

1 Introduction
Increasing evidence suggests that besides DNA regulatory sequences and transcription factors, the spatial and temporal organization of nuclear processes (transcription, reparation, replication, …) as well as chromosome and gene distribution within the nuclear space are central determinants of genome functions. An important goal of the post-genomic systems biology will be therefore to better understand genome function within the architectural framework of the nucleus and integrate different levels of genome regulation [1,2]. However, deciphering the three-dimensional (3D) nuclear organization in a relevant physiological context is a difficult task due to technical constraints and requirements of imaging and computational modeling.
To develop a systems biology view of the nucleus and to give standardized and statistical representations of the nuclear architecture in order to highlight possible organizational rules, we investigated the spatial organization of a nuclear compartment in both plant and animal cells. We set up a multidisciplinary approach combining biological studies, spatial statistics and 3D modeling in the frame of an agroBI-INRA program entitled “Nuclear Architecture: spatial modeling and application to a better understanding of differentiation/dedifferentiation mechanisms”. This article aims at presenting the conceptual background, the main goals and the methodology of our work.
2 Functional nuclear organization: background
2.1 Elements of the nuclear organization
Compartmentalization is one of the fundamental principles regulating organization-function crosstalk in the nucleus. As defined by Misteli, a “compartment” is “a macroscopic region within the nucleus that is morphologically and/or functionally distinct from its surrounding” [3]. The compartments may be either sites highly enriched in a definite set of nuclear proteins (nucleoli, speckles, Cajal bodies, promyelocytic leukemia protein (PLM) nuclear bodies, interchromatin granules, …) or chromatin domains, which we will focus on (Fig. 1). One of the most studied compartments is “constitutive heterochromatin”, a major component of eukaryotic genomes usually formed by large blocks located near centromeres and telomeres and that has a compact organization throughout cell cycle [4–6]. It is opposed to “euchromatin”, which exhibits a more “open” conformation and corresponds to actively transcribed regions of the genome. Euchromatin can be turned temporarily into heterochromatin upon silencing, being thus termed “facultative heterochromatin”. Heterochromatin can be distinguished from euchromatin by cytological properties, compaction levels and epigenetic marks (Fig. 1). However, several studies have shown that transcription can occur in heterochromatin regions in yeast, plants and animals [7–9].
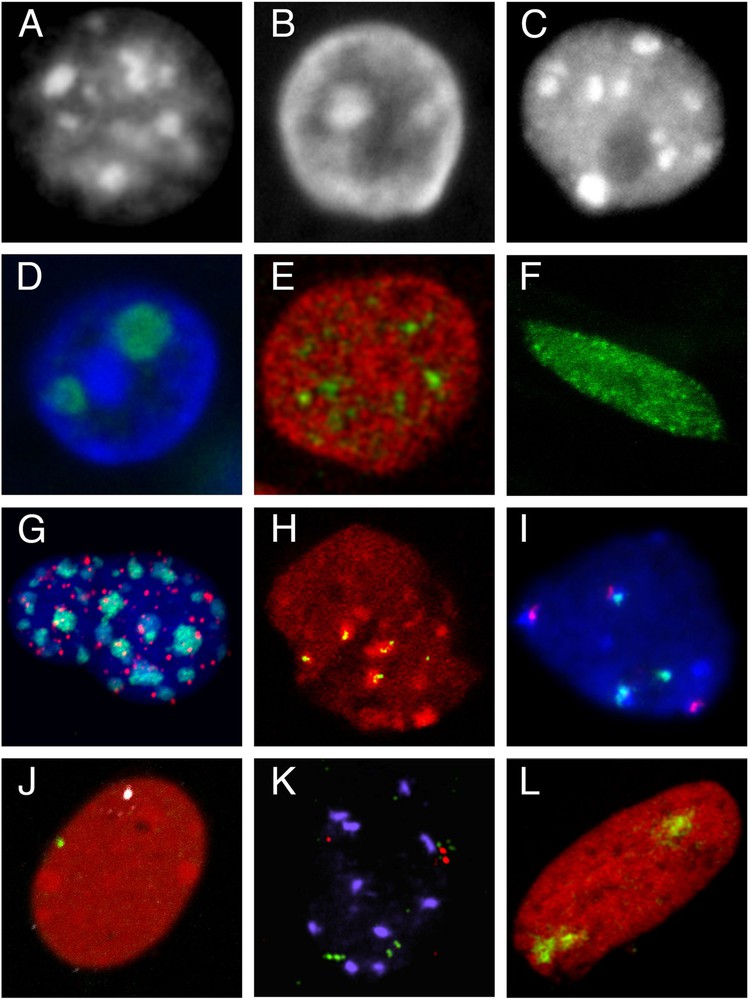
Compartmentalization of the nucleus and elements of its architecture. A–C. Heterochromatin compartments. A. Mouse fibroblast nucleus (Hoechst staining). B. Epithelial cell nucleus from a rabbit mammary gland in lactation (tissue section, DAPI staining). C. Nucleus isolated from an A. thaliana plantlet (DAPI staining). D–F. Compartments defined as regions enriched in nuclear proteins. D. Nucleoli in an epithelial cell nucleus from a rabbit mammary gland in lactation; immunolocalization of B23 protein (green), DNA counterstained with DAPI (blue) (tissue section). E. Splicing speckles in the nucleus of a mouse mammary epithelial cell (cultured HC11 cell line); immunolocalization of SC35 protein (green), DNA counterstained with propidium iodide (red). F. Nuclear localization of LHP1, the plant homolog of Heterochromatin Protein 1 (HP1) in a root cell nucleus from a transgenic line expressing the LHP1-GFP fusion protein. G–I. Chromocenters, centromeres and telomeres. G. FISH with a pancentromeric probe (green) and a LNA telomeric probe (red) in an HC11 cell nucleus; DNA counterstained with DAPI (blue). H. Immunolocalization of pericentromeres (HP1 protein, red) and centromeres (CENP proteins, green) in the nucleus of a rabbit embryo at 8-cell stage. I. 5S (green) and 45S rDNA (red) repeats in an A. thaliana leaf nucleus revealed by FISH; DNA counterstained with DAPI (blue) (kindly provided by F. Tessadori). J–L. Visualization of genomic regions of different sizes by FISH. J. Localization of genes encoding casein (green) and whey acidic protein (white) in the nucleus of HC11 cells after hybridization with gene specific BAC probes and DNA staining with propidium iodide (red). K. Localization of 3 genomic regions (about 100 kbp) with 3 BAC probes (2 in green and one in red) in an A. thaliana leaf nucleus (kindly provided by F. Tessadori). L. Territory of chromosome 15 (green) in the nucleus of a cultured rabbit embryonic fibroblast; DNA counterstained with propidium iodide (red). All images were acquired on confocal microscopes.
Furthermore, large-scale mapping of the human genome has shown a link between chromatin morphology and gene density, with gene-rich regions being present in open chromatin regions and gene-poor regions in more condensed domains, regardless of their activity status [10]. In some animal species such as mouse and, to a lesser extent human, the pericentric heterochromatin of several chromosomes assembles to form large heterochromatic domains called “chromocenters” [11,12]. In Arabidopsis thaliana, discrete and intensely stained heterochromatic compartments are referred to as chromocenters (Fig. 1). They correspond to centromeric and pericentromeric regions and were described as anchoring regions for 0.1–1.5 Mbp euchromatin loops allowing to propose a chromocenter-loop nuclear organizational model [13,14]. Chromocenters can be visualized using FISH [13], immunolocalization [15] or GFP markers [16].
Since the first laser microbeam irradiation experiments [17], it is now well demonstrated that each chromosome of an animal or plant cell's nucleus is confined to a discrete region of the interphase nucleus, referred to as a chromosome territory (CT) and corresponding to a chromatin compartment at a larger scale (Fig. 1) [18–20]. In mammalian cells, the distribution of CT appears non-random, with most gene-rich CTs preferentially localized in the nuclear interior, whereas gene-poor CTs localized toward the nuclear periphery [18,21,22]. Such arrangements are evolutionarily conserved [23,24]. Furthermore, cell or nuclear shapes and chromosome size have been observed to influence the 3D arrangement of CTs: for example, the ellipsoid human fibroblast nuclei present a chromosome size dependent pattern even if the radial distribution of gene-rich and gene-poor CTs was found to be recapitulated at the sub-chromosomal level [25–27]. In A. thaliana, CTs were described as predominantly randomly located within the nucleus. Only chromosomes 2 and 4, which bear nucleolus organizer regions (NOR), were associated more frequently than randomly [19,20]. The relationships between CTs distribution and nuclear organization require therefore further investigation. Parameters such as DNA content, gene density, and transcriptional activity of chromosomal sub-domains might all come into account and raise the question of the mechanism of chromosome positioning.
2.2 Relationship between gene positioning and transcriptional activity
Numerous studies described complex and sometimes conflicting relationships between gene positioning with respect to several compartments, and transcription regulation.
2.2.1 Relative positioning of genes with respect to nuclear compartments
Some studies address the question of comparative positioning inside the nucleus by examining different genes in a given cell, or the same gene in different cell types.
As stated above, gene-rich chromosomes are usually more centrally located than gene-poor chromosomes in human [22], chicken [28], hydra [29], and this trend is observed also for subchromosomal domains [25,30]. The nuclear periphery, which is lined in most higher eukaryotes by a peripheral ring of condensed chromatin, represents generally a repressive zone: some genes are preferentially associated with the nuclear periphery in cells where they are inactive, and more centrally located in cells where they are highly transcribed [31–34]. More generally, late replicating foci, assumed to correspond to inactive chromatin are located at the nuclear periphery. However, location of transcribed genes at the nuclear envelope has also been observed in yeast [35], possibly because of their association with proteins of the nuclear-pore complexes [36]. It is not known whether similar gene activation processes exist at the nuclear periphery in higher eukaryotes. The potential role of the nuclear periphery in genome regulation has become of particular importance in view of human diseases caused by mutations in genes encoding the architectural proteins of the lamina [37].
The relationship between location and transcriptional status of genomic sequences with respect to their CTs is the subject of conflicting reports: non-coding sequences are randomly distributed or preferentially located at the interior of the CT, whereas several genes have been found preferentially located at the periphery of the CT, irrespective of their transcriptional status [38,39]. This peripheral location might facilitate access to the transcriptional machinery located in the inter-chromosomal space [40,41]. Chromatin loops containing large gene clusters (major histocompatibility complex locus, epidermal differentiation complex) can even loop out of their CTs into these interchromosomal compartments under conditions where they are highly expressed [42,43]. On the other hand, some active genes reside in the CT interior [44], suggesting that CTs can be invaginated by infoldings of the interchromosomal compartment [41,45] (reviewed in [46]).
Proximity to centromeric heterochromatin is generally associated with gene silencing, the most striking examples being the association of T-cell specific and developmentally regulated B-cell genes with the Ikaros complex at the centromeric heterochromatin [47] and the beta-globin gene in erythroid cells [48].
2.2.2 Dynamic gene repositioning upon activation/inhibition
Studying the dynamics of genes during the differentiation process has brought other strong arguments for a relationship between nuclear positioning and transcription status [3,49]. For instance, the Hoxb1 locus loops out of its CT during transcriptional activation [50]; movement towards the nuclear periphery was observed upon inactivation [51], or towards the nuclear interior upon neural induction [33]; or gene repositioning was described away of the chromocenters upon activation [48] or towards the chromocenters upon silencing [52]. Finally, active genes widely separated in cis or located on different chromosomes can even colocalize possibly as a result of migration upon activation towards preassembled transcription sites [53,54]. Similarly, the ribosomal RNA genes coalesce in the nucleolus where RNA polymerase I accumulates [55]. More recently, interchromatin granules were suggested to actively recruit genomic loci, thus forming “enhancer hubs” for inter-chromosomal interactions [56].
2.3 Nuclear organization depends on the physiological status of the cell
Besides the above-described changes in gene location, changes in nuclear architecture itself are amongst the most dramatic hallmarks of cell differentiation, development or malignant processes [26,31,49,57,58]. For instance, condensation or morphology of CTs may vary with differentiation in chicken, mouse, or human cells [59,60]. The spatial organization of centromeric heterochromatin in haematopoeitic cells changes with cell types and is ontogenically determined [12]. Clustering of pericentric heterochromatin has been reported during terminal differentiation, e.g. during myogenesis [61], in human neutrophils [62], human and mouse neurons [63] and rat myoblasts [64]. In naturally fertilized mouse embryos, the specific radial (“cartwheel”) organization of chromosomes at 1-cell is progressively modified into a “somatic cell-type” organization during development [65,66]. Interestingly, similar 1-cell type nuclear organization is adopted upon transfer of a somatic nucleus in the oocyte cytoplasm [66,67].
In plants, heterochromatin content varies with differentiation [68–70]. Peculiar heterochromatin organization with smaller chromocenters and additional heterochromatic foci interspersed in euchromatin was observed in endosperm nuclei [71] suggesting a relaxed chromocenter-loop organization of the nuclei. Nuclear compaction of specific loci depends on their transcriptional status in regards to cell differentiation [72] and changes in chromocenter and chromatin organization accompany changes in cell fate and differentiation [70,73].
2.4 Nuclear organization in natural environments
Until now, most data have been gathered on isolated plant cell nuclei or on nuclei from immortalized animal cell lines outside their physiological environment, except for circulating blood cells. Little is known about nuclear organization of cells within their tissues [74]. Few studies compared nuclear organization in primary cells versus cell lines, in cell lines versus tissues, in 2D culture versus 3D cultures and suggested a control of tissue architecture over nuclear organization [75–77]. In addition, few data on nuclear organization were acquired in plant cell nuclei in situ [71,72]. In vivo approaches allowed distinguishing three main types of plant nuclei based on their nuclear shapes (round, elongated and rod-like nuclei), which are associated with various cell-type or tissues [78]. Studies of chromatin dynamics in plants have shown that confinement area for tagged loci is different according to cell types [79]. Further investigation will be required in plant cell nuclei.
2.5 Current models of nuclear organization
Despite being at a very beginning of a system-level view of the nucleus, two major models, a deterministic model and a self-organizing model, emerged [2]. The deterministic model proposes that structure dictates function and is consistent with stable observed structures in the nucleus. However, some experimental evidence does not support this model, such as the loss of prime structural elements without any major consequences on genome function. On the other hand, the nucleus can be seen as a self-organizing system, based on the dynamic interaction of nuclear components and various interplay between structure and function with self-reinforcing mechanism. Indeed, nuclear processes can lead to nuclear changes in global architecture and de novo formation of nuclear structures. For example, supported by theoretical considerations as well as several experimental results from chromosome conformation capture (3C), transcription machinery may function as a main molecular tie that maintain most genomic loops and determine gene activity [80]. 3C experiments have also recently revealed that repeat elements and nucleolus play central roles in the dynamic organization of genome architecture [81]. However, as most probably, the two models are not exclusive and additional forces may have to be taken into considerations to explain the structure/function of the nucleus such as stochastic collisions [82], non-specific entropy-driven crowding forces [83] or “depletion-attraction” forces, highly significant in crowded environments like those in nuclei [14,84,85].
3 Overview of the project
3.1 Aim of the project and main achievements
Two main questions arising from the existing data were at the origin of the project:
- • Are there organization principles, and/or topographical landmarks to describe the nuclear architecture? If existing, are these principles identical in the animal and plant kingdoms?
- • What transformations allow to process from one differentiation state to another? How are co-regulated gene networks positioned with respect to these topological landmarks?
To address these questions, we chose to analyze the centric/pericentric heterochromatin compartment and its three-dimensional nuclear distribution. Indeed, the centric/pericentric compartment is well defined and present in all cell types with some already described variations. Its structure and cohesion are essential for genome organization and proper cell division [70,73,86,87] and it is involved in genome regulation, usually behaving as a transcriptional repressor compartment.
Aiming at producing generic models for the nuclear architecture in physiological environment, we produced images of 3D-preserved nuclei both from animal and plant kingdoms in three different biological systems to be able to extract robust organizational principles. We performed fluorescence or immunofluorescence labeling to reveal the structural elements, generated a homogenous 3D-image collection of cell nuclei acquired in standardized conditions (ICOPAN website/image resource: http://amib.jouy.inra.fr/icopan/) and developed image processing and spatial statistical tools to analyze and model the spatial distribution of the studied compartment.
3.2 Choice of the biological systems
Three biological complex systems were chosen, two animal (rabbit) systems and one plant (Arabidopsis thaliana) system. Rabbit was chosen since: (i) its complete genome sequence (2X coverage) was already available and its 7X coverage was expected to be released in a near future; and (ii) rabbit is phylogenetically closer to primates than rodents. A. thaliana is an excellent plant model for which large molecular genomics tools and genetic resources are available. Furthermore, in the three systems, transitions between totipotency, multipotency and terminal differentiation can be exploited and used to explore nuclear reorganization in relation to modifications of the pattern of gene expression.
Pre-implantation rabbit embryos, the first system, offer the possibility to follow directly a differentiation process within the same intact organism. After the reprogrammation of the parental genomes at fertilization, the first totipotent zygote leads in 5 days to a blastocyst where two different populations of cells coexist, already differentiated trophectoderm cells and totipotent cells of the inner cell mass. The whole process can take place in vitro in an appropriate culture medium. Finely tuned modifications of gene expression occur during this period, together with large restructuring of the nuclear architecture. The advantage of the rabbit is that zygotic genome activation occurs over an extended period of time as compared to the mouse [88].
The second system, the rabbit mammary gland, is subjected to successive cycles of intense development and involution which extend from gestation through lactation to weaning, and thus represents a powerful system to explore the impacts of physiological situations on genome function. During each cycle, genes coding for milk proteins are turned on and off, together with many other genes. However, no studies of the nuclear organization of these epithelial cells in their tissue environment exist so far, only few data deal with mammary cell lines [77,89,90].
A. thaliana plantlets offer a third interesting system, since after digestion of the cell walls, totipotent “protoplasts” can be easily obtained. Upon culture in an appropriate medium, protoplasts can redifferentiate into tissues and new plants. A large reorganization of heterochromatin accompanies the first steps of dedifferentiation/redifferentiation in this system [73].
3.3 Development of modeling and computational tools
To explore the nucleus on a global scale, new tools are required to establish computational models and to unravel principles governing the spatial distribution of nuclear compartments. Indeed most analyses were done so far in 2D, and very rarely in 3D [31,39,91,92]. Few quantitative measures have been performed [93,94]. Standard quantitative analyses of the nuclear organization are essentially based on distances measured on 2D images or on projections of 3D images, and rarely obtained directly in 3D. Distances can be measured between points (centers of compartments), or between points and surfaces (e.g. the nuclear envelope). Sometimes distances are digitized into classes such as, for example, in the so-called erosion analysis used to analyze radial distributions within the nucleus [31].
The testing of complete spatial randomness is commonly used to show that the nucleus is indeed spatially organized. A typical approach consists in choosing arbitrary parameters (distance or angle) and comparing their experimental values with those obtained on patterns simulated according complete spatial randomness. Such an approach based on angles was used to test whether chromosome territories are located independently from each other [27]. In domains such as forestry, ecology, or epidemiology, generic statistical tools have been developed for the analysis of spatial point patterns [95–97]. To date, these tools have only rarely been used to investigate nuclear spatial organizations [98–101]. For example, Beil et al. [101] used the K-function to test whether chromocenters are completely random and to compare chromocenters patterns in undifferentiated and differentiated NB4 human cells. However, the conventional forms of spatial point pattern tools encompass no normalization for nuclear shape variations and are not applicable to repeated experiments.
To take into account the expected spatial heterogeneity of the nuclei and compare the heterochromatin distribution, we developed new tools based on processing and analysis of digital confocal images and a new statistical approach using variants of the spatial point patterns F- and G-functions (Fig. 2). These cumulative distribution functions of nearest-neighbor distances (G-function) or of distances between arbitrary points within the domain of interest (here the nucleus) and their nearest “events” (here nuclear elements) (F-function) indeed provide more comprehensive descriptors than radial distributions. Thus, they allow more powerful testing of hypotheses on spatial distributions. In this study, our F-function variant was the most appropriate tool and allowed us to highlight a non-random distribution of centric–pericentric heterochromatin in differentiated cells from our three biological systems.
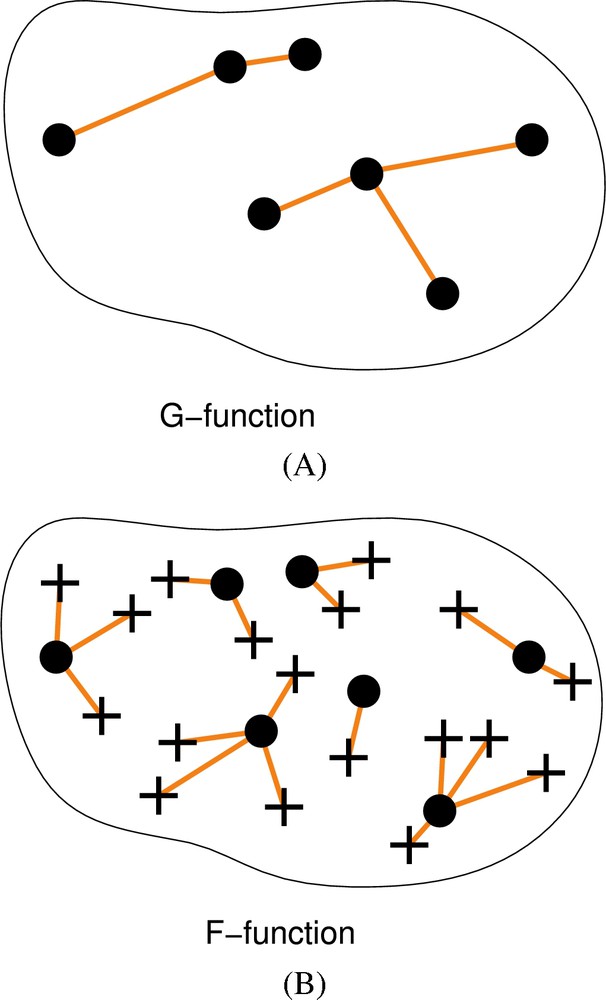
Spatial point patterns and analysis by distance functions. A. The G-function is the cumulative distribution function of point to nearest point distances. Points are shown as black dots and are joined to their nearest neighbor by line segments. B. The F-function is the cumulative distribution function of arbitrary positions to nearest point distances. Sampled positions (shown as crosses) are connected to nearest points (black dots) by line segments.
The use of this F-function variant requires an adapted segmentation method to identify and localize the “interesting” biological objects within images collected on very different types of biological material. Furthermore, the segmentation procedure needs to be automated in order to analyze a large number of images. However, few algorithms are actually available as they may vary according to the specimen. Simple software, especially the commercially available ones, used to extract and calculate 3D nuclear distributions can be used on cells grown in culture, with round shaped nuclei [27] but cannot be used for more sophisticated samples like irregularly shaped nuclei, tissue sections with clumped cells or low-contrasted fluorescent staining. For this project, we set up new automated or semi-automated segmentation algorithms adapted to the different nuclear compartments in the three dimensions of well-preserved nuclei.
4 Conclusion
The frame of the agroBI-INRA program entitled “Nuclear Architecture: spatial modeling and application to a better understanding of differentiation/dedifferentiation mechanisms” allowed a fruitful confrontation between animal and plant biologists, together with statisticians and modelers to define a common multidisciplinary approach. The first results of this joint study will be presented in a forthcoming paper. We believe that comparisons between plant and animals will be highly beneficial to understand the similarities/differences in genome organization, gene regulation and cell differentiation processes.
Acknowledgements
This work was supported by an INRA agroBI Research Program (2006–2008) (http://www.inra.fr/internet/Projets/agroBI/pageagroBI.html). We are grateful to Olivier Grandjean and Lionel Gissot, to the Laboratoire Commun de Cytologie (INRA Versailles), as well as to Pierre Adenot and the RIO MIMA2 platform (INRA Jouy-en-Josas) for technical support in confocal imaging. We thank Eric Biot, Nicole Houba-Hérin, Cathy Hue-Beauvais, Gaetan Lehman, Leila Tirichine, Liu Zichuan for biological experiments and development of image treatment and analysis tools. We thank Professor M.A. Ferguson-Smith and P. O'Brien (Cambridge Resource Centre for Comparative Genomics) for the gift of the rabbit chromosome-specific paints. We are indebted to Christophe Escudé for the LNA telomeric probe and Federico Tessadori for providing images (Figs. 1I, 1K). We thank the Région Ile-de-France for the funding of the Leica TCS SP2 confocal microscope (INRA Versailles) and the Zeiss AxioObserver imaging Apotome system (INRA, Jouy).