

1 Introduction
The major physiological role for white adipose tissue (WAT) fat stores is to supply lipid energy when it is needed by other tissues. This is achieved by a highly regulated pathway whereby the triglycerides stored in the adipocyte are hydrolysed and fatty acids delivered to the plasma. Altered lipolysis could be an element leading to obesity and interindividual variations in WAT lipolysis are of importance for the rate of weight loss. Conversely, excessive lipolytic rates, in conjunction with impairment in fatty acid utilization by muscle and liver, may be a major contributor to the metabolic abnormalities found in subjects with upper body obesity.
This review is composed of two sections. The first section presents the endocrine and paracrine factors controlling the stimulation of lipolysis and the related signal transduction pathways. The second section is devoted to the molecular mechanisms of the hydrolysis of triglycerides that involves lipases, plasma membrane transporters, fatty acid binding proteins and proteins associated with the lipid droplet.
2 Endocrine and paracrine control of lipolysis
2.1 Catecholamines
Catecholamines are important regulators of fat cell lipolysis in human WAT. The neurotransmitter, noradrenaline, and the hormone, adrenaline, regulate lipolysis through lipolytic beta-adrenoceptors and antilipolytic alpha2-adrenoceptors.
2.1.1 Beta-adrenoceptors
Activation of beta-adrenoceptors by catecholamines leads to an increase of cAMP production by adenylylcyclase, which is followed by activation of protein kinase A and hormone-sensitive lipase (HSL) (see Section 3.1). In human fat cells, both beta1- and beta2-adrenergic receptors are known to stimulate cAMP production and lipolysis in vitro [1] and in vivo [2]. The beta3-adrenergic receptor attracted a lot of interest [3]. It is highly expressed in rodent WAT and brown adipose tissue and, unlike the beta1- and beta2-adrenergic receptors, is relatively specific to the tissues. Selective beta3-adrenergic agonists elicit a marked lipolytic and thermogenic response in rodents and hence exert antiobesity and antidiabetic effects [4]. The level of expression and the contribution of the receptor to catecholamine-induced lipolysis in human subcutaneous adipocytes seems however limited [5]. Moreover, the pharmacology of the drugs is complex. One of the beta3-adrenergic agonists, CGP 12177 [6], exerts its effect through a so-called beta4-adrenergic receptor effect, which turns, in fact, to be an atypical interaction of the drug with beta1-adrenergic receptors [7–9]. Confirmation of the lack of beta3-adrenergic effect in humans has also been provided by in vivo studies. Beta3-adrenergic agonists have shown moderate effects [10]. During isoproterenol infusion, there was no evidence for a beta3-adrenergic receptor-mediated increase in human lipolysis, energy expenditure and lipid oxidation [11]. Similar conclusions were obtained using in situ microdialysis [2]. Recent data on mice with gene knockout of the three beta-adrenergic receptors raised the puzzling possibility that catecholamines stimulate lipolysis at high concentrations through an as yet unidentified Gs-coupled receptor distinct from the beta-adrenergic receptors [12]. Interestingly, the pharmacological profile of the residual lipolytic response in WAT of mice without beta-adrenoceptors is somewhat similar to the profile in human subcutaneous adipocytes. The nature and the physiological role of this novel receptor remain to be determined.
It is commonly accepted that the action of catecholamines is impaired in obesity [13,14]. This defect might be an early event since it has been observed in obese adolescents [15]. Using in situ microdialysis, a specific impairment in the capacity of beta2-adrenergic agonists to promote lipolysis has been reported in the subcutaneous abdominal adipose tissue of obese adolescent girls [16]. Conversely, the beta1-adrenergic agonist response is not impaired in obesity. A decreased expression of beta2-adrenoceptors has been reported in fat cells from women with upper body obesity [17].
2.1.2 Alpha2-adrenoceptors
Catecholamines can exert an antilipolytic effect through the stimulation of alpha2-adrenoceptors. Activation of the Gi-coupled receptors leads to a decrease in intracellular cAMP level. The physiological significance of alpha2-adrenergic responsiveness to catecholamines has remained elusive for a long time. In human subcutaneous fat cells where alpha2-adrenoceptors numerically predominate over beta-adrenoceptors, adrenaline-dependent inhibition of lipolysis has been described in vitro. Adrenaline and noradrenaline have a higher affinity for alpha2- than for beta-adrenoceptors suggesting the existence of a role for the alpha-adrenergic pathway in the control of lipolysis in humans [18]. An investigation into the role of this receptor in vivo was achieved by a combination of calibrated exercise to activate the sympathetic nervous system, in situ microdialysis to monitor lipolysis in subcutaneous WAT and use of alpha2-adrenoceptor antagonists. A study in non-obese subjects revealed that adrenaline partly controls lipolysis in human subcutaneous WAT through activation of antilipolytic alpha2-adrenoceptors [19]. Another study was performed in exercising obese and lean men [20]. At rest and during exercise, plasma noradrenaline and adrenaline concentrations were not different between the two groups. The exercise-induced increase in extracellular glycerol concentration that reflects local lipolysis was weakly potentiated by an alpha2-adrenoceptor antagonist infusion in lean subjects, but was strongly enhanced in the obese subjects and reached the concentrations found in lean subjects. Thus, the physiological stimulation of adipocyte alpha2-adrenoceptors during exercise-induced sympathetic nervous system activation contributes to the blunted lipolysis observed in subcutaneous WAT of obese men. These microdialysis studies are supported by earlier in vitro studies showing an increased antilipolytic effect of alpha2-adrenoceptors in obese men [21].
2.2 Natriuretic peptides
Until 2000, the major factors for the physiological control of lipolysis in human WAT were catecholamines and insulin. Our laboratory showed that natriuretic peptides, i.e. atrial and brain natriuretic peptides, well-known for their effects on the cardiovascular and renal system, are also powerful lipolytic agents in vitro and in vivo [22]. This lipolytic pathway is specific of primate fat cells [23]. Natriuretic peptides act through a signal transduction distinct from catecholamines. Activation of natriuretic peptide receptor A, which possesses intrinsic guanylyl cyclase activity, is followed by an increase in the cGMP level and an activation of protein kinase G, which induces phosphorylation and activation of HSL [24]. The lipolysis induced by natriuretic peptides is independent of the fat cell adrenergic and insulin pathways [25,26]. Importantly, the natriuretic peptide lipolytic pathway is involved in the lipid mobilization observed during exercise [27]. Administration of a beta-adrenergic antagonist potentiates the exercise-induced increase in plasma atrial natriuretic peptide level. Therefore, despite their antilipolytic effect, lipid mobilization can occur during beta-adrenergic antagonist treatment. During physiological increases in plasma atrial natriuretic peptide levels, the increased lipid mobilization is associated to an increase in lipid utilization [28]. The unsuspected metabolic role of natriuretic peptides was recently reviewed by Max Lafontan and collaborators [29].
2.3 Other lipolytic pathways
Growth hormone stimulates lipolysis in human adipocytes. Although growth hormone treatments in adults reduce abdominal obesity and improve insulin sensitivity as well as blood lipid profiles, the physiological contribution of growth hormone to the control of human WAT lipid mobilization has remained elusive. Small physiological growth hormone pulses increase interstitial glycerol concentrations in both femoral and abdominal WAT [30]. Moreover, normal nocturnal rise in plasma growth hormone concentrations also leads to site-specific regulation of lipolysis in WAT [31]. Of putative pharmacological interest, C-terminal fragments of human growth hormone have been shown to increase human and rodent fat cell lipolysis in vitro. The molecules induce lipid mobilization and oxidation as well as weight loss after chronic administration in rodents [32,33]. They do not interact with the growth hormone receptor. The mechanism of action remains to be clarified.
Tumour necrosis factor alpha is a macrophage product, also released by fat cells, which is thought to signal the loss of body weight through a decrease in WAT and muscle mass. Stimulation of lipolysis by tumour necrosis factor alpha is not direct, since it becomes apparent only after long-lasting exposure of human and rodent adipocytes to the cytokine [34]. Tumour necrosis factor alpha could regulate lipolysis, in part, by decreasing perilipin protein levels at the lipid droplet surface [35]. Blunting the endogenous inhibition of lipolysis through Gi protein down-regulation is another possible mechanism [36]. It has also been proposed that tumour necrosis factor alpha acting through the second messenger C2-ceramide induces a downregulation of phosphodiesterase 3B, the enzyme catalyzing the degradation of cAMP, and thereby stimulates lipolysis [37,38]. In human fat cells, TNFalpha activates the three mammalian mitogen activated protein kinases in a distinct time and concentration-dependent manner. TNFalpha-induced lipolysis is mediated by p44/42 and Jun kinase but not by the p38 kinase [39].
In humans, many lipolytic peptides active in rodent fat cells (adrenocorticotropic hormone, alpha-melanocyte stimulating hormone, lipotropin...) have no effect. During the neonatal period, thyrotropin also known as thyroid-stimulating hormone shows a lipolytic effect at physiological concentrations that decreases gradually during childhood [40]. Parathyroid hormone has been reported to stimulate lipolysis in human fat cells [41]. The effects of glucagon and glucagon-like peptide-1 are not likely to be of physiological importance. Recently, a lipolytic effect of pituitary adenylate cyclase-activating polypeptide 38 and vasoactive intestinal polypeptide has been described in rat adipocytes [42,43]. These molecules act through a Gs-protein coupled receptor VPAC1, which has equal affinity for both compounds. At present, whether the neuropeptide and the hormone are active on human adipocytes is still unknown.
Cachexia-inducing tumours produce a lipid-mobilizing factor that causes a lipolytic stimulation. Zinc-alpha2-glycoprotein may be such a lipid-mobilizing factor. It is overexpressed in certain human malignant tumours and has also been shown to be produced by adipocytes [44]. Finally, nitric oxide and related redox species have been proposed as potential regulators of lipolysis in rodent and human fat cells [45,46]. The effect of nitric oxide appears complex and is not fully understood (see Section 2.4.3).
2.4 Other antilipolytic pathways
There are two main categories of antilipolytic molecules: agents acting on seven transmembrane domain receptors coupled to Gi proteins and agents activating receptors with tyrosine kinase activity. The major antilipolytic Gi protein coupled receptors involve alpha2-adrenergic receptors (see Section 2.1), nicotinic acid receptors, prostaglandin E2 receptors, A1-adenosine-receptors, and Y1 neuropeptide Y/peptide YY receptors. Insulin and insulin growth factor 1 receptors are the prototypical antilipolytic tyrosine kinase receptors. Other mechanisms of antilipolysis have been described: the nitric-oxide-dependent effect of the vasoactive peptide adrenomedullin and the antilipolytic effect of AMP-activated protein kinase.
2.4.1 Gi-coupled receptors
Nicotinic acid (niacin) has been the first extensively used lipid-lowering agent. It is believed to exert its hypolipidemic action chiefly through inhibition of lipolysis [47]. Despite the old recognition of its antilipolytic properties, the receptor of nicotinic acid has been identified only recently [48,49]. Two homologues of the receptor have been described, which show very high homology. The major difference is a 24-amino-acid extension at the C-terminus. It appears that the shorter homolog, named HM74A, is the high-affinity receptor mediating the antilipolytic effect of nicotinic acid in adipocytes [49,50]. The ketone body, (d)-beta-hydrobutyrate, may be a natural ligand for the receptor [51].
Prostaglandins, especially prostaglandin E2, show a strong antilipolytic effect in fat cells [52]. The effect is mediated through EP3 receptors [53]. Endogenous adenosine produces its antilipolytic effect through adenosine A1 receptor. Adenosine is metabolized very rapidly [54,55]. Nevertheless, substantial amounts of adenosine are found in the interstitial fluid of WAT [56]. Stable adenosine A1 receptor agonists exert potent antilipolytic effect in rat WAT depots [57]. Chronic treatment results in a reduction of plasma free fatty acid and triglyceride levels.
Neuropeptide Y is stored with noradrenaline in the sympathetic nerve terminals. Neuropeptide Y and peptide YY have been shown to exert a strong antilipolytic effect in human and dog fat cells [58–60]. The receptor that mediates the antilipolytic effect of the peptides in human fat cells is the Y1 receptor, which shows a higher affinity for peptide YY than for neuropeptide Y [61]. Co-culture experiments revealed that sympathetic neurons produce NPY which markedly blunts lipolysis stimulated by beta-adrenergic agonists [62]. Moreover, an effect of the gut-produced PYY on fat cells cannot be excluded.
2.4.2 Tyrosine kinase receptors
Fat mobilization is exquisitely sensitive to suppression by insulin [63]. Insulin is more efficient in inhibiting lipolysis than glucose production. In the post-meal situation, the major proportion of the decrease in free fatty acid levels is brought about by insulin antilipolytic effect. Insulin and insulin growth factor show antilipolysis through activation of tyrosine kinase receptors, a decrease in cAMP level, inactivation of protein kinase A and reduced phosphorylation of HSL. The activation cascade is rather complex [64]. When insulin binds to its receptor, the receptor is activated by phosphorylation on tyrosine residues, which causes tyrosine phosphorylation on intracellular substrates such as insulin receptor substrates 1 and 2 and binding of the p85 regulatory subunit of phosphatidyl inositol kinase-3. This binding stimulates the lipid-kinase activity of the enzyme which results in phosphorylation of the phosphoinositol at the D-3 position of the inositol ring. In addition, the serine kinase activity of phosphatidyl inositol kinase-3 autophosphorylates both the p85 regulatory subunit and the p110 catalytic subunit. This step is followed by protein kinase B/Akt phosphorylation and an activation of phosphodiesterase 3B, the enzyme which catalyzes the degradation of cAMP into 5′AMP [65].
2.4.3 Adrenomedullin signal transduction pathway
Adrenomedullin, a peptide secreted from entothelial and vascular smooth muscle cells, has recently been shown to be also produced by human adipocytes [66]. The peptide shows an inhibitory effect on lipolysis induced by the beta-adrenergic agonist, isoprenaline, but not by forskolin, a direct activator of adenylyl cyclase, suggesting that the antilipolytic mechanism lies upstream of adenylyl cyclase activation. Adrenomedullin activates a functional receptor composed of the calcitonin-receptor-like receptor and a receptor-activity-modifying protein. Nitric oxide is a second messenger known to mediate several adrenomedullin effects. In adipocytes, adrenomedullin stimulates nitric oxide production, maybe through endothelial nitric oxide synthase. Oxidative inactivation of the beta-adrenergic agonist could explain the antilipolytic effect mediated by adrenomedullin in vitro.
2.4.4 AMP-activated protein kinase
AMP-activated protein kinase is considered to act as an intracellular sensor of energy. In stress conditions that increase the AMP/ATP ratio such as hypoxia, exercise and long-term starvation, AMP-activated protein kinase is activated to switch on catabolic pathways that produce ATP and switch off anabolic pathways. In adipocytes, activation of AMP-activated protein kinase has been shown to decrease beta-adrenoceptor-stimulated lipolysis [67,68]. The enzyme phosphorylates HSL, although it is not clear at present whether this event explains the antilipolytic effect [69]. The physiological importance of this antilipolytic pathway is not fully understood.
3 Triglyceride hydrolysis
The hydrolysis of the triglycerides stored in the fat cell is a complex phenomenon involving lipases, plasma membrane transporters, fatty acid binding proteins and proteins associated with the lipid droplet.
3.1 Lipases
In the fasting state, regulation of HSL has always been thought to be the key process controlling the release of fatty acids from WAT. However, the rate-limiting role of HSL in WAT lipolysis has been challenged by the data from HSL knock out mice [70–73]. Catecholamine-induced lipolysis is abrogated, but residual basal lipolysis is observed in adipocytes from HSL null mice. These data suggest the existence of non HSL lipases in WAT. Recently, a novel lipase termed adipose triglyceride lipase (ATGL), desnutrin or iPLA2ζ has been identified [74–76]. Using antibodies directed against ATGL, Zimmerman et al. suggest that ATGL is responsible for 75% of the cytosolic acylhyrolase activity in WAT of HSL deficient mice [74]. ATGL could therefore participate together with HSL in WAT lipolysis. Using a highly specific HSL inhibitor, the contribution of HSL and ATGL was recently re-evaluated [77]. The following model for lipase activation can be proposed in human fat cells. ATGL and HSL both possess the capacity to hydrolyse triglycerides in vitro [74,78]. However, only HSL shows a significant diglyceride lipase activity [79]. Although HSL has the capacity to hydrolyze monoglycerides in vitro, monoglyceride lipase, which is not under hormonal control, is required to obtain complete hydrolysis of monoglycerides in vivo [80]. Triglycerides are hydrolyzed at a lower rate than diglycerides indicating that the first step of lipolysis is rate-limiting [81]. As discussed above (see Sections 2.1 and 2.2), stimulation of the catecholamine and natriuretic peptide pathways leads to an increase in cAMP and cGMP levels, respectively [82]. Both protein kinases A and G phosphorylate and activate HSL at least in part through translocation of the enzyme from the cytosol to the lipid droplet [24,83,84]. In line with the role of HSL in triglyceride hydrolysis under stimulated conditions, ATGL is not phosphorylated by protein kinase A and does not appear to be acutely regulated by isoprenaline [74]. Data on human fat cells clearly demonstrate that the two major and independent activation pathways converge on HSL [77]. However, non-HSL lipases contribute to the hydrolysis of triglycerides into diglycerides under basal conditions. Although ATGL seems to play a predominant role in basal lipolysis, it cannot be excluded that other WAT enzymes with the capacity to hydrolyze triglycerides such as carboxylesterase 3 (or triacylglycerol hydrolase) play a role [85].
A defect in HSL expression is likely to play a major role in the impaired lipolysis observed in obese subjects [86]. Indeed, the defect is also observed in first-degree relatives of obese subjects and in adipocytes from obese subjects differentiated in vitro [77,87]. Moreover, there is a strong correlation between lipolytic capacity and HSL expression in human subcutaneous fat cells [88]. The defect may constitute an early, possibly primary, event in obesity, which protects against excessive FFA release. Accordingly, HSL deficiency in mice causes a reduction in plasma FFA levels and favours an antiatherogenic lipid profile [70,71].
3.2 Non-enzymic lipid interacting proteins
Proper activation of lipolysis relies upon proteins that are not directly involved in the catalytic process. Adipocyte lipid binding protein or aP2 is an intracellular fatty-acid-binding protein highly expressed in adipocytes. It interacts with HSL N-terminal region and increases the lipolytic activity of HSL through its ability to bind and sequester fatty acids and via specific protein-protein interaction [89]. A pre-lipolysis complex containing at least adipocyte-lipid-binding protein and HSL exists [90]. The complex translocates to the surface of the lipid droplet upon lipolytic stimulation. Consistent with such a role for the fatty acid binding protein is the observation that knock-out mice exhibit decreased lipolytic capacity [91–93].
Modification of the lipid droplet constitutes another potential mechanism for the control of lipolysis. Perilipins are proteins covering the large lipid droplets in adipocytes [94]. In basal conditions, perilipin suppresses lipolysis by blocking access of the lipases to the lipid droplet. Hormone-induced phosphorylation of perilipin by protein kinase A and probably protein kinase G facilitates the access of lipases to the droplet. As expected from cellular studies, basal lipolysis is highly elevated in perilipin null mice [95,96]. Moreover, HSL translocation to the lipid droplets is impaired during lipolysis in these mice, illustrating the importance of perilipin in this process [97]. The lipid droplet contains specific structural proteins as well as enzymes. Upon lipolytic stimulation, there is a reorganization of the droplet with recruitment of new proteins [98]. The role of these lipid-associated proteins in the lipolytic process awaits further studies.
The final step of the lipolytic process is the efflux of fatty acid and glycerol from the adipocytes. Passive diffusion may explain part of the transport [99]. However, there are several facilitating proteins. Fatty acid translocase/CD36 and fatty acid transport proteins have been shown to mediate fatty acid uptake. Their role in the efflux of the metabolites is poorly documented. Glycerol is transported by aquaporin 7 in fat cells. Mice deficient in the glycerol transporter show impaired glycerol release [100]. The mice develop obesity when aging [101,102]. Fat cells without aquaporin 7 accumulate glycerol. Because of increased glycerol kinase activity, there is a resulting increase in fatty acid re-esterification and triglyceride accumulation that explain adipocyte hypertrophy.
4 Conclusion
The last decade has been marked by the discovery of a number of mechanisms able to clarify the control of fat mobilization (Fig. 1). To date, there are two hormonal pathways, namely catecholamines and natriuretic peptides, stimulating fat mobilization in humans. In contrast, there are numerous antilipolytic pathways that rely on paracrine or endocrine factors. The characterization of ATGL as an adipose tissue triglyceride lipase has prompted a re-evaluation of the roles of the various enzymes in lipolysis. Non-enzymic components such as perilipins and recently characterized transporters such as aquaporin 7 appear also to be essential.
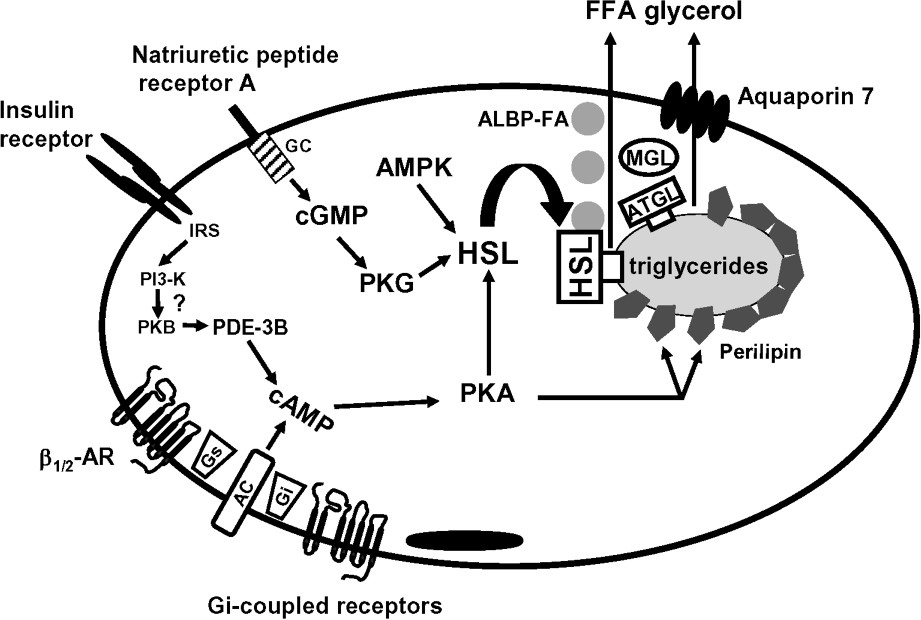
Schematic view of lipolysis in human adipose tissue. AC, adenylyl cyclase; ALBP, adipocyte lipid binding protein; AMPK, AMP-activated protein kinase; AR, adrenoceptor; ATGL, adipocyte triglyceride lipase; FFA, free fatty acid; GC, guanylyl cyclase; Gi, inhibitory GTP-binding protein; Gs, stimulatory GTP-binding protein; HSL, hormone-sensitive lipase; IRS, insulin receptor substrate; MGL, monoglyceride lipase; PDE-3B, phosphodiesterase 3B; PI3-K, phosphatidylinositol-3-phosphate kinase; PKA, protein kinase A; PKB, protein kinase B; PKG protein kinase G.
The reduced lipolysis commonly reported in subcutaneous WAT of obese subjects may involve defects at different steps of the transduction pathways. To date, the best characterized defect is the decreased expression of HSL. It is however still unclear whether altered lipolysis promotes the development and/or stabilization of obesity or is an early protective adaptation to limit excessive fatty acid release.
Acknowledgements
The author's work is supported by INSERM, the 6th framework Integrated Project Hepadip (Adipose tissue and liver in the metabolic syndrome), the French ‘Programme national de recherche en nutrition humaine’ and ‘Programme national de recherche sur le diabète’.