

1 Introduction
All human diseases, including breaking a leg, have a genetic component of large or small import [1]. In addition, the degree and variability of the effect is modulated by gender and ethnic background is responsible for the wide diversity in type and severity of acute and chronic diseases in different ethnicities.
Epistasis or gene-gene interaction plays a major role in susceptibility to common human diseases. This concept has been hijacked by the more recent clinical literature under the banner of modifier genes. In effect, this is a genetic concept that has been around for awhile but, fortunately, it has recently experienced a major renaissance.
Epistasis, the interaction between two or more genes, is a hot topic of current interest in molecular and quantitative genetics. Epistasis describes the interaction between genes that are NOT part of the same locus, and indeed can be very distant of each other in the genome. In other words, it refers to interaction between genes occupying different loci, and the interaction must have a phenotypic consequence: one epistatic gene can ameliorate the expression of another gene (antagonistic epistasis) or synergystic epistasis that reinforces the expression of another gene. Nevertheless, this nomenclature does not define if the phenotypic effect is beneficial or deleterious. In beneficial mutations, synergistic epistasis improves the benefit, and if antagonistic, it has a negative effect. In turn, in deleterious mutations, an antagonist epistasis will diminish the deleterious effect, hence having a positive outcome, while a synergistic mutation, clearly has a negative effect [2].
Complex traits, i.e. those with multiple genetic and environmental determinants, represent the greatest challenge for genetic analysis largely due to the difficulty of isolating the epistasis from other genetic and environmental influences. Mapping the Quantitative Trait Loci (QTLs) that influence complex traits is now possible [3]. However, gene identification commonly involves focusing on single candidate genes or isolated chromosomal regions. To reach the next level, exploration of pleiotropism, epistasis and environment-dependency of genetic effects is required. Genetic interactions and unique environmental features must be as carefully scrutinized as are the single gene effects. According to Philips et al. [3], no one genetic approach is likely to possess all the necessary features for comprehensive analysis of a complex disease. Rather, the entire arsenal of behavioral genomic and other approaches are needed, such as random mutagenesis, QTL analyses, transgenic and knockout models, viral mediated gene transfer, pharmacological analyses, gene expression assays, antisense approaches and importantly, revitalization of classical genetic methods. In our view, classical breeding designs are currently underutilized, and will shorten the distance to the target of understanding the complex genetic and environmental interactions associated with disease. Unique combinations of classical approaches with current molecular genomic approaches offer the best strategy.
Moore's [4] recent working hypothesis is that epistasis is a ubiquitous component of the genetic architecture of common human diseases and that complex interactions are more important than the independent main effects of any one susceptibility gene. This hypothesis is based on the following: First, the idea that epistasis is important is not new. In fact, the recognition that deviations from Mendelian ratios are due to interactions between genes has been around for nearly 100 years. Second, the ubiquity of biomolecular interactions in gene regulation and biochemical and metabolic systems suggest that the relationship between DNA sequence variations and clinical endpoints is likely to involve epistasis. Third, single polymorphisms many times do not replicate independent samples, in both linkage and association studies. Fourth, gene-gene interactions are commonly found when properly investigated.
2 Epistatic component of diseases
2.1 Sickle-cell anemia, the first molecular disease
Sickle-cell anemia (SCA) is the first ‘monogenic’ disease ever described, and constitutes the paradigm for a disease traceable to a single mutation in a single gene. Based on this discovery, Pauling's [5] concept of ‘molecular disease’, opened a new chapter in the history of medicine. Nevertheless, at the phenotypic level, SCA is not a monogenic disease; it is a multigenic disease (a challenging concept at first). Clinically this is evident, because each patient exhibits 3–5 of the about 15 potential complications, and in addition, with significant inter-individual variations in intensity. The latter is the consequence of three types of genes: (a) the gene housing the primary mutation, β globin exhibiting a mutation in position [6Val•Glu]; (b) pleiotropic genes (genes involved in secondary pathophysiologic events beyond the primary mutation, each with the potential of modifying the extent and features of hemolysis, marrow replacement, osteonecrosis, etc.) (Fig. 1); and (c) epistatic genes (polymorphism in pleiotropic genes) that significantly modulates the pathophysiology of the disease in a particular patient). These secondary events are an important part of the phenotype and explain the intense inter-individual differences in the severity of the disease, in spite of all the patients having the same sickle globin gene in the homozygous form.
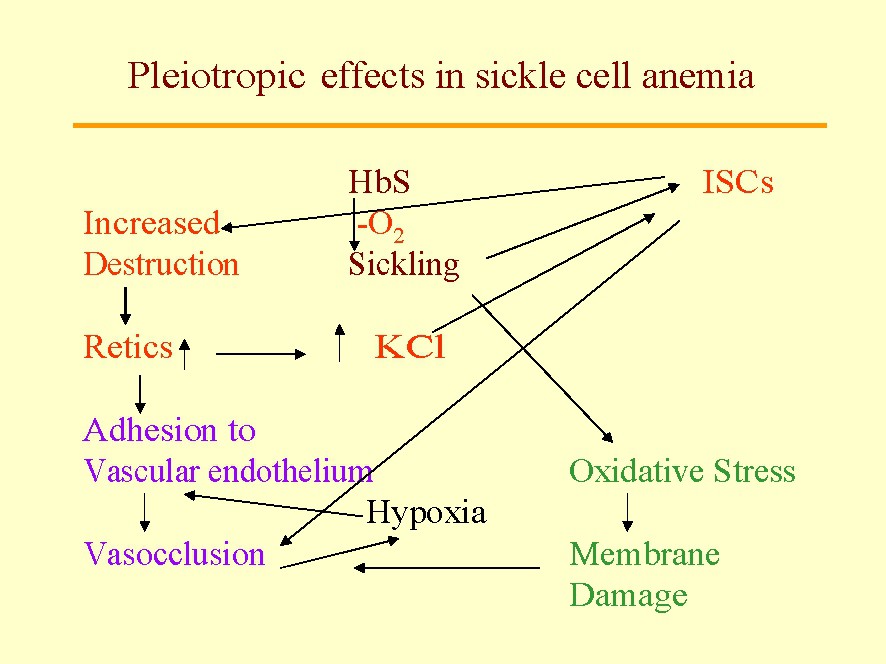
Some pleiotropic effects in sickle-cell anemia: The sickle mutation β6 Glu•Val is an indispensable as the primary genetic event. Nevertheless, this mutation produced a cascade of pleiotropic effects. The first is the induction of sickle polymer formation within the red cells which leads to the formation of irreversibly sickled cells (ISC) among the sickle red cells. The ISCs live only about 3 days in circulation and are big contributors to hemolysis and anemia; they vary in their proportion among patients suggesting the presence of epistasis influence in their generation. We also know that they increase destruction of sickle cells (hemolysis) promoting a marrow response with increased circulating stress reticulocytes, a phenomena, that in turn, increases the activity of the red cell K:Cl cotransport and the Gardos' Channel. This dehydration leads to formation of ISCs. An increase in reticulocytes, sticky red cells, enhances the adhesion of red cells to the vascular endothelium that leads to microvascular vasoocclusion that, in turn, promotes painful crises, silent gray matter infarcts, etc. Also sickling of red cells increases oxidative stress that, in turn, leads to red cell membrane damage and propensity to vascular adhesion and vasoocclusion. Behind all of these pleiotropic events are the potential effects of epistatic genes.
In the last decade, a number of epistatic genes and pleiotropic genes have been defined, and many others are now in the category of potential candidates. Microarray expression technology and high-throughput sequencing promise to accelerate our full multigenic understanding of this disease, not as a Linneus type category, but as genomically individualized concept, as we navigate the new millennium [6].
A complication in which epistatic effects have been studied is cerebrovascular obstruction, a common cause of morbidity and mortality in SCA (HbSS). About 10% of the patients have a clinical stroke before 20 years of age, and another 22% have silent infarction detected by MRI, that many times is accompanied by motor-cognitive deficiencies [7]. The phenotypic variation among patients with HbSS suggests a role for modifier genes and/or environmental influences. Driscoll et al. [7] studied the familial component of clinical stroke in HbSS, estimating the prevalence of clinical stroke among all patients and among SCA sibling pairs at 9 pediatric centers. The sample included 3425 patients with SCD (younger than 21 years), including 2353 patients with SCA. The stroke prevalence was 4.9% for all sickle genotypes: 7.1% for patients with SCA; 1.1% for patients with HbS-β° thalassemia; 0.6% for patients with S thalassemia; and 0% for patients with HbSC. In 207 sibships, more than 1 child had SCA. Of 42 sibships at least 1 sibling had a stroke, and in 10 of the 42, 2 siblings had a stroke. A permutation test indicated that the number of families in which 2 children had strokes was larger than the number expected if strokes were randomly distributed among children in sibships ().
The modulation in hemoglobin F (HbF) levels (an anti-sickling globin) is linked with β-globin gene cluster haplotypes and to gender and other chromosomal sites, significantly influencing the severity of the disease [8]. Coexistence of α-thalassemia with sickle cell disease (SCD, all genotypes) produces hematological and clinical consequences that are beneficial in some complications but deleterious in others. There is little, if any, modulation of the phenotype of SCA by coexistence of G6PD deficiency. Mutations that favor blood coagulation or thrombosis may influence the phenotype of the disease. Improved understanding of the influence of genes involved in modulating the complex pathophysiology of SCD may allow prediction of the phenotype of SCD patients and aid in management decisions [8].
In SCA, HbF concentrations vary by two orders of magnitude. Wyszynski et al. [9] postulated that this variance may be a result of heterogeneity in gene regulatory elements. Accordingly, they searched for single nucleotide polymorphisms (SNPs). More than 180 SNPs were studied in 38 genes of 280 SCA patients. The strongest association with HbF was found with SNPs near a QTL previously localized on chromosome Genetic factors affecting postnatal γ-globin expression are difficult to study in mice, that lacks a gene equivalent t 6q22.3-q23.2. Initially, two SNPs were identified in intergenic portions of this QTL and were associated with about a 20% difference in %HbF. Subsequently, they genotyped 44 additional SNPs between 136.1 Mb and 137.5 Mb on chromosome 6q. Twelve SNPs, associated with a 20%–30% difference in %HbF, were located in the introns of four genes. Haplotypes C–T–T–T in MAP7 and T–C–C in PEX7 were significantly associated with increases in %HbF. Genetic elements abutting the 6q22.3-q23.2 QTL may harbor trans-acting elements that help modulate baseline HbF levels in SCA.
Genetic factors affecting postnatal γ-globin expression are difficult to study in mice due to the lack of an equivalent gene to the human γ-globin. Lin et al. [10] introduced a human β-globin cluster YAC transgene into the genome of FVB/N mice. The β-cluster contained the Greek hereditary persistence of HbF (HPFH) γ allele, resulting in significant postnatal expression of human γ-globin in transgenic mice. To map genes affecting postnatal γ-globin expression, a 20-centiMorgan (cM) genome scan was performed, followed by high-resolution marker analysis of promising loci. From this analysis, Lin et al. [10] mapped a locus within an 18-cM interval of mouse Chromosome (Chr) 1 (LOD = 4.3) that contributes 10.9% of variation in γ-globin level.
The S+S-Antilles transgenic mouse has renal defects similar to those seen in SCA patients: congested glomeruli, medullary fibrosis, renal enlargement, vasoocclusion, and a urine concentrating defect. Rybicki et al. [11] have used gene expression microarrays to identify genes highly up-regulated in the kidneys of these mice and validated their expression by real-time PCR. Kidney hypoxia, as demonstrated by the presence of deoxyHb, was detected by blood oxygen dependent magnetic resonance imaging (BOLD-MRI). Some of the up-regulated genes included cytochrome P450 4a14, glutathione-S-transferase α-1, mitochondrial hydroxymethylglutaryl CoA synthase, cytokine inducible SH-2 containing protein, retinol dehydrogenase type III, arginase II, glycolate oxidase, Na/K ATPase, renin-1, and alkaline phosphatase 2. An increase in enzyme activity was also demonstrated for one of the up-regulated genes (arginase II). These pleiotropic genes can be integrated into several different pathophysiological processes: a hypoxia cascade, a replacement cascade, or an ameliorating cascade, one or all of which may explain the phenotype of this disease. The authors conclude that microarray technology is a powerful tool to identify genes involved in renal disease in SCA and that the identification of various metabolic pathways may open new avenues for therapeutic intervention. The next stage is to search for SNPs in humans with SCA that present with diverse forms of renal damage.
Hydroxyurea (HU) is the only FDA approved drug for the treatment of SCA, and recently it has been proven that it reduces mortality [12]. Nevertheless, many patients die of sickle complication, while taking HU. Bakanay et al. [13] reported that of the 226 patients treated with HU, 38 died (34 of sickle-related causes). Acute chest syndrome was the most common cause of death (35%). The patients involved in this group were significantly older when the HU cohort was instituted; they had significantly higher serum BUN and creatinine. Also, they were more anemic and the sickle gene was more likely linked to the Bantu and Cameroon hyplotypes, which appeared to be acting as carriers of synergistic epistatic interactions with the sickle gene. The Bantu haplotypes are known to be associated with lower HbF expression [14].
Table 1 is a summary of the epistatic effects on sickle-cell anemia.
Summary of the epistatic effects on sickle-cell anemia
| Genotype | Epistatic effect | Epistasis |
| SS (SCA) | Modulation in the Hb F levels | Gender: females have higher HbS than males with SCA |
| SS (SCA) | Modulation in the Hb F levels | β-gene cluster haplotypes (HP): Senegal and Arab India, HP have lower HbF than Benin, Bantu and Cameroon HP.A locus within an 18-cM interval of mouse Chromosome 1 contributes 10.9% of variation in -globin level |
| SS, SC, SE | Reduction of hemolysis | Reduction of MCHC and hence, reduction of delay time of polymerization of HbS |
| Reduction in incidence of most complications | ||
| SS and SCD | Incidence of cerebral stroke | Families in which 2 children had strokes had a higher number than expected if they were randomly distributed among children in sibships (p=0.0012) |
| SS and Thalassemia | Reduction of HbS in sickle cells and potentially lowering the extent of polymerization | (AT)x(T)y, 500 bpto the human-globin gene is a silencer for this gene. BP1 binds most tightly to DNA of the Indian HP, and the Benin HP, that exhibits weaker affinity for BP1, and the weakest to DNA of the Bantu HP, associated with clinically more severe sickle cell symptoms |
| SS, SCD | An increased frequency of the variant alleles in the HSMBP1B genotype patients who had not suffered severe infections | Mannose-binding protein (MBP) is a serum lectin involved in the innate immune response to infections. Children with SCD have an increased susceptibility to meningitis, septicaemia, and osteomyelitis. Apart from known variant alleles, three novel ones have been found, that reduces incidence of severe infections [40] |
2.2 Epistasis in other diseases
The genetics of human systemic lupus erythematosus (SLE) has been under intense studies during the past decade. Although the complexity inherent to polygenic, multifactorial diseases is challenging, several new insights have been obtained in the past several years using linkage and association studies of families containing SLE patients as well as case-control studies of populations [15]. In addition, recent advances in our understanding of the human genome and emerging technology have provided new tools in analyses of complex traits, such as SLE.
Immunoglobulin GM and KM genes have been associated with antibody responses to a variety of antigens. A promoter-region polymorphism of IL-6 gene (−174 G/C) has been shown to be associated with antibody responses to heat-shock proteins HSP60 and HSP65. To detect possible epistatic effects on the autoimmune responses to HSP60 and HSP65, 176 healthy Caucasian subjects from Finland were genotyped for several allelic determinants of GM, KM, and IL-6 genes by PCR-RFLP and IgG antibodies to HSP60 and HSP65 measured by ELISA [16]. Significant interactive effects of GM f,z and IL-6–174 genotypes were noted for both anti-HSP60 () and anti-HSP65 () antibody levels. Since these autoantibodies have been implicated in susceptibility to coronary heart disease and carotid atherosclerosis, the associations might be relevant to the etiology of these diseases.
The biological activity antigens of the ABH and Lewis blood group family are still unresolved. Genes involved in their biosynthesis and their tissue distribution in humans and other mammals, has allowed Marionneau et al. [17] to propose that selective forces may maintain or propagate these oligosaccharide antigens. The ABO, α-1,2 fucosyl-transferase and α-1,3 fucosyl-transferase enzyme families have been generated by gene duplications. Members of these families contribute to biosynthesis of the antigens through epistatic interactions. The authors suggest that these highly polymorphic genes provide intraspecies diversity that allows coping with diverse and rapidly evolving pathogens. In contrast, the genes of low frequency polymorphism are expected to play roles at the cellular level, although they may also be dispensable at the individual level. In addition, some members of these three gene families are expected to be functionally redundant and may either provide a reservoir for additional diversity in the future or become inactivated. The role of the ABH and Lewis histo-blood group antigens in pathologies such as cancer and cardiovascular diseases is merely incidental and devoid of evolutionary impact.
The variable clinical manifestations of cystic fibrosis (CF) suggest the influence of epistatic (modifier) genes. For example, meconium ileus is present in approximately 10–15% of neonates with cystic fibrosis; however, the genetic and/or environmental factors involved have not been determined. Rholfs et al. [18] propose the HFE gene as a candidate modifier locus for CF based on the following: (1) the suggestion of an association between the HLA loci and CF phenotypes; (2) the location of the HFE gene near the HLA loci, and (3) the similarity between the gastrointestinal manifestations of hereditary hemochromatosis (HC) and CF. The frequency of the C282Y and H63D mutations in a group of 89 CF patients homozygous for δ F508 and with meconium ileus status known, was determined. The carrier frequency of C282Y in CF patients with meconium ileus was significantly different from that of our unaffected control group (19.4% versus 7.7%). However, the difference between the meconium ileus and the nonmeconium ileus groups was not significant (19.4% versus 10.3%). There was no difference in the frequency of the H63D among the three groups that were studied. These data are suggestive, but not definitive, of a relationship between the development of meconium ileus or other gastrointestinal diseases in CF and the HFE gene.
Susceptibility to complex autoimmune diseases (AIDs) (not to be confused with AIDS) is a multigenic phenotype affected by a variety of genetic and environmental or stochastic factors [19]. Linkage analyses in the identification of non-major histocompatibility complex (non-MHC) susceptibility alleles have proved difficult, predominantly because of extensive genetic heterogeneity and possible epistatic interactions among the multiple genes required for disease development. Despite these difficulties, progress has been made in elucidating the genetic mechanisms that influence the inheritance of susceptibility, and the pace of gene discovery is accelerating. An intriguing new finding has been the colocalization of several AID susceptibility genes in both rodent models and human linkage studies. This may indicate that several susceptibility alleles affect multiple AIDs or alternatively, that genomic organization has resulted in the clustering of many immune system genes.
Vestibular dysfunction leads to dizziness and imbalance. Genes play an important role in its etiology, but the genetics are complex and poorly understood. Cryns et al. [20] have analyzed the complex inheritance pattern in the Epistatic Circler mouse that shows circling behavior indicative of vestibular dysfunction. This phenotype exists in a fraction of the F2-generation from a crossing between C57L/J and SWR/J mouse strains. Genetic investigation indicates that the circling behavior is caused by a major recessively inherited gene derived from the SWR/J strain (the Ecs-gene) in combination with at least three different epistatic genes derived from C57L/J (the Ecl-genes). Genetic mapping made it possible to localize the Ecs-gene to chromosome 14 and the Ecl-genes to chromosome 3, 4, and 13.
Interactions between glucocorticoid receptor (GRL), lipoprotein lipase (LPL), and adrenergic receptor (ADR) genes on plasma insulin and lipid levels are poorly understood. Ukkola et al. [21] undertook a cross-sectional study based on 742 individuals from phase 2 of the Quebec Family Study (QFS) cohort. Gene markers were identified by Southern blot or PCR. Plasma glucose and insulin in the fasted state and during an oral glucose tolerance test were determined, and insulin and glucose areas were computed. Triglyceride and cholesterol in plasma and lipoprotein fractions were determined enzymatically. The results showed that GRL and LPL variants had independent effects on plasma high-density lipoprotein cholesterol (HDL-C), and two β2-ADR variants were related to total cholesterol concentrations. The α2-ADR gene Dral polymorphism was the only variant that had an independent effect on the plasma insulin area. Gene-gene interaction (epistatic) effects were found between GRL and α2-ADR genes for low-density lipoprotein cholesterol ([LDL-C] ) and between GRL and LPL genes for HDL-C, although the p value was weak (). Stronger interaction effects involving GRL, LPL, and ADR markers were observed for the plasma insulin area ( to 0.025) but not the glucose area. After correction for multiple tests, the findings remained essentially unchanged for the insulin area but became non-significant for the lipid phenotypes. In conclusion, multiple interactions among GRL, LPL, and ADR gene markers contributed to insulin metabolism and perhaps to lipid levels, but no significant effect was found for each gene separately. The LPL locus appeared to determine the pattern of interactions with ADR and GRL loci. These results suggest that multiple epistasis effects could play a role in the etiology of risk factors for common chronic diseases.
3 Ethnicity and founders' effect: impact on epistasis
In the search for epistasis, we need to be cautious about another complicating factor that represents an added potential pitfall: epistatic genes tend to be diverse in different ethnic groups, and therefore admixed populations tend to be more complex.
As previously discussed, the diverse interaction between epistatic genes discussed above in different ethnic groups and in admixed populations will add another hurdle in the generation of useful sets of data in genetic diseases found around the world. A case in point is sickle-cell anemia: the gene seems to have originated independently in at least five times and sites: Atlantic West Africa (linked to the Senegal haplotype), Central West Africa (Benin haplotype), the sickle gene that arose exclusively among the Eton people in Cameroon (Cameroon haplotype), and finally, the Arab-India haplotype linked to the sickle mutation, found among the Adavasi of India and among Arabs in the eastern oasis of Saudi Arabia [22–24].
The Atlantic slave trade and the Arab slave trade dispersed these genes to the Americas and the Middle East. While the Benin haplotype linked to the sickle gene migrated to Sicily, Portugal, Greece and Turkey several hundreds of years ago, more recently, a more genetically diverse sickle gene has migrated (with the immigrant flow of ‘guest workers’) to several countries of Europe, including France, Germany, Spain and Italy. The occurrence of this gene flow and the forthcoming admixture with the host populations has resulted in important genetic consequences. For example, African-Americans, are now, in average about 25% Caucasians [25]. Hence, epistatic genes will vary in patients with SCA, in each of these admixed populations, according the extent and origin of the admixing genetic influx.
Finally, founder's effect has to be considered. This term refers to the potential distorting effect on gene frequency due to by the migration and resettling of a relatively small group of people from a particular ethnicity into a larger community. The migrants might carry only a subset of the epistatic genes found in the population from which they came. This particular phenomenon might be operative in the enclaves of sickle cell carriers, for example, found on the islands of Greece, Turkey and southern Chile [26–28].
Initial efforts have been made recently to understand and measure the ethnic admixture/hemoglobinopathy relationship. A range of estimates for sickle cell and β-thalassemia have been derived for the different ethnic groups living in the UK [29]. In England approximately 3000 affected babies (0.47%) carry the sickle cell trait and 2800 (0.44%) carry the β-thalassemia trait annually: with approximately 178 (0.28‰ conceptions) affected by sickle cell disease (SCD) and 43 (0.07‰) by β-thalassemia. About 140–175 (0.22–0.28‰) affected infants are born annually, with SCD and from 10 to 25 (0.02–0.04‰) with β-thalassemia.
The Department of Health in Britain has undertaken a linked antenatal and neonatal screening program for hemoglobinopathies by 2004 in a comprehensive national plan for the National Health Service in Britain; includes the investigation of the effectiveness of ethnic origin as a basis for selection [30]. Two previous reports in Britain emphasized the importance of generating a standardized instrument for collecting ethnicity data and recommended early development of such work. Nevertheless, substantial variability in practice and the quality of data resulted in the occurrence of mis-classification as high as 20% against a recommended target of fewer than 5.5%. Hence, further work to produce the proper instruments is necessary.
In the US, four interesting efforts are worth mentioning. Gregg et al. [30] studied the resistance to activated protein C (APC) as the most common risk factor for venous thromboembolism, a major cause of morbidity and mortality with an incidence of about 1/1000 per year. The Arg 506 to Gln mutation in exon 10 of the coagulation factor V gene (factor V-Leiden), an autosomal dominant trait, is responsible for >90% of the APC resistance cases. Initial studies suggested that this mutation is restricted to European Caucasians with an allele frequency in European and American Caucasians of 4.4%, making it one of the most common monogenic disorders in this population. A limited number of other ethnic populations have been tested, and the mutation has been found only rarely. Gregg et al. [30] in a multiethnic survey of 602 Hispanic–Americans found them to have the highest frequency of the factor V-Leiden mutant allele, 1.65%, while African–Americans had lowest frequency (0.87%). No factor V-Leiden mutations were found in 191 Asian–Americans or in the 54 Native Americans tested. Hence, the factor V-Leiden mutation segregates in populations with significant Caucasian admixture and is rare in non-European groups. This ethnic stratification may be useful in developing cost effective, selective screening programs to identify those at risk for thromboembolism who can benefit from prophylactic therapy.
William et al. [31] traced European–American admixture in the Gila River Indian Community and its mode of entry. Among the 9616 residents, 2015 claim only partial Native American heritage. A procedure employing 23 alleles or haplotypes at eight loci was used to estimate the proportion of European–American admixture, , for the entire sample and within six categories of Caucasian admixture calculated from demographic data, md. The genetic analysis estimated a total European–American admixture of 0.054 (95% confidence interval: 0.044–0.063).
An estimate from demographic records was similar (0.059). Regression of on md yielded a fitted line , (). When total European–American admixture is partitioned between the contributing populations, Mexican-Americans provided 0.671, European-Americans 0.305, and African-Americans 0.023. They concluded that accurate, reliable estimates of genetic admixture are possible from allele and haplotype frequencies, even when there is little demographic information for the population.
Of particular interest is the effort of Smith et al. [32] that embarked on admixture mapping (mapping by admixture linkage disequilibrium or MALD) to localize the genes that cause disease in admixed ethnic groups such as African Americans, with approximately 100 times fewer markers than are required for whole-genome haplotype scans. However, it has not been possible up to now to perform powerful scans with admixture mapping because the method requires the availability of a dense map of validated markers known to have large frequency differences between Europeans and Africans. To create such a map, the authors screened through databases containing approximately 450 000 single-nucleotide polymorphisms (SNPs) for which frequencies had been estimated in African and European population samples. They confirmed the frequencies of the most promising SNPs in a multiethnic panel of unrelated samples and identified 3011 as a MALD map (1.2 cM average spacing). They estimated that this map is approximately 70% informative in differentiating African versus European origins of chromosomal segments. This map provides a practical and powerful tool, which is freely available, without restriction, for screening of disease genes in African American patient cohorts. The map is especially appropriate for those diseases that differ in incidence between the parental African and European populations.
4 Environmental/genetic interactions
There is little doubt, around the world, that poverty reduces the chance of proper medical care and reduces the lifespan of patients suffering from many diseases, including the hemoglobinopathies. Poverty also leads to malnutrition that is clearly detrimental. In the case of SCA, exposure to cold weather, high altitude and un-pressurized aircrafts are associated with precipitation of painful crises and in the cases of S/β-thalassemia and sickle trait, of spleen infarcts. Sickle trait individuals are susceptible to death in hard training exercise as are common in military training [33].
Concurrent diseases, such as respiratory infections were common causes of death in infants, as substantiated by the considerable increase in survival rates in the first decade of life when pneumococcal vaccines and penicillin prophylaxis were introduced in the US for patients with sickle syndromes [34,35]. Concurrent sleep apnea, promotes painful crises and possibly acute chest syndrome in these patients. Iron deficiency is considered a problem, but excess iron administration is contra-productive. Iron supplement in moderation is not a problem and might even decrease malaria infection [36].
An interesting question is the effect of malaria around the world on the survival of hemoglobinopathies. The prevalence of malaria parasitemia seems to be normal or possibly lower in asymptomatic SCA subjects than in AA controls [37]. These observations could be explained by HbS-containing red cell features that protect the carriers, not from getting malaria, but from dying of it and from severe manifestations. Mechanisms involved have been reviewed by Nagel et al. [38] and extended recently by Hebbel [39].
5 Conclusions
Epistasis is, most probably, the principal mechanism that explains the inter-individual phenotypic variability of genetic diseases. For example, the case in point is sickle-cell anemia. We observed that among the main 15 complications for which these patients are at risk, only about five or fewer complications are apparent in each patient. This phenotypic diversity negatively impacts on quality of life and survival. Finally, small enclaves of patients with sickle-cell anemia can have a significant founder's effect (see above).
The increasing capability of defining epistatic genes in this disease, as a consequence of the technical advances generated by the genomic era, unfolds the exciting potential of being able to define the plethora of epistasis in individual patients and, hence, being able to advance the therapeutic strategies that are in concert with the risks involved. Fortunately, it is becoming apparent that the same is true for most genetic diseases.
The admixture of ethnic groups is a major issue in defining epistatic effects. Although ethnic admixture existed since the earliest times, it was strongly promoted by the Age of Discovery, with its transatlantic voyages, and the active Atlantic and Arab slave trades. In the last century, this phenomenon has exploded with increases in travel facilities, increased communications, and the drastic consequences of the last two World Wars that devastated Europe. Fortunately, ideas as how to develop methodologies to quantitate and identify the effect of epistatic genes, characteristics of each of the ethnic groups participating in the admixture, are emerging.
Nevertheless, we need to be cautious about another complication that represents a potential pitfall: epistatic genes tend to be diverse in different ethnic groups and their complexity increases in ethnically admixed populations. As the diverse interaction between epistatic genes, discussed above, in different ethnic groups and admixed populations add another hurdle to the generation of useful sets of data in genetic diseases present around the world. A case in point is sickle-cell anemia: the gene seems to have originated independently at least four times (Atlantic West Africa (linked to the Senegal haplotype), Central West Africa (Benin haplotype), and the sickle gene that arose exclusively among the Eton people in Cameroon (Cameroon haplotype), and finally, the Arab-India haplotype linked to the sickle mutation, found among the Adavasi of India and among the Arabs in the eastern oasis of Saudi Arabia [39,40].
The Atlantic and the Arab slave trades (propel by different needs and with different consequences) dispersed these genes to the American and to the Middle East. While the Benin haplotype linked to sickle gene migrated to Sicily, Portugal, Greece and Turkey several hundreds of years ago, more recently, a more genetically diverse sickle gene has migrated (with the flow of ‘guest workers’) to several countries of Europe, including France, Germany, Spain and Italy. The occurrence of this gene flow and the forthcoming admixture with the host populations has resulted in important genetic consequences. African-Americans, for example, are now, on average about 25% Caucasian, as mention before [25]. It is necessary to conclude that epistatic genes will vary in patients with sickle-cell anemia in each of these admixed populations according to the extent of the gene influx to their original genetic make-up.