

1 Introduction
Facing the 21st century biodiversity crisis, the 6th mass extinction [1,2], it is essential to speed up the inventory and description of organisms. The global biodiversity assessment is far from being achieved since only about from 10% to 30% of living eukaryotic organisms have been properly discovered, named, and described [2–4]. Tropical regions harbour the most important part of this biodiversity. Among them, Southeast Asia has been proposed as one of the 25 world hotspots for biodiversity conservation [5]. Amphibians are one of the most endangered animal classes [6,7], mainly because of their sensitivity to environmental changes (e.g., habitat destruction, climatic change such as increasing dryness, or appearance of new pathogenic agents such as the chytrid fungus Batrachochytrium dendrobatidis) due to their dependence on water and to their skin permeability.
Recent studies based on DNA barcoding and morphology (integrative taxonomy) provided evidence for numerous cryptic species in amphibians, e.g., [8–10], increasing from about 100 to 300 the annual description of new species during the last decade [11]. Some authors (e.g., [3]) have suggested that only half of the extant amphibian species are known at present. Therefore, the global amphibian decline could be worse than currently estimated. Facing the extinction risk for numerous species (e.g., in the genus Atelopus; [12,13]), a rapid inventory of amphibians worldwide is urgently needed, not only with the aim of getting a testimony of the diversity of life on Earth for future generations (as museum collections), but also to elaborate rigorous, efficient and viable conservation strategies and management action plans. Setting up such conservation planning requires accurate information about species, such as a thorough delimitation of species boundaries, as well as knowledge of distribution areas and ecological requirements of species. Unfortunately, most of this information is missing, particularly for tropical herpetofauna.
Biodiversity surveys generally overlook anuran larvae. However, tadpoles present several advantages: they are usually easy to detect and to collect in the field (especially for the pond-dwelling species) as they are very conspicuous and live in delimitated habitats, present in large number, and they sometimes are the only evidence of the occurrence of secretive species or simply of species outside their reproductive period. Indeed, after breeding, numerous amphibian species leave the reproduction site and, though they could remain in the surroundings, silent adults are difficult to locate. Additionally, in such cases, it can be difficult to understand which areas are really crucial for these species, in particular for breeding — but such information can be obtained from larval surveys and crucial waterbodies are then properly flagged and protected. On the contrary, tadpoles stay in average two months in a rather well delimitated habitat before metamorphosing (several years for some altitudinal stream-dwelling species). As a consequence, tadpoles are key organisms for amphibian surveys and identifying tadpoles is necessary to improve amphibian species inventories. We suggest that ignoring this part of the anuran life-cycle would result in missing a significant part of the biodiversity of this group in time-limited field surveys as it is generally the case (see the Rapid Assessment Programs, e.g., [14]). Furthermore, the biology of tadpoles is highly relevant in ecological and systematic studies, as well as in conservation decisions. Diverse aspects of the whole life-cycle, including eggs deposition site(s), larval morphology and the habitat type(s) used for development, should be known.
Tadpoles of tropical species have been until recently overlooked from most of the scientific works, from basic field surveys to evolutionary hypotheses through traditional taxonomy, systematics, and ecology. A large number of frogs still lack detailed descriptions of their larval forms because tadpoles are difficult to identify, especially in tropical and subtropical areas, where several closely related species can be found in sympatry. Until the last decade, only two main methods were available to assign with some reliability a species name to a tadpole: both collecting and rearing spawn from an identified mating pair of frogs or keeping tadpoles alive until their metamorphosis. The first method depends on the encounter of mating pairs and their disposition to lay eggs in captivity, and is time demanding in rearing tadpoles to an appropriate developmental stage. As environmental effects might influence tadpole morphology [15–17], the study of captive reared specimens should be used with caution. The second method too is time-consuming, and the identification of young froglets might be uncertain. Furthermore, the tadpoles, which are very sensitive to environmental conditions, such as those collected from altitudinal and torrent habitats, cannot be carried out of the habitat, and require cold and very oxygenated water for rearing, which is often difficult to obtain in field-laboratory conditions. Hence, until recently the literature about tadpoles was scarce, inaccurate and sometimes mixing several species as very few field naturalists took trouble in rearing tadpoles with the intention of properly identifying and describing them, e.g., [18]. In addition, published misidentifications added confusion in the interpretation of larval characters in an evolutionary context (see for example Table 12.2 in [19]). Finally, the continuous change of larval characters during ontogeny increases the difficulty of species identification with the old literature or descriptions based on only one developmental stage.
The molecular taxonomy approach provides a simple method to identify tadpoles, e.g., [20–24]. Hebert et al. [25,26] proposed to standardize this molecular approach in using the 5’ region of the mitochondrial cytochrome c oxidase subunit 1 (COI) gene. DNA barcoding has been successfully applied to the identification of larvae of diverse animal groups, e.g., caterpillars [27], larval communities of Diptera [28], zooplankton [29], association of adults and larvae in multi-species assemblages [30], larvae of fish with application to fish aquiculture [31]. Although COI barcoding is, in theory, applicable to all animals, the 16S rRNA gene has been preferred in most taxonomic studies on amphibians [10,32–36]. In agreement with that, the number of sequences found for amphibians in GenBank (25 April 2014) was 5290 for COI and 33772 for 16S. For this study, we newly generated 397 16S and 343 COI sequences from respectively 109 and 86 larval and 288 and 257 adult anuran amphibians from 65 localities in Southeast Asian, and analysed these using both phylogenetic and distance-based methods.
The aims of this study are:
- • to compare the performance of COI and 16S for molecular taxonomy of Asian amphibians (PCR amplification success, levels of nucleotide divergence, ability to recover monophyletic species and genera);
- • to estimate the percentage of taxonomic misidentification for tadpoles collected in the field;
- • to determine how tadpoles can improve the assessment of biodiversity.
2 Material and methods
2.1 Choice of taxa and sampling strategy
Field work was carried out from 1997 to 2006 by three different collectors in several parts of Southeast Asia (Cambodia, Laos, Nepal, Thailand, Vietnam; Fig. 1; Supplementary Table S1). A piece of thigh muscle or of liver for adult frogs, the tip of the tail for small tadpoles and a piece of caudal muscle taken at the base of the tail for large tadpoles were collected and immediately preserved in 99% alcohol. The samples were deposited in the tissue bank of the amphibians and reptiles team from the Muséum national d’histoire naturelle, Paris, and from the Thailand Natural History Museum, Pathum Thani. A few Chinese samples available to us have also been included. All tissues belong to the representatives of five well-represented families in Southeast Asia, i.e. Dicroglossidae Dubois, 1987, Megophryidae Bonaparte, 1850, Microhylidae Günther, 1858, Ranidae Rafinesque, 1814 and Rhacophoridae Hoffman, 1932. The nomenclature used in this paper follows the most recent major systematic work [37] as well as the online database on world's amphibians [38], except for the following cases: the species Leptobrachium echinatum is considered here valid, as its nomenclatural status is not yet resolved [34,39,40]; Hylarana guentheri and Pelophylax lateralis are here allocated to Humerana following evidence of previous molecular work and personal observation of the humeral gland that characterises the genus Humerana [41]. For a review of the literature used to identify the adult forms, see Supplementary Information S1. The tadpoles have been identified in the field without field guides or keys, as close as possible to the species-level.

(Colour online.) Distribution of collection localities for the samples analysed in this study. Names and geographic coordinates are listed in Supplementary Table S1. Numbers 1 to 7 refer to areas where an equivalent effort has been undertaken to collect both life-stage forms. In the area 1 (Luang Prabang Province) two localities close to the town Luang Prabang were omitted (Luang Prabang and Kouangi Waterfall) because no effort has been made to collect tadpoles in these places. The inserted histogram shows for each of the seven chosen localities the number of species collected in their adult stage (first column), the number of species collected in their tadpole stage (second column), and the number of species for which both adults and tadpoles were collected (third column). Note that in the localities 1, 3, and 5, and 4, a species for which no free-living tadpoles exist (Raorchestes parvulus and Limnonectes limborgi, respectively) has been collected, thus inducing a slight bias in the comparison of the number of collected tadpoles relatively to the number of adults.
All available tadpole samples were sequenced. For adults, usually a single sample was selected for each species and each locality.
2.2 Choice of genes
To evaluate the potential and the efficiency of different genes as DNA barcoding markers, we have chosen two widely employed mitochondrial genes: the mitochondrial 16S rRNA gene, which is advocated for amphibians as standard DNA barcoding marker [32], and the subunit I of the cytochrome c oxidase (COI) gene, which has been proposed as universal marker [25,26].
2.3 Molecular techniques
Total genomic DNA was extracted from 99% ethanol-preserved fresh tissues (muscle or liver) using a cetyltrimethylammonium bromide (CTAB) protocol [42]: the tissue was homogenized in 700 μL of CTAB with 1 μL of proteinase K for digestion, deproteinized in 700 μL of chloroform/isoamyl alcohol 24:1 (CIA), and precipitated with ethanol absolute at –20 °C. Centrifuged DNA pellets were resolved in 200 μL of demineralised water.
PCR amplifications were done using primers specifically designed for anuran amphibians as the universal COI primers [43] resulted in very poor amplification (data not shown). A 713 bp fragment of the mitochondrial COI gene was amplified using the following set of primers: AH_CO1A_S (5’-CTACAAYCCRCCRCCTRCTCGGCCAC-3’) and AH_CO1A_AS1 (5’-TADACYTCDGGRTGDCCAAARAATCA-3’). A fragment of approximately 410 bp of the mitochondrial 16S rRNA gene was amplified with the primers AH-16S_S (5’-CGCCTGTTTACCAAAAACATCGCCT-3’) and AH-16S_R (5’-TGCGCTGTTATCCCYRGGGTAACT-3’). Amplifications were performed in a total reaction volume of 30 μL, containing 1.5 μL of each primer at 10 μM, 0.4 μL of RedTaq DNA polymerase (Qbio Appligen) with 3 μL of the buffer supplied by the enzyme manufacturer, 3 μL of dNTP (6.6 mM) and 1 μL of DNA extract. The following polymerase chain reaction (PCR) cycling procedure was used: 94 °C for 4 min; 94 °C for 30 s for denaturation, 50 °C for 30 s for annealing and 72 °C for 45 s for extension for 30–60 cycles; 72 °C (7 min) for final extension. The PCR products were visualized in a 1.5% agarose gel and were sequenced in both forward and reverse directions at the Genoscope (Évry, France). Both strands were assembled and checked using Sequencher 4.7 software (Gene Codes Inc.). The sequenced gene fragments correspond to the positions 3967-4412 and 7389-8101 of the mitochondrial genome of Xenopus laevis [44] (GenBank accession number NC_001573) for the 16S and COI genes, respectively. All DNA sequences obtained for this study were deposited in GenBank (accession numbers #####-##### to be added upon manuscript acceptance) and stored in the Barcode of Life Data Systems (BOLD) [45].
2.4 Phylogenetic analysis and genetic distances
The sequences were aligned by eye under Se-Al v2.0a11 Carbone (A. Rambaut; available at http://tree.bio.ed.ac.uk/software/seal/). The final COI gene sequence alignment was 706 bp long. The COI sequences were translated into amino acids to check that no insertion, deletion or stop codon occurred in any sequence. In the 16S alignment, a matrix was built for each family to conserve a maximum of homologous nucleotides (alignments of phylogenetically remote taxa would result in larger very variable zones). All ambiguous regions were excluded from the analysis to avoid erroneous hypotheses of primary homology. The final length of the sequences varies from 317 to 380 bp, depending of the family. Distance calculations are then done from these five matrices, independently. This results in minimizing genetic distances but in a conservative way. Nucleotide divergences were calculated using uncorrected pairwise distances (p-distances). For graphic purpose, a NJ tree was built from a global alignment of all 16S sequences. Removing the very variable areas lead to a 246 bp long sequence matrix.
The trees were constructed using a neighbour-joining (NJ) algorithm in Paup 4.0b10 [46] with Kimura 2-parameter (K2P) [47] distance model. The support of nodes was estimated using bootstrap [48] with 1000 bootstrap replicates. For a graphic representation, most similar sequences were clustered using the Automatic Barcode Gap Discovery (ABGD) [49]. The trees displayed in this study do not intend to represent phylogenetic relationships of the species included due to the rather short segments of mitochondrial genes. They only aimed to visualize graphically the genetic distances of the sample. Then more phylogenetically oriented algorithms (ML or BI) were not used here. Furthermore the use of large DNA barcoding datasets of short sequences can lead to problematic results due to overparameterization with such algorithms.
To check for the accurateness and applicability of DNA barcoding to larval identification, we assessed the maximum intrapopulation genetic divergence of any tadpole with conspecific individuals, the maximum interpopulation genetic divergence of any tadpole with conspecific individuals and the minimum genetic divergence with the closest heterospecific sequence. This test has been carried out only for the 16S gene as this marker had the best amplification success and so the largest sample size. Due to the low number of tadpole sequences relative to the adult sample, tadpole haplotypes have been compared to both conspecific adults and tadpoles (except for other tadpoles collected in the same lotic water point, which could be siblings; on the contrary, tadpoles taken from different points of the same stream were considered members of a population and not siblings). In the case of tadpoles coming from the same pool the haplotype of only one individual was used in this test. This test aimed at assessing the percentage of cases where a tadpole clusters with another species (failure in identification) and the percentage of cases where a tadpole shows a smaller genetic distance to a conspecific sample from another locality than to a syntopic conspecific. The analysis comprised the comparisons of 105 tadpole haplotypes, nine allowing only intrapopulational comparisons (no sample from other locations available) and 26 allowing only interpopulational comparisons (single specimens from one locality).
3 Results
3.1 Comparison of the 16S and COI mitochondrial genes for the DNA barcoding of Southeast Asian anurans
The DNA of 412 specimens was extracted, among which 121 tadpoles and 291 adults (Megophryidae 17 tadpoles and 39 adults; Microhylidae eight tadpoles and 43 adults; Dicroglossidae 15 tadpoles and 60 adults; Ranidae 11 tadpoles and 83 adults, and Rhacophoridae 67 tadpoles and 68 adults). In total, we obtained sequences from 83 recognized species along with a number of unallocated specimens.
3.1.1 Amplification success
Amplification success was 96.4% and 83.7% for the 16S and the COI respectively. Failures for the 16S concern in 80% of the cases tadpole tissues, the other concern old DNA extract (> 5 years) of adult tissues. All samples that failed to amplify for the 16S gene failed for the COI gene too, suggesting a low DNA quality of these samples.
3.1.2 Analysis of the clusters recovered in the phenetic trees
None of the two gene trees recovered all families as monophyletic. Microhylidae are recovered as monophyletic in the two trees, as well as the Megophryidae in the 16S tree and the Ranidae in the COI tree. However, the very short 16S matrix of 246 bp of 397 sequences could explain that all families are not monophyletic (Fig. 2).
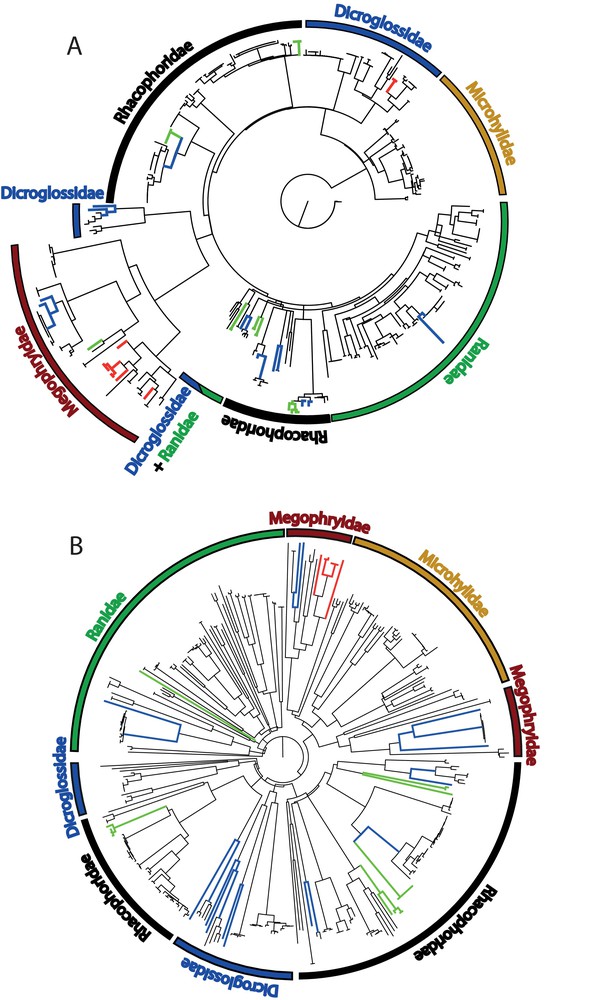
(Colour online.) Neighbour-joining phylograms using K2P distances for the 16S barcode sequences (A) and the COI barcode sequences (B). Blue branches indicate taxa with deep intraspecific genetic divergences that potentially represent several distinct species. Green branches indicate species represented only by their larval forms. Red branches indicate candidate species. Note that the 16S tree was built from a unique alignment of all samples leading to an only 246-bp-long matrix after removing very variable areas. Then some of the deeply divergent conspecific lineages are less marked here than in trees built with intrafamilial matrices (see Suppl. Figs. S1–S5). On the contrary, the COI tree was built with a full length sequences matrix of 706 bp. For detailed intrafamilial phylograms and bootstrap supports, refer to Suppl. Figs. S1–S5.
Using a separate matrix for each family, nine supraspecific clades are recovered, with a high bootstrap support (>70%) in both gene trees (Suppl. Figs. S1–S5). Additionally, five and one supraspecific clusters are recovered in the 16S and COI trees only, respectively (details in Supplementary Information S2). Some discrepancies are observed between the two phylograms: the genus Leptobrachium and the species Xenophrys sp. 1 are paraphyletic in the COI analysis; the 16S analysis failed to recover monophyletic groups within the Feihyla vittatus complex.
3.1.3 Analysis of the uncorrected “p” pairwise distances
The uncorrected-p distances in the 16S and COI fragments range from 0 to 5.0% and 0 to 14.5% in intraspecific comparisons, and from 1.5 to 24.1% and 5.4 to 27.0% in interspecific comparisons, respectively (Table 1; Supplementary Figs. S6–S10). The intraspecific and interspecific distances overlap in the two genetic markers except for the members of the Microhylidae, in which a clear gap is observed, probably due to the reduced number of species included (Table 1; Supplementary Fig. S7). Some of the intraspecific values are particularly high, due to various problems (Supplementary Information S3), among which a probable case of introgressive hybridization in a tadpole of Rhacophorus kio.
Intra- and interspecific uncorrected pairwise sequence divergences for each family and for each of the two genetic markers (16S and COI gene portions).
| Megophryidae | Microhylidae | Dicroglossidae | Ranidae | Rhacophoridaea | |||||||
| Intraspecific (%) | Interspecific (%) | Intraspecific (%) | Interspecific (%) | Intraspecific (%) | Interspecific (%) | Intraspecific (%) | Interspecific (%) | Intraspecific (%) | Interspecific (%) | ||
| Uncorrected p-distances | 16S | 0–5.0 | 2.2–21.2 | 0–3.5 | 5.6–14.6 | 0–4.8 | 3.3–24.1 | 0–4.9 | 1.8–16.0 | 0–4.4 | 1.5–19.8 |
| COI | 0–14.2 | 6.5–27.0 | 0–10.2 | 15.3–22.1 | 0–14.5 | 14.3–25.6 | 0–11.9 | 7.5–25.0 | 0–12.1 | 7.3–27.0 |
The lowest interspecific sequence divergence values are observed mostly between Polypedates leucomystax and Polypedates impresus, and Chiromantis vittatus and C. cf. vittatus for the 16S gene (both pairs 1.5%), whereas Xenophrys sp. 2 and Xenophrys sp. 3 are the less divergent species for the COI gene (6.5–6.6%). The highest intraspecific values are found in Rhacophorus rhodopus, which exhibits two deeply divergent lineages that potentially represent distinct species (“p” distances 4.4% and 12.1% for the 16S and COI genes, respectively). A specimen of Hylarana erythraea shows as well an important genetic divergence from other conspecific sequences (4.9% for the 16S and 11.8% for the COI).
3.2 Allocation of tadpoles to species
The tadpoles have been collected in the field by three different collectors with different expert assessment methods. Tadpoles belonging to a subsequently described species following revision of cryptic species complexes are not considered as erroneously named in the field if they were identified as tadpoles of the initial species. All the tadpoles assigned to a different species by the DNA barcode that originally in the field have been subsequently checked morphologically to ensure the validity of their new identity. A total of 19.8% of the tadpoles have been identified to the generic level (although 3.6% of them belonged to an undescribed species at the time of collection), 2.7% to the familial level and 13.5% of the tadpoles had no identity allocated at all in the field. Errors of identification occurred at different taxonomic levels: 4.5% of tadpoles have been attributed to a wrong family, 7.2% to a wrong genus and 4.5% to a wrong species. In total 48.6% of the identifications of tadpoles in the field were erroneous or incomplete. Furthermore, DNA barcoding revealed that three tadpole series (0906Y1-5, 0936Y1-4 and 0937Y1-7) out of eight collected in the same pool and identified as homogeneous morphologically were actually composed of two or three different species. All species belonged to the Rhacophoridae, genera Rhacophorus and Chiromantis.
The genetic distances observed between tadpole sequences and intrapopulational conspecifics range from 0 to 0.0063, i.e., less than 1% of divergence (except for the sequence 0957Y, whose distance to the other one can reach 0.0176), whereas those observed between the tadpole sequences and interpopulational conspecifics range from 0 to 0.0440 (the maximum value is due to a cluster within Rhacophorus rhodopus flagged as a candidate species; removing these samples, the extreme value lowers to 0.0381). Distances between tadpoles and their closest heterospecific sequences were 0.0147–0.1206 (Fig. 3). In no case is the maximum intraspecific sequence divergence either intra- and/or interpopulational higher than the lowest interspecific genetic divergence. Also, in no case is the maximum intrapopulational genetic divergence higher than the highest interpopulational genetic divergence.
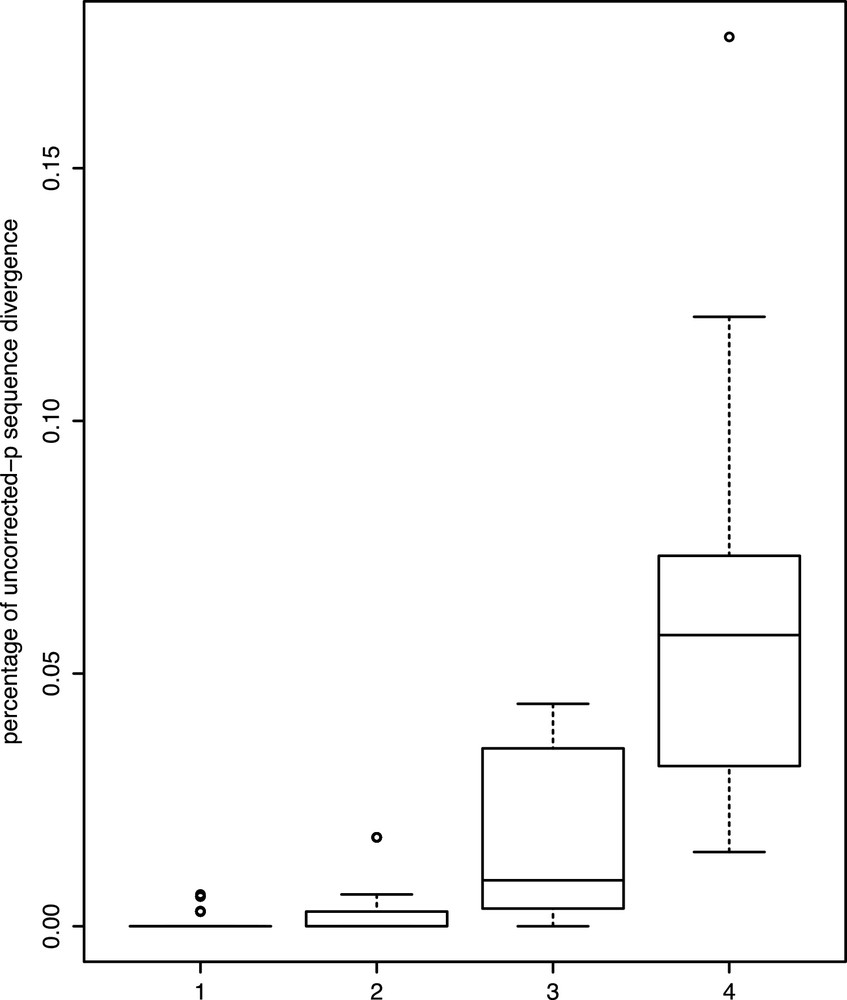
Boxplots of uncorrected pairwise distances for the 16S barcode marker for (1) the minimum intraspecific genetic distances between a tadpole and an adult or a tadpole from another water body from the same population; (2) the maximum intraspecific genetic distances between a tadpole and an adult or a tadpole from another water body from the same population; (3) the maximum intraspecific genetic distances between a tadpole and an adult or a tadpole from another population; (4) the minimum interspecific genetic distances between a tadpole and the closest heterospecific sequence. Boxes represent the median (black horizontal line) and the first and third quartiles, and the error bars represent roughly 95% percentiles; dots represent outliers.
No tadpole sequence of the 16S or COI barcode of our dataset clusters with a heterospecific sequence when GenBank sequences are added (data not shown).
3.3 Biodiversity revealed by tadpoles
Estimation of the biodiversity of the adult and tadpole forms has been assessed for seven well-prospected areas (Fig. 1). Four different situations were encountered:
- • all the tadpoles were collected with their conspecific adults (areas 1 and 3);
- • only a small proportion of adults and tadpoles were conspecific (areas 2 and 6);
- • the number of species in the larval form collected surpassed the number of species in the adult stage (area 6);
- • in the area 7 the same number of tadpoles and adult (in term of species) have been collected though only about one third are conspecific.
Seven clusters composed of only tadpoles were found in the analysis of the 16S sequences (Fig. 2, Suppl. Figs. S4–S5). One of them is composed of four Rhacophorus sp. sequences from Vietnam and Thailand corresponding to two different species. Tadpoles clusters allocated to Rhacophorus rhodopus and Feihyla cf. vittatus 2 are deeply divergent (4.4%) or around the threshold value of 3% defined by Fouquet et al. [9], i.e. 2.4–3.2%, respectively, and can be considered as unconfirmed candidate species. Although only 27.6% of the sequences in this study are from tadpoles, they allowed the identification of one confirmed candidate species, the detection of an undetermined tadpole, and of four unconfirmed candidate species.
3.4 New lineages revealed by DNA barcoding
Although not fundamentally the goal of this study, DNA barcoding of our tissue bank revealed the presence of 17 confirmed and/or unconfirmed candidate species, i.e. an increase by 20.5% of the initial pool of species. Furthermore, several species contained in our dataset have been described or identified but not yet scientifically named (eight species), or synonyms have been resurrected since the design of this study (two species).
4 Discussion
4.1 Comparison of the 16S and COI mitochondrial genes for the DNA barcoding of Southeast Asian anurans
The commonly used 16S gene primers amplify the whole range of vertebrates with a 100% amplification success under different laboratory conditions [32]. In our study all failures (3.6%) concern tadpole tissues taken at the extremity of the tail or old (>5 years) DNA extracts. The fins of tadpoles are composed of a double layer of skin overlying loose connective tissue [50], the collagen inhibiting PCR [51], while the muscle pieces from the tail tip were usually very small. As additional evidence for failure in amplification due to these conditions, none of the DNA samples that failed to amplify for the 16S gene resulted in a positive amplification for the COI gene. The amplification success obtained with our newly developed COI primers parallels those of previous studies on amphibians [52–54], but is globally between 80–90%.
Clustering of congeneric sequences is important, especially if sequences of an organism under study are lacking from the reference database. This clustering then gives essential taxonomic information. More supraspecific monophyletic taxa are retrieved in the 16S phenograms (although the clusters are less statistically supported by the bootstrap) than in the COI trees. The lower support of the clusters in the 16S tree is most probably due to the much shorter sequences compared to COI gene segment although a similar result has been found in Hynobiidae salamanders [53]. Che et al. [55] noticed that COI barcode failed to cluster individuals in their respective groups at the generic and higher taxonomic levels. More worrying Xia et al. [53] showed than 16S sequences cannot diagnose some taxa (19.4% of the species failed to be recovered), whereas COI sequences recover all species as monophyletic. In our study this concerns only a species complex, the Feihyla vittatus species complex. A striking discrepancy between the two markers is the clustering of the sequence of another species (X. jingdongensis) with the two sequences of Xenophrys sp. 1 in the COI NJ tree, contrary to what is observed in the 16S NJ tree. An independent cladistic analysis based on morphology confirmed the conspecificity of these two specimens [56]. This discrepancy cannot be explained at this stage.
An important prerequisite of the DNA barcoding is the ability to accumulate a huge amount of data in databases and in alignment matrices for edition of phenograms (NJ trees). While the generation of a protein-coding COI gene matrix is straightforward due to the conservation of the reading frame and keeps all information, the alignment of 16S sequences requires laborious and painstaking attention to alignment due to the numerous insertions/deletions occurring within the highly variable loop regions. Any new sequence added to the alignment requires further work to ensure the primary homology of the nucleotides, becoming more and more difficult with the addition of taxonomically remote taxa. So our 410-pb raw 16S sequences were reduced to 317–380 bp in intrafamilial matrices and to only 246 bp in the global matrix gathering all samples. It should be noted that, although anecdotic, the identification of the ambiguous zones is subjective and the range of these zones can vary among authors. As a result, the percentage of the genetic distance can vary as well as the composition of the clusters in the graphic representations (pers. obs.).
The COI gene is the ideal candidate for a global DNA barcoding with hundreds of thousands of sequences encompassing large taxonomic and geographic sample (see [57] for the amphibians), whereas 16S sequences can be used for rapid assessment of the tadpole biodiversity of a site, ensuring total amplification of the samples.
4.2 New lineages revealed by DNA barcoding
Exploration of amphibian biodiversity by DNA barcoding has already been realized in Madagascar [10,54] and in French Guyana [9], and revealed numerous cryptic species with an about two-fold increase in species number in both countries. Several recent studies in Southeast Asia integrating molecular phylogenies and morphological studies revealed numerous cryptic species within species complexes previously understood as single species [8,58,59]. We here detected additional potentially new lineages that need now accurate screening of their representatives by an integrative approach to determine their taxonomical status. We expect such futures studies will increase the number of species of our sample by more than 20%.
4.3 Allocation of tadpoles to species
Field identification of tadpoles is extremely difficult, mainly because of their homogeneous morphology, small size, and absence of keys. Furthermore, differences between congeneric species are usually very subtle. In the present approach, we compared the field identifications with the assignations obtained with DNA barcoding. About half of tadpoles sampled in this study were erroneous or incompletely allocated to species or genus names. Even with a trained experience in larval morphology, it is extremely difficult to make identifications in the field. In these conditions, a significant proportion of the collected tadpoles have been “conservatively” allocated to the generic level only to avoid hazardous specific allocations that could further stay linked to the specimen and be misleading for further studies. Errors occurred even at the familial level in the three collectors.
In this study, three series of tadpoles each considered as taxonomically homogeneous proved to be composed of two or three different species. All these species belonged to two close genera that were very difficult to identify without appropriate tools. In these cases, the use of the DNA barcoding of only one representative by series can overlook the real biodiversity of a locality or even of a single pool as presently. Tadpole series should then be carefully examined in the laboratory and/or several specimens should be DNA barcoded within each series.
All sequences from tadpoles have been successfully allocated to their respective species except for a possible case of introgressive hybridization (see below). In an intensive comparison between tadpoles and adults of two geographic sites separated by a distance of about 250 km in Madagascar a tadpole sequence yielded a higher BLAST score with a conspecific adult sequence from the other site rather than from the same site in only 2% of the cases [32]. Although in our results this case is more frequent (data not shown), in all cases but one tadpoles and adults from the same locality displayed less than 1% of genetic divergence (0.63% exactly) and in no case minimum genetic distance between a tadpole and a conspecific adult sequence was higher than with the closest heterospecific sequence. Thus DNA barcoding is a very efficient tool for the specific allocation of tadpoles and is able to overcome the difficulties linked to their identification especially when intensive sampling of both adult and larval forms has been carried out in a location. A risk of misidentification exists with the phenomenon of introgressive hybridization whose extent is still unknown in tropical amphibian communities. Only one case of introgressive hybridization has been suspected in this study because tadpoles have been morphologically screened. It concerns a tadpole of Rhacophorus kio that shares the haplotype of R. rhodopus with 100% of similarity. Blind DNA barcoding would have overlooked this problem and wrongly assigned this specimen to another species.
4.4 Biodiversity revealed by tadpoles
The results presented here show that in several cases tadpoles were collected in the field without conspecific adults (Figs. 1–2, Suppl. Figs. S4–S5). It represents six lineages involving six different localities; some of these lineages clustering tadpole samples from up to three different localities. On the other hand, in Phetchabun and Prachuap Khiri Khan (Thailand), two and three tadpole lineages were collected without conspecific adults, respectively. These results strengthened the idea that tadpoles must be carefully collected in the field to increase and refine the estimation of the batrachological biodiversity of a site. As shown here, even with relatively high collecting effort, it is thus possible to collect tadpoles without getting any conspecific adult. In these cases, DNA barcoding cannot achieve its goal by failing molecular identification of these samples when compared to the local database of the collected adults. Using more supplied databases as GenBank or BOLD can (at least in part) overcome this problem. A cluster of four unidentified Rhacophorus tadpole sequences collected in the same site has been compared to the full set of GenBank sequences. Two sequences have been identified as Rhacophorus maximus, whereas the other two remained unidentified (the two sets of sequences diverge of 4.4% uncorrected-p distance).
The whole larval cycle and associated ecological requirements must be fully known to efficiently protect amphibian fauna. DNA barcoding of tadpoles should be of great help in this task.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We are grateful to the following people for technical help and discussion: Didier Geffard and Dario Zuccon (UMS 2700, MNHN, Paris), Sandra Goutte, Violaine Nicolas and Nicolas Puillandre (UMS 7205, MNHN, Paris), Alodie Snirc (université Paris-Sud XI, Paris). SG has been financed by the BQR of the département de systématique et évolution of the MNHN, Paris. The fieldwork of AO and SG was supported by PPF “Faune et Flore du sud-est asiatique” and PPF “État et structure phylogénétique de la biodiversité actuelle et fossile” from the MNHN, Paris. For logistical assistance, we warmly thank the Institute of Ecology and Biological Resources (Vietnam), Frontier Vietnam Forest Research Project (UK/Vietnam), Maison du Patrimoine, Luang Prabang (Laos), and the Phongsaly Forest Conservation and Rural Development Project (Laos). Thanks are addressed for their help in the field to Sandra Dos Santos, Pierre Guédant, Chantip Inthara, Steven Swan, Jack Tordoff, Kim Valkone, Ian C. Bickle.