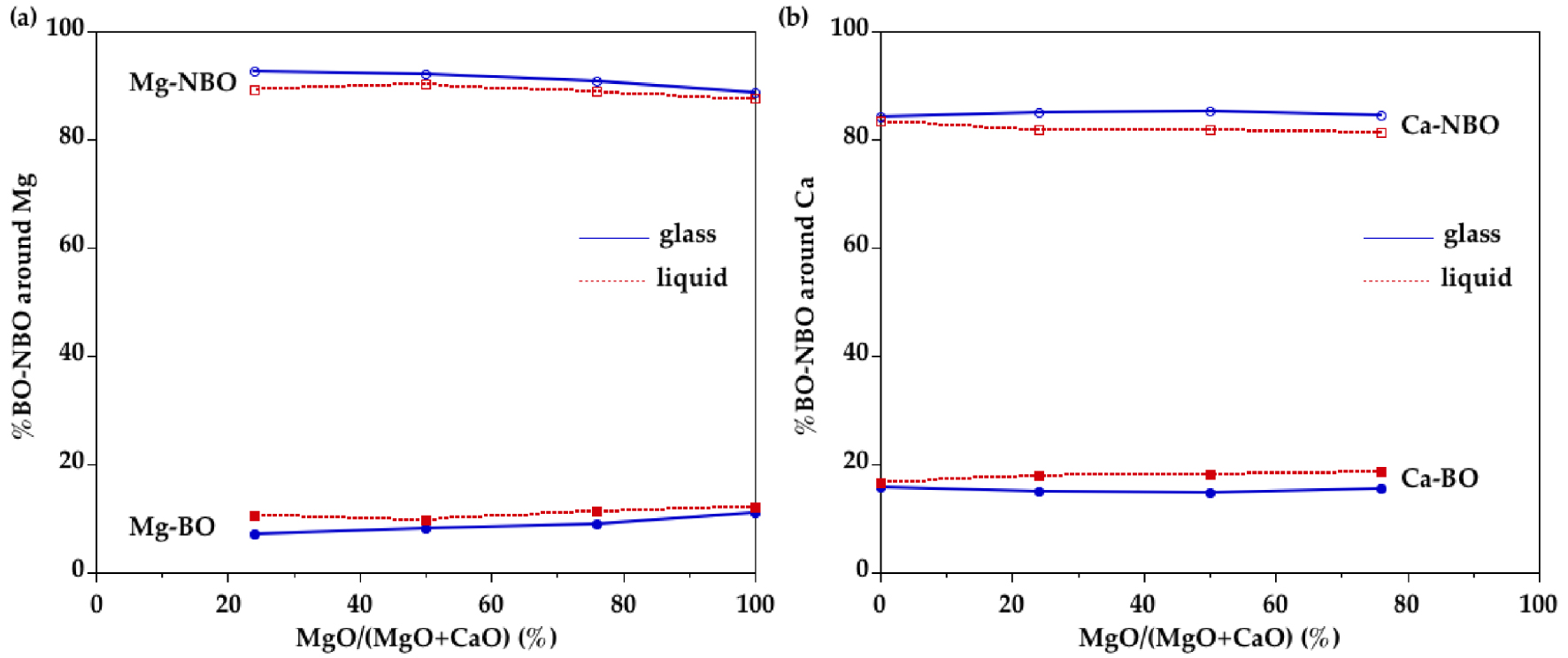
Proportion de voisins ONP et OP autour de (a) Mg et (b) Ca, telle que déterminée dans la première couche de voisins O dans les modèles EPSR de verres et de liquides.
Très peu d'études ont été réalisées in situ pour comprendre l'environnement du Mg dans les liquides silicatés. George et Stebbins ont obtenu des spectres RMN de 25Mg jusqu'à 1743 K pour un diopside liquide et pour un liquide proche des compositions CMS12.38 (mais avec 6%mol Al2O3) [George et Stebbins 1998] et Fiske et Georges sur CMS29.14 à 1673 K [Fiske et Stebbins 1994]. Pour les liquides, George et Stebbins ont observé un changement avec l'addition de Ca (ou d'alcalis) avec un déplacement de la position du pic vers des fréquences plus élevées indiquant une diminution du NC moyen (Mg–O). Cependant, ces changements sont minimes. On observe une forte dépendance du NC(Mg–O) en fonction de la température, le NC étant sensiblement plus bas dans le verre [Fiske et Stebbins, 1994]. Les déplacements chimiques en RMN à haute température sont en accord avec un CN compris entre cinq et six et significativement différents de celui attendu pour une coordinence quatre [George et Stebbins 1998]. Ce comportement est cohérent avec l'augmentation du NC du verre au liquide, comme déduite de la présente analyse des données de diffraction. Les ajustements gaussiens de la PDF aux rayons X à haute température ne montrent aucun changement de coordinence de Mg pour la composition de l'enstatite (CMS00.50) [Wilding et al. 2010], alors qu'une petite augmentation de la coordinence de Mg–O de 4.8 dans le verre à 5.1 dans le liquide pour un échantillon CMS37.00 (37.5MgO-62.5SiO2) a été révélée en utilisant plusieurs fonctions gaussiennes pour ajuster la contribution de Mg–O [Benmore et al. 2020].
5.4. Évolution de l'environnement de Ca en fonction de la température
Comme pour le Mg, l'analyse des modèles EPSR indique que les cations sont localisés dans une large distribution de sites (figure 8b). L'environnement de Ca est principalement constitué de [6]Ca avec à la fois du [5]Ca et du [7]Ca dans les verres. La spéciation du site est redistribuée à des sites à coordinence plus élevée dans les liquides (figure 8b) : moins de [5]Ca et de [6]Ca et plus de [7]Ca et de [8]Ca. La coordinence moyenne de Ca est plus élevée que celle de Mg, conformément à sa plus faible intensité de champ (définie comme le rapport z∕d2, où z désigne la charge cationique et d la longueur de la liaison cation-oxygène). L'évolution de la première distance Ca–O dans les PPDF Ca–O montre un comportement similaire à celui des PPDF Mg–O pour une température croissante : raccourcissement de la longueur moyenne des liaisons Ca–O de 2.30 ± 0.02 Å à 2.25 ± 0.01 Å et une distribution élargie et asymétrique du côté des r élevés (figure 5 et figure supplémentaire S7b). Le NC(Ca–O) moyen est augmenté de ∼5.8 dans les verres à ∼6.3 dans les liquides. Ceci est cohérent avec les simulations DM du verre/liquide de CaSiO3 qui indiquent à la fois des courtes et longues distances de Ca–O dans le liquide [Skinner et al. 2012]. Dans cette dernière étude, un ajustement gaussien symétrique du premier pic Ca–O conduit à une diminution du NC de Ca–O de 6 dans le verre à 5 dans le liquide [Skinner et al. 2012]. Cependant, en utilisant des fonctions gaussiennes supplémentaires dans l'ajustement, le NC(Ca–O) ne varie pas avec la température (6.6 dans le verre et 6.5 dans le liquide) pour un échantillon CMS20.00 (20CaO-80SiO2) mesuré à l'aide de DRX à haute température [Benmore et al. 2020]. Cela souligne à nouveau qu'un environnement déformé ne peut pas facilement être capturé par un simple ajustement gaussien.
5.5. Distribution de Ca et Mg
Pour comprendre la connexion des sites de Mg avec le réseau silicaté tétraédrique, nous avons calculé le nombre de ONP et OP autour des cations (figure 11). Une proportion importante de voisins O sont ONP (∼60 %) et cette proportion ne diminue que faiblement en fonction de la température. La proportion de OP dans la première couche de coordination autour de Ca est plus élevée que pour Mg, indiquant une association préférentielle de Ca avec OP. Ceci est attribué à la plus faible intensité de champ de Ca par rapport à Mg et donc à sa plus faible capacité à équilibrer la charge négative autour des sites ONP. Un site [4]Mg implique de solides forces de liaison et, par conséquent, un atome OL qui est connecté à deux SiO4 et un MgO4 serait surlié de façon importante. Ainsi, l'oxygène reliant Mg à Si est préférentiellement un atome ONP. La proportion élevée de liaisons Ca–OP peut s'expliquer par le fait que les atomes de Ca se trouvent en partie dans les régions riches en silicates plus polymérisés. Cette différence de comportement entre Mg et Ca est corroborée par une étude de RMN de 17O indiquant que le mélange de cations Ca–Mg peut ne pas être entièrement aléatoire le long du joint MgSiO3-CaSiO3 [Allwardt et Stebbins 2004]. Cependant, l'arrangement ordonné Ca–Mg est faible par rapport à d'autres mélanges de cations qui associent des cations présentant des différences de charge et de rayon plus importantes [Cormier et al. 2010]. L'augmentation des NC de Ca et Mg dans les liquides est associée à des proportions plus élevées de OP dans la première couche de coordination, mais l'association préférentielle de OP autour de Ca est préservée.
La connexion des cations avec le réseau silicaté peut être sondée plus en détail à partir des PPDF Si–Mg et Si–Ca (figure 5 et figure supplémentaire S7c, d). Dans les PPDF de Si–Mg, le premier pic est à 3.2 Å dans les verres et à 3.3 Å dans les liquides. Le décalage est faible mais apparent (figure supplémentaire S7c). Ces distances déterminées à partir des ajustements EPSR indiquent principalement des polyèdres partageants des sommets. En effet, quelques exemples de partage d'arêtes entre MgO4 et SiO4 dans les modèles EPSR donnent des distances Si–Mg à 2.7–2.9 Å. Dans les PPDF Mg–Si de la figure 12a (voir également la figure supplémentaire S8a), un épaulement à 2.75 Å est perceptible dans le premier pic à 3.28 Å et correspond au partage d'arêtes entre les tétraèdres SiO4 et les sites [5]Mg et [6]Mg. Ceci est également cohérent avec une étude numérique récente par DFT (sigle pour Density Functional Theory) sur les verres d'aluminosilicate Ca–Mg [Gong et al. 2021]. L'intensité à la position correspondant au partage d’arêtes est plus élevée dans les liquides tandis que dans les verres l'intensité correspondant au partage des sommets domine. De même, dans les PPDF Mg–Mg (figure 12c et figure supplémentaire S8c), l'intensité correspondant au partage d’arêtes entre les polyèdres de Mg est plus élevée dans les verres que dans les liquides, et ces derniers montrent des intensités plus élevées aux niveaux des partages de Mg–Mg par faces et par sommets. Cette variation dans les PPDF Mg–Si et Mg–Mg indique une variabilité moindre dans les verres par rapport aux liquides avec principalement le partage des sommets Mg–Si et le partage des arêtes Mg–Mg à température ambiante.
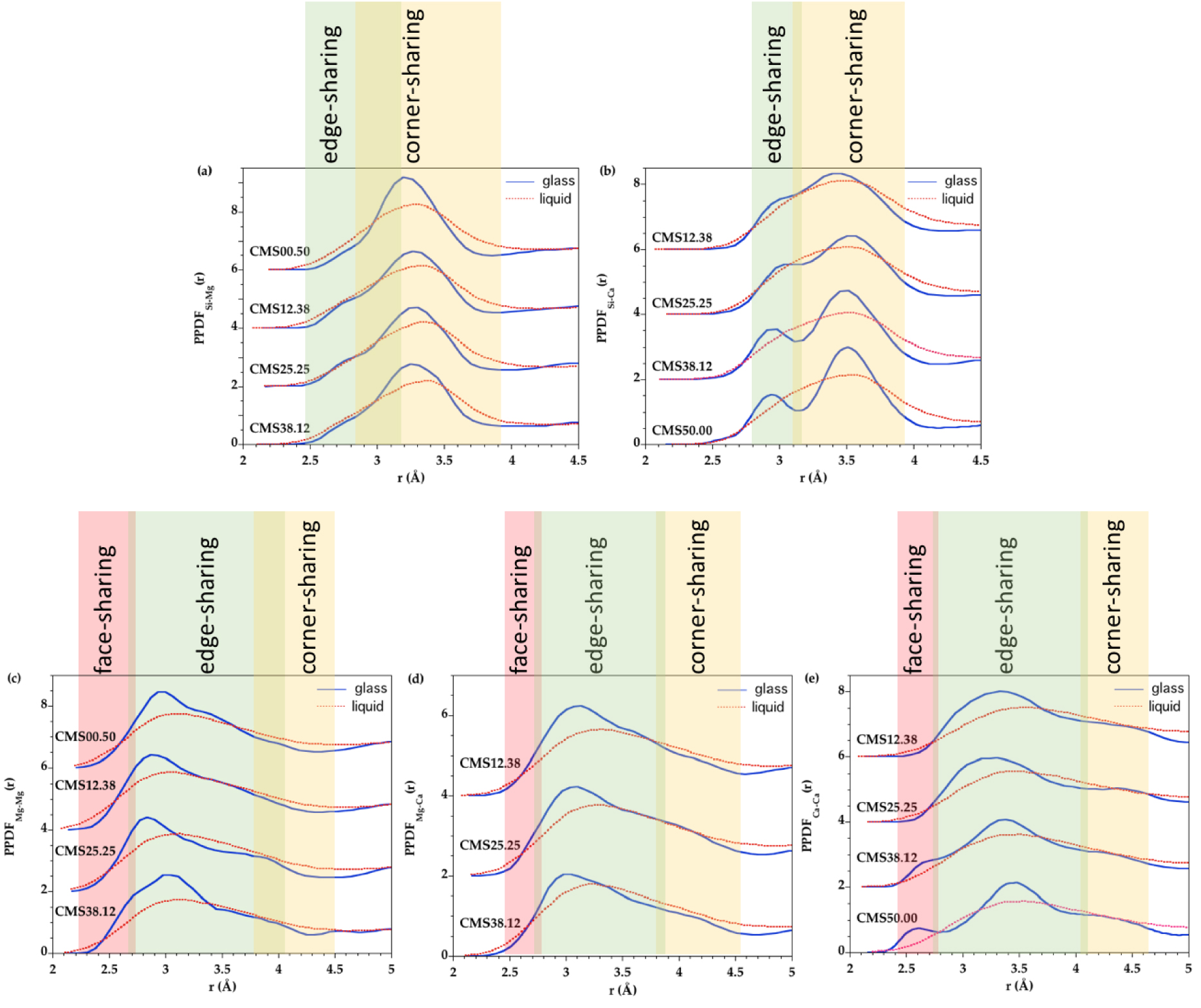
Fonctions de distribution partielle de paires EPSR pour le verre (courbe bleue) et le liquide (courbe rouge en pointillés) pour les différents échantillons, mettant en évidence les régions de longueur de liaison qui sont caractéristiques des partages par faces, par arêtes et par sommets pour les paires (a) Si-Mg, (b) Si–Ca, (c) Mg–Mg, (d) Mg–Ca et (e) Ca–Ca.
Deux pics distincts sont observés dans le PPDF Ca–Si des verres (figure 12b et figure supplémentaire S8b) à 2.95 Å et 3.50 Å qui sont attribués à la connexion par partage d'arêtes et de sommets respectivement entre les tétraèdres SiO4 et les polyèdres Ca, [Gong et al. 2021], avec une dominance de liaisons par partage des sommets. L'intensité correspondant au partage d'arêtes Si–Ca décroît avec la température, un comportement différent des liaisons Si–Mg. Les PPDF Ca–Ca montrent un petit pic à 2.60 Å qui diminue avec moins de teneur en CaO, ce qui indique une proportion significative de partage des faces Ca–Ca dans le verre CMS50.00 (figure 12e et figure supplémentaire S8e). De telles liaisons par partage des faces sont caractéristiques des verres riches en Ca car elles disparaissent dans les liquides correspondants. La grande proportion de partage de face Ca–Ca dans le verre suggère des nano-hétérogénéités et un regroupement de Ca [Benmore et al. 2010], qui peuvent résulter d'une séparation de phase liquide-liquide partielle lors du refroidissement [Benmore et al. 2020].
Lors du refroidissement, une diminution de la population de Ca–Ca partageant des sommets et une augmentation de Ca–Ca partageant des arêtes ont été précédemment suggérées sur la base de l'analyse des distributions des angles de liaison dans des simulations DM de verre/liquide CaSiO3 [Benmore et al. 2010]. Ce résultat est confirmé dans la présente étude basée sur l'analyse des PPDF de tous les verres et cette tendance est observée pour les liaisons Ca–Ca et Ca–Mg (Ca–Mg a des intensités plus faibles au niveau du partage des sommets et Ca–Mg a des intensités plus élevées au niveau du partage des arêtes avec une diminution de la température). Pour les compositions intermédiaires, la contribution Ca–Mg (figure 12d) est bien définie, en accord avec un mélange entre les deux cations différents.
La proportion croissante de polyèdres Ca/Mg partageant des arêtes avec une température décroissante entraîne une augmentation de la polymérisation et du regroupement Ca/Mg. L'agrégation Ca/Mg augmente plus rapidement au voisinage de la température de transition vitreuse, ce qui peut expliquer en partie la forte augmentation continue de la viscosité dans le liquide surfondu à l'approche de la Tg [Benmore et al. 2010, Skinner et al. 2012].
5.6. Structure des liquides silicatés
Les verres diffèrent des liquides en raison des contributions configurationnelles qui apparaissent dans les propriétés thermodynamiques au-dessus de Tg et qui impliquent un changement substantiel de la structure avec la température. Dans les verres/liquides silicatés, un changement dans la distribution de Qn a été déduit des études de Raman et de RMN, indiquant que la réaction de dismutation 2Qn ⇄ Qn−1 + Qn+1 est décalée vers la droite dans les liquides [Mysen et Frantz 1993, Stebbins 1995]. Bien que cette contribution soit importante, la capacité thermique résultante ne peut pas complètement expliquer la capacité thermique configurationnelle mesurée, indiquant l'apparition d'autres modifications structurales [Stebbins 2008]. Dans les modèles EPSR, les changements dans la distribution de Qn sont faibles (moins de Q3 et plus de Q1 et de Q0 à haute température). Les changements structuraux induits par la température peuvent être divers : relaxation conformationnelle du réseau couvrant l’ordre à moyenne distance, dilatation thermique du site, changement de coordinence ou distorsion locale [Calas et al. 2006, Henderson et al. 2006, Majérus et al. 2004]. Pour le système CMS, la présente analyse met en évidence un désordre considérable autour des cations Mg et Ca à la fois à courte et à moyenne échelle, car dans les liquides, les deux cations ont tendance à entrer dans des sites plus grands avec un changement dans la connexion entre les polyèdres (figures 8 et 12). À haute température, la mobilité accrue des cations leur permet d'explorer des sites énergétiquement moins favorables avec une coordinence plus élevée qui favorise une augmentation de l'entropie [de Ligny et Westrum 1996].
Nos résultats indiquent que les structures des verres/liquides des deux membres terminaux présentent des différences significatives bien que les PPDF de Mg–Ca bien définis indiquent un mélange substantiel entre les deux sous-structures. Le NC moyen est différent entre Mg et Ca avec des sites à coordinence élevées pour Ca et l'augmentation de la température entraîne une localisation des cations dans des sites à coordinence encore plus élevée. En conséquence probable des polyèdres cationiques plus grands, des modifications majeures sont observées, en fonction de la température, dans les liaisons cation–cation ou cation-silicium pour tenir compte de l'empilement aléatoire de polyèdres MgOx et CaOx distincts. La plus grande diversité des connexions explique une augmentation de l'entropie configurationnelle dans les liquides. L'organisation structurale évolue continuellement en fonction de la composition atomique entre les deux membres terminaux et, de ce fait, les changements sont compatibles avec le mélange idéal Ca/Mg attendu [Neuville et Richet 1991].
Les processus dynamiques tels que l'écoulement laminaire sont contrôlés par le mécanisme de rupture des liaisons Si–O survenant lors de l'échange d'espèces Qn, comme proposé pour les liquides silicatés riches en SiO2 [Farnan et Stebbins 1994]. Dans les liquides davantage dépolymérisés, il a été suggéré que la rupture des liaisons Ca–O et Mg–O et la mobilité des cations pourraient participer de manière significative aux processus de relaxation structurale et d'écoulement [Nasikas et al. 2012]. En effet, une corrélation entre les mouvements de Mg et la viscosité a été confirmée dans une étude RMN à haute température [George et Stebbins 1998], ce qui indique que ce cation est fortement lié au réseau silicaté. La dynamique cationique pourrait être encore renforcée par l'augmentation de l'entropie dans les liquides résultant de diverses connexions MgOx et CaOx. Aucune étude n'a étudié en détail la relaxation près de Tg et la question reste ouverte de savoir si un comportement de découplage se produit comme dans les systèmes aluminosilicates [Gruener et al. 2001] et s'il existe un comportement de découplage différent entre les systèmes contenant du Mg et du Ca.
À l'approche de la transition vitreuse, il a été démontré que les polyèdres à arêtes partagées CaOx ont tendance à se regrouper dans le réseau silicaté de CaSiO3 [Benmore et al. 2010, Skinner et al. 2012]. La présente étude suggère des hétérogénéités cationiques croissantes similaires pour l'ensemble du joint MgSiO3-CaSiO3. L'augmentation de l’ordre à moyenne distance proche de Tg empêche le liquide de trouver un bassin minimum correspondant à la structure cristalline dans son paysage d'énergie potentielle. De telles hétérogénéités liées à l'organisation Mg/Ca pourraient également limiter la mobilité des cations, augmenter la viscosité et favoriser ainsi la formation du verre. La présence d'hétérogénéités intrinsèques dans la structure du verre a été rapportée récemment dans de nombreux systèmes aluminosilicates présentant un mélange de cations (aluminosilicate Na–K [Le Losq et al. 2017, Le Losq et Neuville 2013] ; aluminosilicate de Ca–Mg [Allu et al. 2018] et leur étendue spatiale peut être affectée par la vitesse de trempe [Dargaud et al. 2012].
6. Conclusions
On a étudié la structure d'une série de verres et de liquides CaSiO3–MgSiO3 par diffraction des neutrons et des rayons X en utilisant la lévitation aérodynamique. Des données expérimentales ont été ajustées en utilisant la méthode EPSR pour obtenir des modèles atomistiques à petite et moyenne échelle. La coordinence de Mg et de Ca a été déterminée à température ambiante et à haute température. Les résultats révèlent des sites avec une coordinence plus élevée dans les liquides que dans les verres. Cette évolution du NC implique des modifications de la connexion entre les polyèdres Ca et Mg et la connexion des polyèdres Ca et Mg avec les tétraèdres SiO4. Plus précisément, la diminution de la température augmente la proportion de sommets partagés pour Si–(Mg,Ca), d'arêtes partagés pour Si–Ca, de faces partagées pour (Mg,Ca)–(Mg,Ca) et d'arêtes partagées pour (Mg,Ca) et diminue la proportion de sommets partagés pour (Mg,Ca)–(Mg,Ca). Ces changements suggèrent des hétérogénéités cationiques sur l'ensemble de la série de verres. Les compositions riches en Ca présentent un partage de faces Ca–Ca et Ca est plus lié à OP que Mg, ce qui indique que les cations Ca2+ sont légèrement localisés de manière préférentielle dans les régions de silicate polymérisé. Les résultats de ces modifications de l’ordre à moyenne distance permettent de mieux comprendre les changements de configuration en fonction de la température.
Conflits d’intérêts
Les auteurs n'ont aucun conflit d'intérêts à déclarer.