

1 Introduction
Apoptosis is essential for the development of multicellular organisms and for their correct functioning. This process is defined by specific biochemical and morphological changes, including fragmentation of DNA, compaction of the chromatin into dense structures, blebbing of the plasma membrane, exposure of phosphatidylserine residues in the outer leaflet of the plasma membrane, and engulfment of the apoptotic cells by phagocytes. These hallmarks of apoptosis are due to the activation of the aspartate-specific proteases that are known as caspases [1,2]. In many apoptotic responses, activation of caspases requires the release of mitochondrial apoptogenic factors such as cytochrome c, an event made possible thanks to permeabilization of the outer mitochondrial membrane by pro-apoptotic members of the Bcl-2 family.
Proteins that belong to the Bcl-2 family are characterized by the presence of at least one of the four Bcl-2 homology domains (BH1, BH2, BH3 and BH4). They are further classified into three groups, according to their pro-survival or pro-apoptotic nature and to the occurrence of the different BH domains. Anti-apoptotic Bcl-2-like proteins (e.g., Bcl-2, Bcl-xL, Bcl-w, Mcl-1 or A1/Bfl-1) have the four BH domains, pro-apoptotic multidomain proteins (e.g., Bax, Bak or Bok/Mtd) lack BH4, and pro-apoptotic BH3-only proteins (e.g., Bid, Bim/Bod, Bad, MAP-1, Bmf, Bik/Nbk, Blk, Noxa, Puma/Bbc3, or Hrk/DP5) only possess a BH3 domain. Some members of the Bcl-2 family also have a hydrophobic C-terminal region that allows their localization on intracellular membranes [3]. Heterodimerization between individual members of the family is an important mechanism that controls their activity [4–7].
Under normal conditions, Bak is a transmembrane protein of the outer mitochondrial membrane, while Bax is isolated both as a cytosolic protein and as a peripheral protein of the mitochondria. As will be detailed below, Bax could be maintained in an inactive conformation through interactions with cytosolic retention factors. Following some cytotoxic signals, the conformation of Bax and Bak changes, and their N-terminal domains, which are buried in the inactive proteins, become accessible to antibodies. Bax translocates to the mitochondria, integrates into the outer mitochondrial membrane, and both Bax and Bak oligomerize, leading to the permeabilization of the outer membrane (for a model, see Fig. 1). The mechanism of activation of Bax and Bak remains largely unknown, even though there is no doubt about the key-role played by BH3-only proteins. Once activated, at the transcriptional or the post-translational level, BH3-only proteins bind to and activate Bax and Bak (this is the case for Bid, Bim-s or Puma) and/or bind to and inactivate anti-apoptotic proteins (this is the case for Bad or Noxa) (Fig. 2). Incubation of isolated mitochondria with recombinant Bax and tBid, the active form of Bid generated by proteolytic cleavage leads to the oligomerization of Bax and to cytochrome c release, independently of an effect of tBid on Bcl-2-like proteins [8,9]. As these events no longer occur if mitochondria are pre-incubated with proteinase K [10], additional proteins seem to be required [11], even though other reports suggest that tBid and liposomes are sufficient to trigger Bax activation [12,13]. During these last years, increasing evidence has implicated other proteins in the regulation of apoptosis, in particular in the regulation of proteins of the Bcl-2 family. The purpose of this review is to present these new actors and to briefly discuss the models suggested for the mechanism of permeabilization of mitochondria.
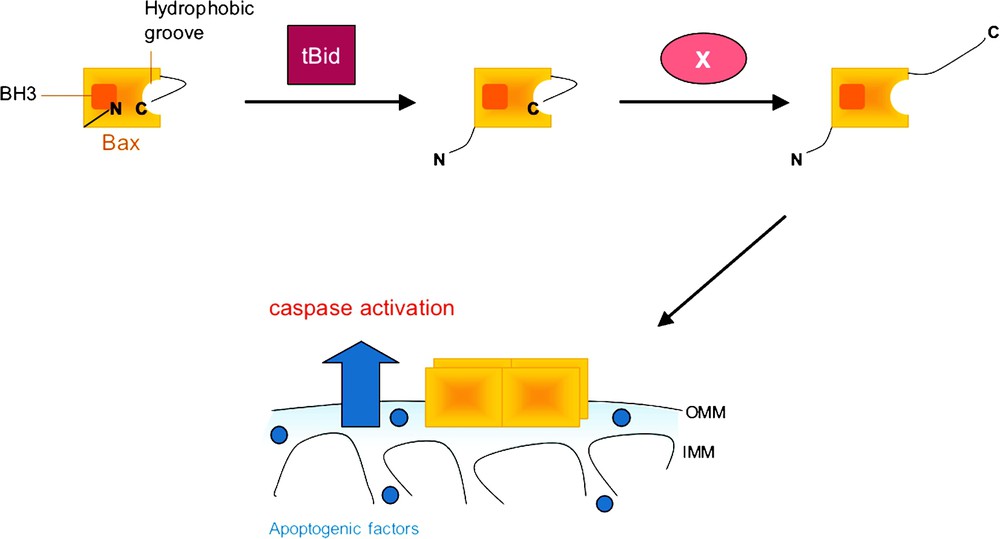
Multistep model for Bax activation. Under resting conditions, Bax is usually cytosolic, with its C-terminal tail being buried in a hydrophobic pocket formed by its BH1, BH2 and BH3 domains. Following some cytotoxic insults, the conformation of Bax changes: through the binding of a BH3-only protein such as tBid or Puma, the N-terminal region of Bax is unmasked; through an interaction with an unknown factor X, the C-terminal region of Bax is released, allowing its translocation to mitochondria where it may interact with cardiolipin. Exposure of the BH3 domain of Bax results in the recruitment of other Bax molecules. Oligomerization of Bax follows, which leads to the permeabilization of the outer mitochondrial membrane and caspase activation. IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane.
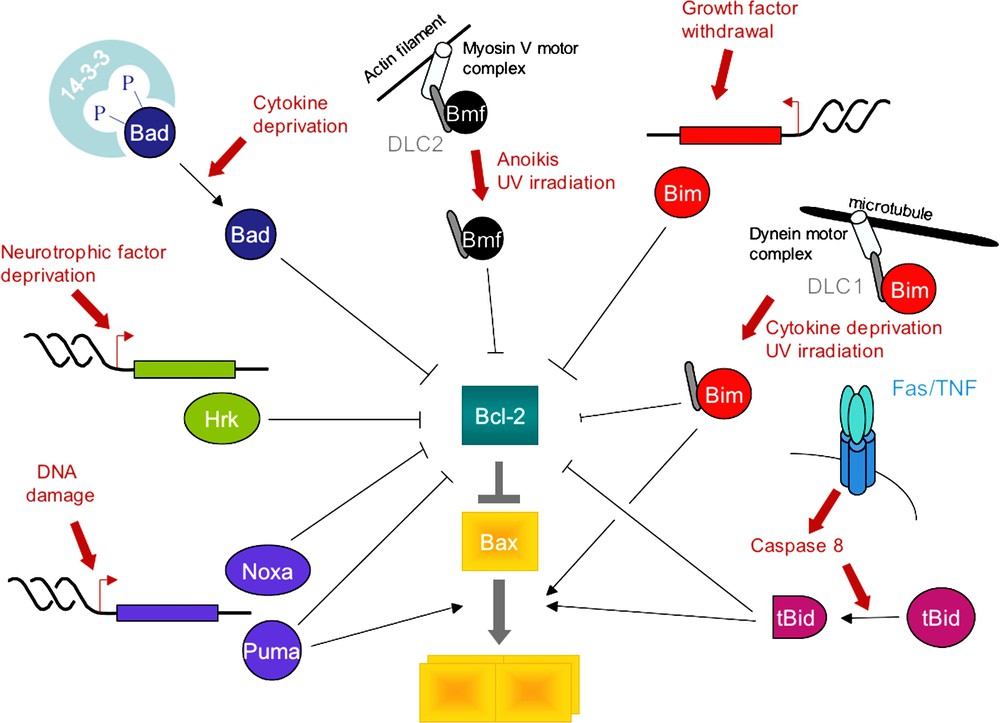
The role of BH3-only proteins in apoptosis. BH3-only proteins are activated at the transcriptional level (Noxa, Puma, Hrk, Bim) or at the post-translational level (Bid is cleaved by caspase 8 and myristoylated; Bad is dephosphorylated and dissociates from 14-3-3 proteins; Bmf is released along with dynein light chain 2 (DLC2) from the actin cytoskeleton to which it binds through the myosin V actin motor, while Bim bound to DLC1 detaches from the dynein motor complex and from microtubules) [126]. Through their BH3 domain, BH3-only proteins then either bind to and activate Bax (tBid, BimS, Puma) or bind to and inactivate Bcl-2-like anti-apoptotic proteins (Bad, Noxa, Puma, Hrk, Bmf) [4,53,54].
2 Proteins that prevent the activation of Bax and Bak
As mentioned above, under resting conditions, Bax exists as a soluble protein in the cytosol. Some reports indicate that it is sequestered through interactions with inhibitory proteins, even though gel filtration analysis suggests that it is monomeric. Proteins that have been proposed to act as Bax- or Bak-inactivating proteins are discussed (Fig. 3).
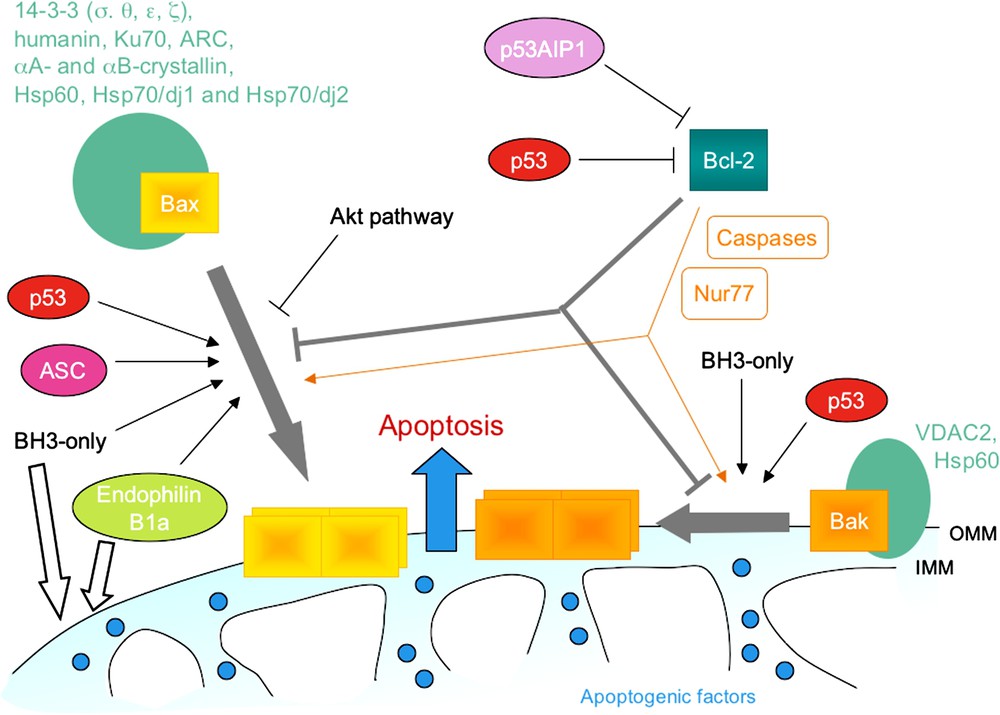
Activation of Bax and Bak is a highly regulated process, inhibited by anti-apoptotic members of the Bcl-2 family and promoted by BH3-only proteins. Numerous factors were identified as cytosolic retention factors for Bax (14-3-3 isoforms, humanin, Ku70, ARC, αA- and αB-crystallin, Hsp70/dj1 and Hsp70/dj2, and Hsp60) and some as inhibitors of Bak (VDAC2, Hsp60). Phosphorylation of Bax through an Akt-dependent pathway also prevents its translocation to mitochondria. Activation of Bax and Bak was reported to be directly potentiated by endophilin B1, p53 and ASC, even though no direct interaction between p53 and Bax could be detected [62]. Some proteins can also promote cell death through an inhibition of pro-survival Bcl-2 proteins (p53, p53AIP1), while others even convert them to killing factors (Nur77-binding or caspase-cleavage). Finally, some proteins might also contribute to permeabilization of the outer mitochondrial membrane through a direct modulation of its properties. A role in the remodelling of mitochondrial membranes was suggested for tBid and for endophilin B1.
2.1 14-3-3 proteins
14-3-3σ was the first 14-3-3 isoform identified as a Bax-interacting protein. Its expression in human colorectal tumour cells enforces a G2 arrest and prevents apoptosis when they are exposed to DNA-damaging agents [14]. Since then, three other members of the family were also proposed to sequester Bax in the cytosol [15]. Caspase cleavage of 14-3-3θ and phosphorylation of 14-3-3 proteins by the c-Jun NH2-terminal kinase (JNK) were suggested as mechanisms promoting the dissociation of Bax, allowing its translocation to mitochondria [15,16]. In agreement, inhibition of proteins of the 14-3-3 family leads to apoptosis [17], but the mechanisms involved could be numerous, as they are also known to bind to several other proteins involved in survival and in death signalling, including another member of the Bcl-2 family, Bad [18].
2.2 Humanin
The small peptide humanin was initially identified as a secreted protein that prevents cell death induced by overexpressing a mutant amyloid precursor protein [19]. Later, overexpression of humanin was also shown to inhibit Bax-dependent apoptosis, and its down-regulation to sensitise SF268 cells to staurosporine and to serum-deprivation-induced apoptosis [20]. More recently, cytosolic humanin was also shown to bind to two other members of the Bcl-2 family, BimEL and Bid [21,22]. The authors indicate that it remains uncertain if humanin is a physiologic inhibitor of these pro-apoptotic proteins, as it is not clear if it is endogenously expressed in the cytosol; nevertheless, humanin might be an interesting tool to study the mechanism of activation of Bax and for the development of therapeutic compounds to prevent inappropriate cell death.
2.3 Ku70
Ku70 was first characterized as part of the Ku70/Ku80 heterodimer that is involved in DNA double-strand repair and in the rearrangement of T-cell receptor genes. A small fraction of Ku70 localized in the cytosol was unexpectedly shown to bind to Bax and cells expressing a Ku70 antisense RNA and Ku70-/- mouse embryonic fibroblasts were reported to be more sensitive to apoptosis than wild-type cells [23]. A small pentapeptide was identified as the Bax-binding motif and protects cells from apoptosis to the same extent as Bax-deficiency [24,25]. Association between Ku70 and Bax is inhibited by acetylation of lysine residues of Ku70, an event that occurs in 293T cells after UV irradiation [26]. Histone deacetylase inhibitors, which are under clinical trial for cancer treatment, promote the dissociation between Ku70 and Bax and induce Bax-dependent apoptosis in neuroblastoma cells [27].
2.4 Apoptosis regulator with a CARD (ARC)
ARC, a small protein highly expressed in the skeletal muscle and cardiac tissue, was initially identified as a pro-caspase-2- and pro-caspase-8-binding protein that attenuates cell death triggered by death receptors [28]. However, it was also recently shown to bind to Bax and to inhibit Bax-induced cytochrome c release from mitochondria isolated from rat heart [29]. Down-regulation of ARC by antisense ARC oligonucleotides leads to spontaneous exposure of the N-terminal domain of Bax and would seem to increase cell death, as revealed by trypan blue exclusion [30]. ARC was thus suggested to be a general apoptosis inhibitor, acting both at the level of the extrinsic and of the intrinsic apoptotic pathways [30]. However, the relative contributions of these mechanisms to the pro-survival activity of ARC need to be further analysed.
2.5 Heat-shock proteins (Hsps)
Hsps are a family of chaperone proteins that are induced under stress conditions to help cells limit the damage and allow them to recover. Several Hsps were identified as apoptosis regulators, mostly as inhibitors of apoptosis. They were reported to act at multiple steps of the cell death pathways, both extrinsic and intrinsic [31]. Very recently, some Hsps have been characterized as inhibitors of pro-apoptotic members of the Bcl-2 family. The small Hsp αA-crystallin is highly expressed in vertebrate lens, while its isoform αB-crystallin has a broader tissue distribution. Expression of αA-crystallin in human lens epithelial (HLE) cells protects them from staurosporine-, UV-, anti-Fas and tumour necrosis factor α (TNFα)-induced apoptosis [32], and expression of αB-crystallin in breast cancer cells or prostate carcinoma cells protects them from etoposide- and TNFα-induced apoptosis [33]. Moreover, in various cancer cell lines, the expression level of αB-crystallin could be correlated to resistance to TRAIL (TNF-related apoptosis-inducing ligand)-mediated apoptosis [34]. Several anti-apoptotic mechanisms were proposed. αB-crystallin prevents complete maturation of pro-caspase-3, and both crystallins prevent the up-regulation of Bak that results from staurosporine-treatment of HLE cells [33,35]. In addition, they were recently reported to bind to the pro-apoptotic proteins Bax and Bcl-XS and to prevent their translocation to mitochondria following staurosporine-induced apoptosis in HLE cells [35].
Similarly, the cytosolic chaperone pairs Hsp70/dj1 and Hsp70/dj2 were shown to bind to Bax and to inhibit its mitochondrial translocation in RAW 264.7 macrophages exposed to lipopolysaccharide and interferon-γ, a treatment that leads to apoptosis through the production of NO and the endoplasmic reticulum-dependent up-regulation of the protein CHOP [36]. However, as for crystallins, other anti-apoptotic mechanisms have also been proposed, including inhibition of the recruitment of pro-caspases to the apoptosome complex, downstream of cytochrome c release [37,38], and inhibition of TNF-induced Bid activation through the blockage of JNK [39].
Finally, overexpression of Hsp60 and/or Hsp10 protects cardiac myocytes from ischemic injury and from doxorubicin-mediated apoptosis [40,41]. These chaperonins are principally present in the mitochondrial matrix, but a small fraction (15–20%) resides in the cytosol. They were reported to interact with proteins of the Bcl-2 family, as well as to change their expression level through post-translational mechanisms. In cells overexpressing Hsp60 and/or Hsp10, the expression level of the anti-apoptotic proteins Bcl-2 and Bcl-xL was increased, while the expression level of Bax, Bak and Bad was decreased [41,42]. Down-regulation of Hsp60 by antisense oligonucleotides decreases viability of cardiac myocytes and reciprocally modifies the expression levels of Bcl-2 proteins [41,42]. Direct interaction between Hsp60 and Bax and Bak was detected by co-immunoprecipitation [41,42]. In addition to its anti-apoptotic activities, Hsp60 was shown to accelerate in vitro the maturation of caspase-3 and could thus promote apoptosis under certain circumstances [43].
2.6 Hexokinases
One mechanism through which the kinase Akt promotes cell survival is by promoting an increase of hexokinases I and II at the mitochondria [44]. In the presence of glucose, overexpression of hexokinase I attenuates apoptosis of Rat1a cells exposed to a low dose of UV in the absence of serum [44], and overexpression of hexokinase II inhibits Bax-mediated apoptosis in HEK 293 cells and tBid-induced N-terminal exposure of Bax and cell death in Rat1a cells [45]. Reciprocally, overexpression of tBid promotes dissociation of hexokinase II from mitochondria [46]. As hexokinases also prevent mitochondrial-binding of a recombinant form of Bax that lacks its C-terminal tail, they were proposed to prevent apoptosis by occupying its binding sites or by sterically hindering its access to contact sites [45]. However, the catalytic activity of hexokinases participates in protection, since glucose, which does not modify the extent of hexokinase-binding to mitochondria, is also necessary [44]. In addition, promoting the dissociation of hexokinases from mitochondria leads to cytochrome c release, kills Bax/Bak double knockout cells by apoptosis and in a Bcl-2-independent manner, indicating that they have a role in maintaining mitochondrial integrity, and that the mechanism of cytochrome c release that occurs in these cells is different from the one mediated by Bax and Bak [47]. How hexokinases interfere with Bax and Bak activation and cytochrome c release needs to be clarified further.
2.7 Voltage-dependent anion channel 2 (VDAC2)
VDAC2 was recently identified as a Bak-interacting protein that stabilizes Bak in an inactive conformation [48,49]. Down-regulation of VDAC2 sensitises cells to multiple apoptotic stimuli, and VDAC2-/- mouse embryonic fibroblasts undergo more spontaneous apoptosis than wild-type cells [48].
2.8 Relevance of these interactions
The requirement of the above-mentioned proteins to maintain Bax and Bak in inactive conformations has often been questioned. Indeed, their down-regulation never convincingly led to spontaneous apoptosis, which always required an additional cytotoxic signal, and phenotypes of knockouts do not suggest massive apoptosis. Bax could be bound to multiple retention factors. Dissociation from several of these proteins could be necessary for its translocation to mitochondria, and active mechanisms that promote exposure of its N-terminal domain, release of its C-terminal tail, and oligomerization are certainly required. How these proteins inhibit apoptosis needs to be studied further, as for most of them multiple mechanisms were proposed. Moreover, care must be taken when interpreting results from biochemical experiments, as Bcl-2-like proteins are known to be sticky and their conformation to be sensitive to the experimental conditions, in particular to the presence of non-ionic detergents.
Sequestering Bax or Bak is not the only mechanism that was proposed for Bax/Bak-inactivating proteins. In the case of Hsp60, post-translational modulation of the level of expression of Bcl-2 family proteins was also suggested. Destabilization of Bax was reported as well in lung cancer cells exposed to nicotine, which increases their resistance to cisplatin-mediated apoptosis [50]. In addition to these mechanisms, Bax was recently shown to be regulated by phosphorylation. Phosphorylation of Bax on Ser184 can be detected when neutrophils are treated with various anti-apoptotic stimuli and when lung cancer cells are exposed to nicotine [50,51]. In both cases, the Akt pathway is involved, and Akt can phosphorylate Bax in vitro [50,51]. A Ser184Glu mutant of Bax only localizes to the cytosol and its overexpression does not lead to cell death [52]. A Ser184Ala mutant of Bax constitutively localizes to mitochondria, is a more potent apoptosis inducer than wt Bax, while nicotine does not promote the survival of cisplatin-treated cells expressing this Bax mutant [50]. Phosphorylation of Bax on Ser184 was proposed to facilitate its heterodimerization with anti-apoptotic Bcl-2 family members [51].
3 Proteins that promote the activation of Bax and Bak
The mechanisms that promote conformational rearrangements of Bax and Bak and lead to their oligomerization and to permeabilization of the outer mitochondrial membrane are not completely understood. In addition to three BH3-only factors (tBid, BimS, and more recently Puma) [8,9,53–55] and to the distantly related protein MAP-1 [56], other unrelated proteins were identified as Bax- and/or Bak-interacting factors that directly promote their activation and cytochrome c release (Fig. 3). These observations are detailed below.
3.1 Endophilin B1
Endophilin B1 (also named Bif-1) was initially identified in a yeast two-hybrid screen for Bax-interacting proteins [57]. Endophilin B1 translocates to mitochondria when Cos7 cells are treated with staurosporine, and its overexpression accelerates interleukin 3 withdrawal-induced apoptosis of FL5.12 cells [57,58]. Upon growth factor deprivation, the association between endophilin B1 and Bax transiently increases, but endophilin B1 cannot bind to the active conformation of Bax that is induced by the detergent Nonidet P-40 [57]. Endophilin B1 displays high homology to endophilins A that are involved in synaptic vesicle endocytosis and can tubulate liposomes in vitro. Moreover, endophilin B1 was recently shown to play a role in the maintenance of mitochondrial morphology [58]. As will be detailed below, during apoptosis, mitochondrial membranes are not only permeabilized, but also remodel, and endophilin B1 could provide a link between these two processes.
3.2 p53
The level of the tumour suppressor protein p53 increases in cells under various stress conditions, such as DNA damage, growth factor withdrawal, or in the presence of cytotoxic drugs, and leads to growth arrest or to apoptosis [59]. p53 is a nuclear transcription factor that can stimulate the expression of pro-apoptotic genes such as those encoding the Bcl-2 proteins Bax, Noxa and Puma, the death receptors DR5/KILLER and Fas/APO-1, and the PIGs (p53-induced genes), some of which are related to the oxidative status of the cell. In addition, p53 can also suppress the expression of anti-apoptotic genes, including Bcl-2, Bcl-xL or survivin. However, p53 can also promote cell death through transcription-independent mechanisms, and the relative contributions of both pathways depends on the cell type, the context, and the nature and the intensity of the stress signal [59,60]. For example, apoptosis of the colorectal cancer cells DLD-1 induced by overexpression of p53 is prevented by transcription and translation inhibitors [61]. On the other hand, UV-induced apoptosis of mouse embryonic fibroblasts requires p53, but still occurs in the presence of wheat germ agglutinin, which inhibits nuclear import of p53 and the induction of p53-regulated genes [62].
Following certain cytotoxic signals, a small fraction of p53 translocates to mitochondria [63]. Overexpression of mitochondrial-targeted p53 leads to apoptosis [63,64] and incubation of isolated mitochondria with purified p53 results in cytochrome c release [64,65]. The precise mechanism through which p53 promotes the permeabilization of mitochondria is not clear, as both anti- and pro-apoptotic Bcl-2 proteins were suggested to be directly regulated by p53. p53 was reported to co-immunoprecipitate with Bcl-2 and Bcl-xL in mitochondria isolated from ML-1 and RKO cells treated with the DNA-damaging agent camptothecin and from RBL-2H3 cells treated with eugenol [64,66]. In addition, p53 displaces tBid and Bax from glutathione S-transferase-Bcl-xL, while a fifty times higher concentration of either of them is required to displace p53 [62]. Thus, p53 could promote the permeabilization of mitochondria by sequestering anti-apoptotic proteins, allowing the release of their pro-apoptotic counterparts. However, in mitochondria isolated from Saos2 cells stably transfected with a temperature-sensitive variant of p53, and in the two cancer cell lines LNCaP and MCF7 treated with camptothecin and UV respectively, p53 was shown to co-immunoprecipitate with Bak, but not with Bax or with Bcl-xL [65]. Incubation of isolated mouse liver mitochondria with p53 decreases the association between Bak and Mcl-1, and leads to the oligomerization of Bak and to the release of cytochrome c [64,65]. On the other hand, another group reported that addition of p53 to isolated mouse liver mitochondria does not result in cytochrome c release unless recombinant Bax is added [62]. Furthermore the release of cytochrome c that follows incubation of mouse liver mitochondria with cytosolic extracts from Saos2 cells overexpressing p53 is abolished when Bax is depleted from the extracts by immunoprecipitation, but not when p53 is immunodepleted [67]. According to these studies, p53 would lead to the permeabilization of mitochondria by directly activating multidomain pro-apoptotic proteins, disrupting their association with Bcl-2-like proteins. The discrepancies between the above-mentioned results could reflect differences between the cell types, the experimental conditions of the assays, and the purification procedures of p53 and Bcl-2 proteins.
3.3 ASC and p53AIP1
Upon DNA-damage, p53 also up-regulates the expression of several pro-apoptotic proteins that localize at the mitochondria. Among these, the apoptosis-associated speck-like protein (ASC) and the p53-regulated apoptosis-inducing protein 1 (p53AIP1) were shown to interact with proteins of the Bcl-2 family. Overexpression of ASC leads to cell death, while its down-regulation inhibits apoptosis that has been induced by overexpressing p53 in Saos2 cells, by treating IMR90-E1A cells with etoposide or MCF7 cells with camptothecin [68]. Endogenous ASC co-immunoprecipitates with Bax in DNA-damaged cells, but even though ASC-induced apoptosis is reduced in Bax-/- cells, ASC can still lead to apoptosis independently of Bax and Bak, suggesting alternative caspase-activating pathways [68]. An additional role for ASC in the maturation of caspase-1 in pathogen-infected macrophages has also been reported [69]. Overexpression of p53AIP1 leads to cell death and its down-regulation can inhibit apoptosis, but p53AIP1 rather seems to act through the neutralization of Bcl-2 [70,71].
3.4 Nur77/TR3/NGFI-B
The nuclear orphan receptor Nur77, also known as TR3 or NGFI-B, possesses mitogenic as well as pro-apoptotic activities. It is involved in activation-induced T-cell apoptosis and in the death of several types of cancer cells in response to apoptosis-inducing agents [72]. Its effects on cell proliferation require DNA transcription and functional DNA-binding and transactivation domains, while it mainly seems to trigger apoptosis at the level of the mitochondria, promoting cytochrome c release through transcription-independent mechanisms [72,73]. Nuclear export of Nur77 is mediated by another nuclear receptor, RXRα (retinoid X receptor-α) with which Nur77 normally cooperates to regulate transcription [74–77], and mitochondrial targeting seems to depend on Bcl-2 [76]. At the mitochondria, Nur77 directly promotes a conformational change of Bcl-2 that results in the exposure of its BH3 domain and a reduced accessibility of its hydrophobic pocket [78]. These rearrangements were proposed to convert Bcl-2 to a pro-apoptotic protein. Down-regulating the expression of Nur77 or of Bcl-2 inhibits the death of peripheral blood lymphocytes treated with the phorbol ester TPA and the calcium ionophore ionomycin, and of gastric cancer cells treated with 3-Cl-AHPC [78]. Killing of adenocarcinoma H460 cells that have low levels of Bax however still requires the expression of Bak [78]. Individual contributions of Bax and Bak to Nur77-mediated apoptosis still need to be determined. Similar conversion of anti-apoptotic Bcl-2-like proteins to killer proteins was previously reported upon caspase-cleavage and removal of an N-terminal fragment [79,80].
3.5 Other proteins relocalize to mitochondria
In addition to all of the above-mentioned proteins, others involved in signalling, such as the kinases LKB1 [81], PKCδ [82] or JNK [83], were also shown to translocate to mitochondria, even though their targets have not yet been identified. Several caspases also translocate, not only to accelerate apoptosis by increasing the killing potential of proteins of the Bcl-2 family, but they are also known to be responsible for the late increase in ROS and loss of mitochondrial potential that are often observed during apoptosis [84,85]. Finally, the nuclear protein histone H1.2 [86], the chloride intracellular channel mtCLIC/CLIC4 [87–89] and the cytoskeletal protein cofilin [90] were all reported to relocalize to mitochondria and to promote cytochrome c release. Even if no interaction with proteins of the Bcl-2 family could be detected, overexpression of Bcl-2 or Bcl-xL prevents cell death induced by overexpressing each of these three proteins.
Although the number of proteins reported to participate in the activation of Bax or Bak is increasing, the precise mechanism that results in their oligomerization is still unknown. Are BH3-only proteins sufficient? Do they require cooperation with other factors, specific for the pro-apoptotic stimulus? Or are there unidentified proteins that mediate these conformational changes? These questions are still to be answered.
4 Permeabilization of the mitochondria
In addition to understanding how Bcl-2 family proteins are regulated and integrate information regarding the complex state of a cell, understanding how cytochrome c and other apoptogenic factors sequestered in the mitochondrial intermembrane space are released is crucial. Several models have been proposed and extensively debated [91–96]. They are briefly recapitulated.
4.1 Rupture of the outer mitochondrial membrane
According to this model, during apoptosis, water and solutes would enter the matrix, causing swelling of the mitochondria. Since the inner membrane has a larger surface area than the outer membrane, matrix expansion would ultimately result in breaking the outer membrane. This would cause the passive efflux of the whole content of the intermembrane space into the cytosol. Two models were proposed to explain matrix swelling. In the first one, Bax-like proteins would promote closure of VDAC. Hyperpolarization of the inner membrane would follow, because the exchange of mitochondrial ATP for cytosolic ADP would no longer occur. Indeed, this antiport is normally mediated by a macromolecular complex formed by the outer membrane protein VDAC and the adenine nucleotide translocator (ANT) that resides in the inner membrane. Hyperpolarization of the inner membrane is predicted to lead to osmotic swelling of the matrix. The second model involves opening of the permeability transition pore (PTP), a high-conductance channel that is mainly formed by the apposition of the VDAC and ANT channels at contact sites between the outer and inner mitochondrial membranes and that also includes the matrix chaperone cyclophilin D. Opening of the PTP would trigger a rapid increase in the permeability of the inner membrane to molecules of mass : the membrane potential would drop and the matrix would swell. In this model, the channel would open following interaction of Bax with ANT.
4.2 Cytochrome c-conducting channels
A major breakthrough in the understanding of the mechanisms of action of Bcl-2 family members was the discovery that the structure of Bcl-xL, Bax and Bid is similar to the structure of the transmembrane domain of the bacterial colicins and of the diphteria toxin. Since the transmembrane domain of these toxins forms a pore, it was hypothesized that Bcl-2 family members could also be pore-forming proteins. This was confirmed for Bax, Bcl-xL and Bid, which form channels of various conductances across synthetic lipid membranes, the largest ones being formed by Bax [97–100]. These data suggest that a Bax oligomer (maybe a tetramer) might form a pore large enough for apoptogenic factors to exit mitochondria. Anti-apoptotic proteins could either prevent formation of the Bax pore or modify the structure of the pore such that it would be unable to allow the efflux of mitochondrial proteins. Alternatively, Bax was proposed to modulate opening of resident mitochondrial channels such as VDAC and to stimulate their opening.
4.3 The roles of lipids in Bax/Bak-induced mitochondrial permeabilization
Over the past years, it has become clear that lipids also play a central role in apoptosis. Indeed mitochondrial lipids undergo important rearrangements that could be important for the process of mitochondrial permeabilization. Cardiolipin is a mitochondria-specific phospholipid that is essential for the activity of several proteins of the electron transport chain, including cytochrome c, and of several mitochondrial carriers, including ANT [101]. Peroxidation of cardiolipin was reported to occur during apoptosis, and could be necessary for the complete release of cytochrome c from mitochondria [102–105]. Cardiolipin was also proposed to be present in the mitochondrial outer membrane, or to redistribute to the outer membrane during apoptosis [106,107], where it could interact with proteins of the Bcl-2 family. Indeed, cardiolipin seems to be required for the recruitment of tBid to mitochondria, in particular at contact sites where it would be enriched [108]. Knocking-down PGS (phosphatidylglycerophosphate synthase) prevents synthesis of cardiolipin, PGP (phosphatidylglycerophosphate) and PG (phosphatidylglycerol), and abolishes tBid-induced release of cytochrome c from isolated mitochondria [108]. Recruitment of tBid could also be mediated by MCL (monolysocardiolipin), as both cardiolipin and MCL can be co-immunoprecipitated with Bid after the stimulation of the death receptor Fas [109].
Cardiolipin not only seems to be important for mitochondrial binding by tBid, but was also shown to be required for the permeabilization of pure synthetic liposomes by activated Bax, even though its presence does not modify the extent of Bax oligomerization induced by tBid or the amount of Bax inserted in the liposomes [12,13]. In some studies, PG could also substitute for cardiolipin [13,110], but in others it could not [111]. Interestingly, oligomers of a recombinant form of Bax lacking its C-terminal tail no longer permeabilize outer membrane vesicles filled with cytochrome c if the contact sites are removed from the vesicles [112]. In addition, the number and the surface of the contact sites of mitochondria isolated from rod photoreceptors of mice were reported to increase after exposure of the mice to lead, which promotes apoptosis of these cells [113]. The unique proteic and/or lipidic composition of contact sites therefore seems to be important for the recruitment and the action of proteins of the Bcl-2 family, partly perhaps because of their enrichment in cardiolipin.
Rearrangement of the mitochondrial membranes was also observed in isolated mouse liver mitochondria incubated with tBid, and in cells undergoing apoptosis triggered by a series of insults [114]. This remodelling was suggested to be required for increasing the pool of cytochrome c available for release, but could also change the properties of the mitochondrial outer membrane and thus modulate the activities of members of the Bcl-2 family. Cristae remodelling also occurs when tBid is added to mitochondria isolated from Bax/Bak double knockout cells [114], but it is prevented when it is added to mouse liver mitochondria that have been pre-incubated with the cardiolipin-specific dye 10-N-nonyl acridine orange (NAO) [114,115]. This treatment abolishes tBid-induced cytochrome c release, but does not prevent the oligomerization of Bak [115]. While the total amount of tBid bound to the mitochondria is not modified by NAO, NAO prevents its association with contact sites [115]. Thus, at least two functions of tBid are important for cytochrome c release: the BH3-dependent induction of Bax or Bak oligomerization, and the BH3-independent association of tBid with cardiolipin that could increase the amount of cytochrome c available for release and/or could modify the properties of the mitochondrial outer membrane such that Bax and Bak oligomers can trigger its permeabilization. As mitochondrial remodelling was reported in cells undergoing tBid-independent apoptosis, other proteins, perhaps also members of the BH3-only sub-family, should possess a similar activity.
As mentioned previously, in the absence of cardiolipin, activated Bax does not permeabilize artificial liposomes [12,13]. This observation suggests that the properties of this lipid, in particular its propensity to adopt a non-lamellar phase under conditions of reduced electrostatic repulsion between its negatively-charged headgroups, as seen in the presence of divalent cations, might be important for the process [116]. Formation of non-lamellar structures happens during membrane fusion and fission events [116,117] and was also suggested to occur at the plasma membrane of cells undergoing apoptosis or necrosis [118]. Changes in membrane curvature have indeed been reported to modulate the capacity of activated Bax to permeabilize synthetic liposomes. However, permeabilization of synthetic liposomes by activated Bax was mostly reported to be potentiated by lipids with an intrinsic positive curvature (with a large hydrophilic part in comparison to their hydrophobic region) [13,110]. This would suggest the formation of lipid pores, similarly to those that are formed by the peptide magainin [119]. According to results from another group, the process is more complicated, and their observations cannot be simply explained by the intrinsic curvature of lipids [111,120]. Clearly, how the composition of liposomes affects their permeabilization by oligomeric Bax needs to be clarified further, as well as the properties of cardiolipin that makes it essential for Bax activation.
During apoptosis, mitochondrial membranes reorganize: they fragment and cluster in the perinuclear region [91,121]. Expression of a dominant-negative mutant of dynamin-related protein 1 (Drp1), a component of the mitochondrial fission machinery, inhibits mitochondrial fragmentation and delays apoptosis, even though it does not prevent the activation of Bax [121,122]. Moreover, formation of clusters of Bax and Bak occurs at mitochondrial scission sites, to which Drp1 is also recruited [122]. Permeabilization of mitochondria by activated Bax and Bak might require cooperation with components of the fission machinery, because vesicle fusion and fission events involve changes in membrane curvature [117]. It is also possible that fission proteins are necessary for cristae remodelling (see above). An inhibition of the mitochondrial fusion apparatus also seems to contribute to the fragmentation of mitochondria that occurs during apoptosis [123,124].
Fission proteins might be recruited to sites enriched in Bax and Bak because of the modifications (lipid composition and membrane curvature) that activated pro-apoptotic proteins seem to confer to microdomains of the outer mitochondrial membrane. Alternatively, proteins such as endophilin B1 could mediate these interactions, as it possesses an SH3 domain through which it could recruit components of the mitochondrial fission apparatus [125]. Endophilin B1 could also be responsible for some of the changes that mitochondrial membranes undergo during apoptosis.
5 Conclusion
Proper execution of apoptosis is crucial for the normal functioning of an organism. Regulatory mechanisms are therefore complex. They often converge on Bax and Bak, which are central for permeabilization of the outer mitochondrial membrane and release of apoptogenic factors, which control the activation of caspases. The number of factors proposed to directly interact with Bax and Bak and to control apoptosis has recently boomed. It is possible that, depending on the cell type and the precise context, different regulatory mechanisms are used, but as long as the precise steps that lead to Bax and Bak activation are not understood, it will be difficult to unify these observations. Multiple events occur at the mitochondria when the intrinsic pathway of apoptosis is activated. Proteins translocate to the mitochondria, proteins are released from them, they fragment, and they cluster in the perinuclear region. Permeabilization of mitochondria is therefore a very intricate process, probably involving cooperation between proteins of the Bcl-2 family, mitochondrial lipids, components of the fission apparatus and the cytoskeleton. Understanding its precise mechanism will require integrating all of these parameters and discriminating between causes, consequences, and by-products of the process.
Acknowledgements
We would like to thank E.A.C. Lucken for proofreading the manuscript.