

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Le développement de la pandémie de COVID-19 est exploré dans une myriade d’articles. Malgré cette abondance, et en raison de notre anthropocentrisme, il est exceptionnel que les études publiées se placent du point de vue du virus. Bien sûr, de nombreux travaux se penchent sur le détail de la composition et de la structure du génome du virus SARS-CoV-2, des protéines qu’il code et de sa parentèle. Pourtant, les études portant sur la façon dont le virus exploite le métabolisme de son hôte cellulaire sont très rares. C’est que l’urgence de trouver le moyen de contrôler la maladie conduit à mettre l’accent sur la vaccination ou plus généralement sur le système immunitaire de l’hôte. On sait bien, hélas, que s’il a été parfois relativement facile de trouver un vaccin à la fois efficace et inoffensif contre une maladie répandue, le contraire est vrai aussi. Il existe encore des maladies très graves et très communes pour lesquelles il n’existe pas de vaccination possible pour l’instant. Vacciner efficacement suppose, en particulier, que la descendance d’un agent pathogène reste suffisamment longtemps la même pour ne pas conduire aisément à l’évitement de la réponse immune déclenchée par le vaccin. Les coronavirus sont des virus formés d’un génome long et d’une enveloppe. La longueur du génome aurait pu conduire à un taux de mutation très élevé, mais ces virus, évitant ainsi la contrainte universelle du cliquet de Muller — voir Encadré, p. 1 — ont recruté une fonction spécifique de correction des erreurs de réplication [1]. Cela fait que, s’ils ont tendance en effet à voir apparaître des variants génétiques au cours du temps, le nombre de ces variants reste assez faible. Ce taux de mutation peut paraître très limité, mais le nombre des particules virales engendrées au cours d’une infection est énorme, alors que la population humaine actuellement reconnue comme infectée va bientôt atteindre vingt millions de personnes. Il s’en suit que le taux de substitution par nucléotide – bien sûr très hétérogène en raison de la pression de sélection sur certaines positions — est de l’ordre de 8 × 10−4 changement par site et par an [2].
Ici, nous avons mis cette situation en regard du théorème fondamental de la sélection naturelle proposé par Fisher, qui relie l’évolution de l’adéquation à l’environnement (« fitness ») et la variance génétique [3]. Nous nous sommes efforcés d’utiliser les traces de l’évolution de la fitness du virus — constatées sous la forme de séquences génomiques — en présence des contraintes biochimiques qui biaisent les choix disponibles. Nous avons dû prendre en compte, cependant, le fait que les termes du problème ne sont pas aussi explicites qu’on aurait pu le souhaiter : la fitness n’est pas connue, pas plus que l’étiquette temporelle (estimée sur des arbres phylogénétiques ou tout simplement prise comme le temps physique), et la fréquence de certaines souches dans les arbres phylogénétiques peut être moins due à la sélection naturelle qu’à l’hétérogénéité de l’intensité d’échantillonnage et de séquençage. Cela a motivé notre utilisation de procédures robustes par rapport à ces incertitudes. Néanmoins l’avantage d’un telle analyse est de nous permettre de proposer des projections sur l’évolution du virus. Il s’agit donc là d’un moyen explicite permettant d’alimenter des modèles épidémiologiques ou cliniques.
Dans ce contexte, il nous a semblé d’un grand intérêt d’explorer le détail des mutations du virus SARS-CoV-2 au fil du temps, dans les différents endroits où COVID-19 s’est répandue, en mettant en évidence les lignées pertinentes en relation avec le métabolisme de l’hôte. Cela devrait nous permettre d’anticiper en partie l’avenir de la descendance du virus, avec des conséquences importantes pour le contrôle de la maladie. L’analyse des contraintes qui régissent l’accès au métabolisme des nucléotides qui composent le génome du virus nous a montré que le contenu en cytosine (C) de son génome est soumis à une forte pression négative, conduisant à une déplétion systématique, au fil du temps, de la cytosine monophosphate [4]. Une analyse rudimentaire a pu faire croire en un effet causal majeur de l’« édition » du contenu en C du génome par des désaminases de la famille APOBEC [5, 6], mais nous savons aujourd’hui que c’est l’organisation du métabolisme des pyrimidines, et plus particulièrement de la cytosine triphosphate [7], qui a l’effet critique sur l’évolution correspondant (Figure 1).
Le CTP contrôle toutes les étapes métaboliques cruciales nécessaires à la synthèse d’un virus SARS-CoV-2 fonctionnel. (1) le CTP est un précurseur du génome du virus ; (2) les lipides de son enveloppe proviennent de précurseurs liponucléotidiques à base de cytosine ; (3) toutes les molécules d’ARN de transfert produites par l’hôte doivent être complétées par un triplet CCA à leur extrémité 3′OH ; et (4) la glycosylation post-traductionnelle des protéines virales, en particulier de sa protéine spicule, doit être ancrée par un groupement dolichyl-phosphate dans le réticulum endoplasmique (RE) et la dolichol kinase dépend spécifiquement du CTP. Voir le texte et la référence [7] pour plus de détails.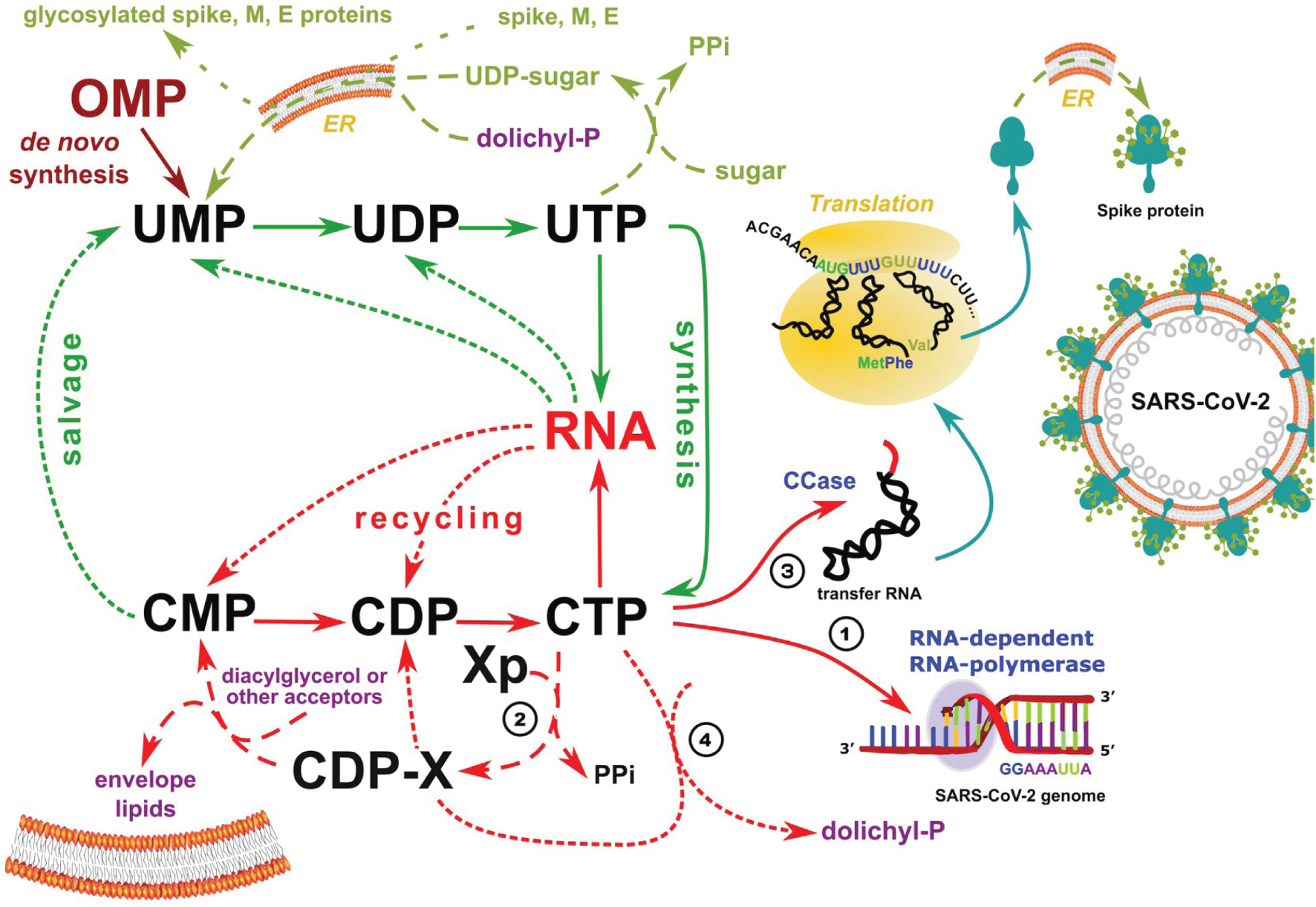
En effet, en raison de l’extrême dissymétrie de la réplication du virus — qui recopie de 50 à 100 fois sa matrice complémentaire [8] — un effet de l’édition du génome par ces enzymes très dépendantes du contexte ne serait important que lors de la modification d’un C en U sur l’ARN négatif, ce qui conduirait à un enrichissement majeur en A du génome viral, ou encore de transitions U → C dues à l’action d’une désaminase d’une classe différente et qui agit sur l’ARN double brin, nommée ADAR, et qui désamine l’adénine en inosine [9]. APOBEC et ADAR sont des enzymes très spécifiques et cela ne cadre guère avec les transitions C → U très répandues que nous continuons à observer au fur et à mesure de l’évolution du virus. Ici, nous nous sommes concentrés sur la dynamique de la perte de C dans le génome, et nous avons cherché les lieux et les causes des changements dans cette dynamique. Dans un premier paragraphe nous résumons les raisons métaboliques de ce phénomène remarquable. Ensuite, dans le corps de l’article nous montrons que la contrainte sur le contenu en C du génome entraîne des filiations spécifiques qui révèlent l’existence de fonctions importantes du virus ainsi que le rôle de la réponse de l’hôte.
2. Une exigence métabolique universelle, qui règle la synthèse du triphosphate de cytidine (CTP), oriente l’évolution du virus
Que savons-nous de la synthèse des composants qui permettent la genèse d’une particule virale (un virion) ? Lors d’une infection virale les cellules arrêtent généralement de se multiplier. Toutes leurs ressources sont rapidement détournées au profit de la multiplication du virus. Or, la croissance est une propriété universelle de la vie. Cela signifie que, presque toujours — les neurones différenciés sont une exception — le métabolisme cellulaire est organisé de façon à permettre la croissance cellulaire dès que l’occasion de se multiplier se présente. Au moment où il infecte une cellule — à nouveau, à l’exception de celles qui ne se multiplient pas — tout virus aura donc à gérer la pression métabolique qui organise la disponibilité des composants nécessaires à sa construction. Dans notre espace physique habituel (tridimensionnel), croître introduit une contrainte inévitable. La cellule doit mettre ensemble la croissance de son cytoplasme (à trois dimensions par conséquent), celle de la membrane qui l’entoure (deux dimensions) et celle de son génome (à une dimension, car les acides nucléiques sont des polymères linéaires). Or c’est un métabolisme commun, déployé pour l’essentiel dans le cytoplasme, qui produit les matériaux de construction de ces trois compartiments majeurs. Nous avons donc là une question semblable à celle que se posent les économistes lorsqu’ils évoquent la question de la croissance « non-homothétique » [10]. Mais la vie s’est développée en plusieurs étapes durant plus de 3,5 milliards d’années à partir d’un métabolisme primitif et on aurait pu craindre que chaque organisme ait trouvé une solution idiosyncratique à cette contrainte. De façon inattendue il semble bien qu’un métabolite unique, le nucléotide cytidine triphosphate (CTP) a été utilisé à cette fin [4, 7].
Le rôle unique du CTP apparaît en quatre endroits essentiels dans le métabolisme cellulaire, et ces lieux sont essentiels pour la formation de nouveaux virions. (1) Il s’agit du précurseur immédiat d’un des quatre nucléotides formant le génome du virus ; (2) le CTP est requis pour la synthèse des précurseurs liponucléotidiques de l’enveloppe virale ; (3) les ARN de transfert humains sont synthétisés à partir de 415 gènes ne codant pas leur extrémité CCA 3′OH-terminale — cette séquence est synthétisée par une nucléotidyltransférase à partir de CTP [11] ; et pour finir (4) la « décoration » des protéines par des glycosylations compliquées se fait en parallèle avec la traduction dans le réticulum endoplasmique via l’ancrage de substrats par le dolichyl-phosphate, produit par une kinase qui utilise le CTP, et non l’ATP, comme donneur de phosphate [12]. Par ailleurs, le métabolisme intermédiaire repose sur une organisation originale du métabolisme des pyrimidines, qui utilise systématiquement l’uridine triphosphate (UTP) ce qui limite considérablement l’accès au CTP (Figure 1). Il s’en suit que les erreurs de réplication accidentelles vont tendre à remplacer la cytosine par l’uracile dans le génome.
3. Évolution générale du virus SARS-CoV-2
En utilisant les données de séquence rassemblées dans la base de données SARS-CoV-2 GISAID (https://www.gisaid.org) nous avons, comme d’autres [13, 14], reconstitué un arbre phylogénétique de l’évolution du virus. Comme les séquences du génome viral ainsi que la date d’identification de ces séquences sont connues avec une assez grande précision, cet arbre permet d’explorer la filiation ordonnée des mutations qui apparaissent au cours du temps. En particulier, sauf si l’on peut soupçonner un événement de recombinaison due à l’infection d’un même patient par deux virus ou plus, lorsque deux mutations identiques apparaissent dans des branches distinctes de l’arbre, on peut faire l’hypothèse qu’il s’agit de convergence évolutive [15]. Les raisons de cette convergence sont discutées à l’occasion de l’analyse de telle ou telle mutation. Une deuxième observation, qui nécessite d’être mise en perspective (voir plus loin) est que la forme de l’arbre n’est pas du tout homogène. On remarque en effet la présence d’« efflorescences » où, à un nœud particulier de l’arbre, un grand nombre de branches apparaissent, démontrant une apparition « explosive » de nouvelles mutations (Figure 2). Nous avons mis au point une approche statistique nous permettant de les caractériser de façon explicite.
Un exemple d’efflorescence détecté par notre approche statistique. Au niveau du nœud N0, on recense 25 états différents sur les 40 échantillons du sous-arbre et un nombre élevé de ramifications. Ce comportement diverge significativement de celui des autres sous-arbres. Chaque pays est représenté par une couleur distincte au niveau des feuilles de l’arbre.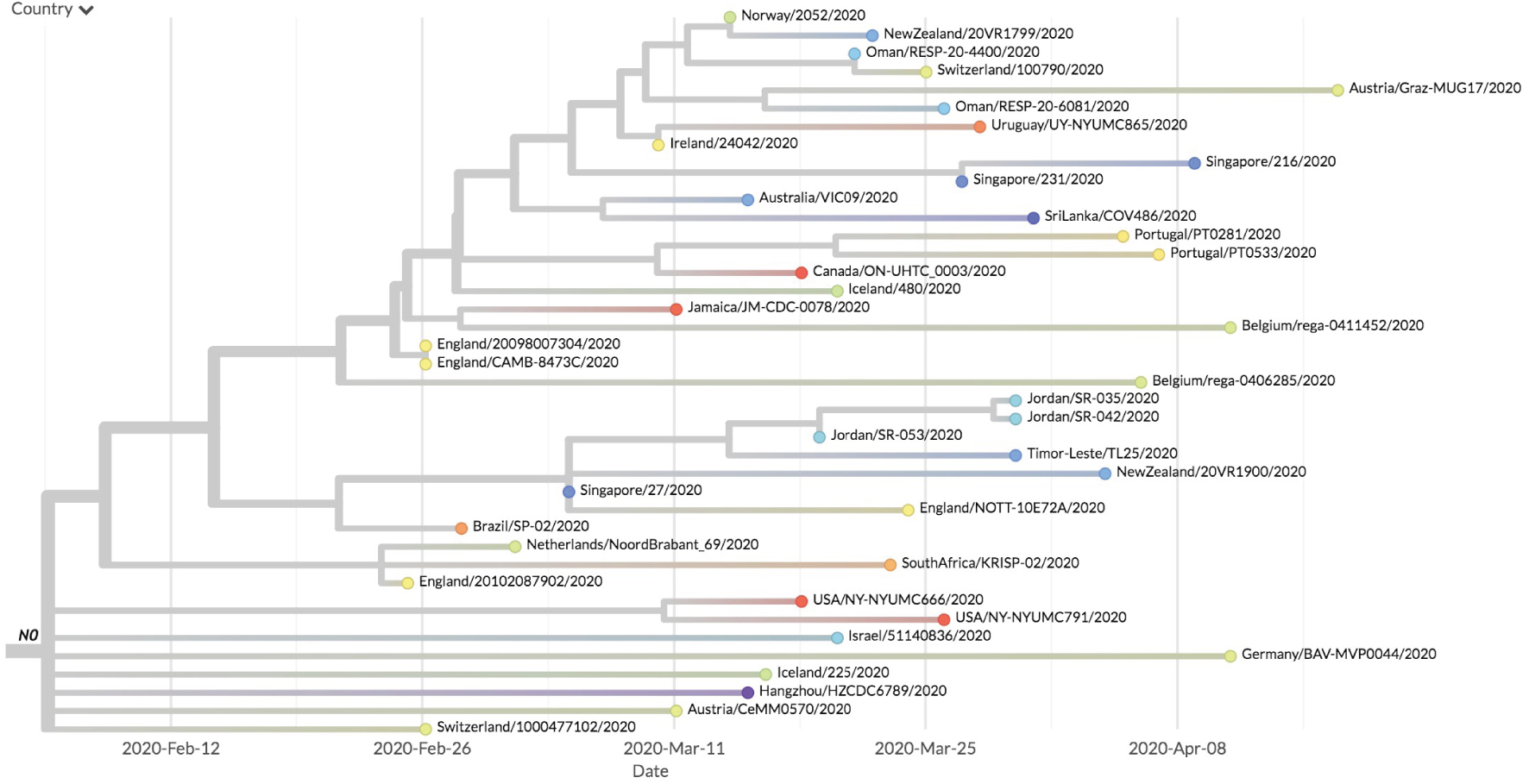
Les causes de ces efflorescences sont multiples, mais l’altération de fonctions virales importantes peut en être l’origine, et nous avons retenu quelques cas de ce genre pour plus ample discussion (voir Matériels et Méthodes, pour une description statistique de ce que sont les efflorescences).
4. Description et analyse de l’évolution du contenu en C du génome
D’une façon générale, le génome des coronavirus tend à évoluer en adaptant son contenu en C à son hôte. Plus spécifiquement SARS-CoV-2 évolue vers des formes moins riches en C au fur et à mesure que l’épidémie se développe [7]. Cependant cette évolution n’est pas homogène.
Dans les deux jeux de données étudiés, les transitions entre pyrimidines sont représentées à 77% par des transitions de cytosine vers uracile. Ces transitions représentent 48% de l’ensemble des substitutions recensées dans le premier jeu (respectivement 49% dans le second). Un important déséquilibre peut également être relevé au niveau des transversions, sachant que plus de 73% d’entre-elles concernent une substitution de purine en pyrimidine sur le premier jeu (respectivement 74%). Cependant, seulement 20% de ces 73% mènent à l’apparition d’une cytosine (respectivement 17%), dénotant une nouvelle fois une tendance à favoriser la génération d’uracile, démontrant par là que la contrainte majeure du processus mutagène est la disponibilité de la cellule en chacun des nucléoside triphosphates. Cette inhomogénéité se remarque encore au niveau de l’arbre. Au niveau de la branche B4 (20% des échantillons), la tendance est fortement marquée à perdre peu de C par rapport au reste de l’arbre (Figure 3).
De façon intéressante, c’est aussi la branche contenant en moyenne les souches avec le moins de divergence par rapport à la souche d’origine du virus. À l’inverse, sur la branche B1, la perte de C paraît plus importante. Le processus de mutation du virus semble aussi s’y accélérer, avec un taux de transversions 20% plus important que le reste de l’arbre (et des taux de transitions également plus élevés, mais dans des proportions plus anecdotiques). Enfin, en ce qui concerne la branche B3, siège principal des efflorescences, une diminution de 29% du taux de transition des pyrimidines et de 30% du taux des purines comparativement au reste de l’arbre est à souligner.
Cette inhomogénéité peut être la conséquence de nombreuses contraintes :
Carte en dégradés de couleurs des pertes en C depuis la séquence d’origine. Les branches 1 et 4 se démarquent visuellement par leurs valeurs extrêmes.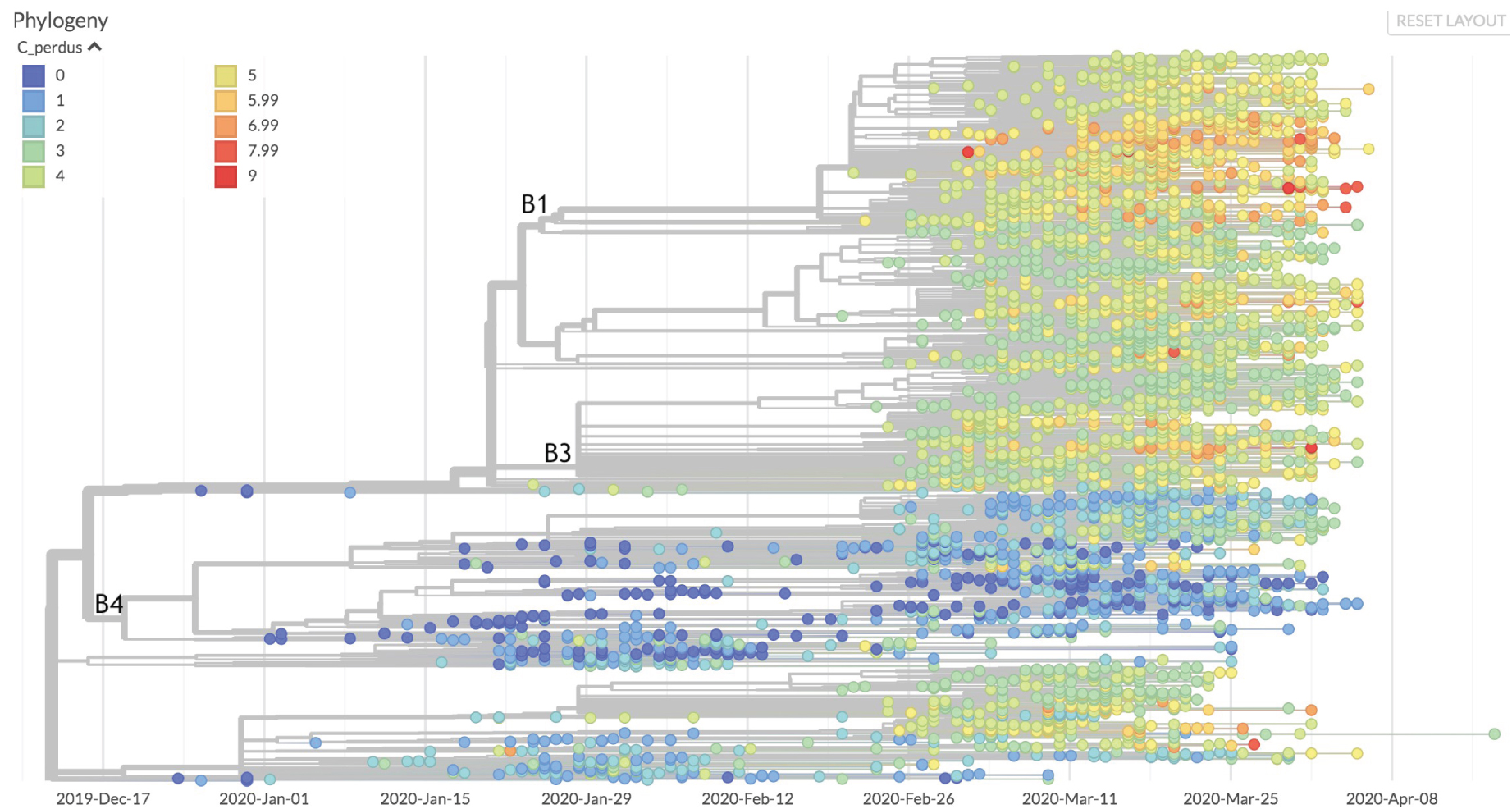
(1) La structure même du génome, qui doit se replier dans une enveloppe-capside compacte impose à certaines régions de conserver une séquence riche en C. C’est le cas des régions qui contrôlent le démarrage de la réplication [8] ou la transcription, AACGAAC par exemple [16]. Dans le cas des régions traduites, la pression sur la présence de C varie en fonction de sa position dans les trinucléotides du codon. Lorsque C est situé à la première position d’un codon, il est utilisé pour introduire l’arginine, la glutamine, l’histidine, la leucine ou la proline dans les protéines. L’histidine et la glutamine sont codées dans des familles à deux codons, discutées plus bas. Pour l’arginine, la pression de sélection est plus faible car les codons CGN peuvent être remplacés par des codons AGR — nous utilisons ici les conventions décidées par l’IUPAC pour l’étiquetage des nucléotides ou des acides aminés, par exemple N pour aNy, R pour puRine, etc. (https://www.bioinformatics.org/sms/iupac.html). La pression de sélection sur la teneur en leucine est à nouveau plus faible, car en plus des codons CUN, cet acide aminé peut être introduit à l’aide des codons UUR. Dans la deuxième position du codon, C est à nouveau utilisé pour coder la proline, mais aussi la thréonine (ACN), l’alanine (GCN) et la sérine (UCN). Là encore, ce dernier acide aminé échappe à une grande partie de la contrainte imposée par la disponibilité en C car il peut aussi utiliser les codons AGY. Enfin, la troisième position des codons est beaucoup moins contraignante car elle peut être remplacée par U mais aussi par A ou G dans les familles à quatre codons (alanine, proline, thréonine, valine). Les familles à deux codons UGY, AGY et NAY sont discriminées selon un axe pyrimidine / purine. Une pyrimidine est utilisée pour maintenir la même nature du résidu codé, car le codon utilise un U ou un C à l’extrémité 3′ (aspartate, asparagine, cystéine, histidine et tyrosine). Enfin, l’isoleucine est codée par trois codons (AUH), et la terminaison U ou C est prise en compte par les ARNt appropriés [17] ;
(2) La fonction de certaines protéines du virus peut imposer la présence de certains acides aminés dans leur séquence. Par exemple, le résidu proline codé par les codons CCN n’est pas strictement un acide aminé, mais est essentiel pour le repliement des domaines clés de protéines virales [18] ;
(3) Mettant mieux encore en relief l’importance du CTP, l’immunité innée antivirale a recruté au cours de l’évolution l’activité d’une enzyme, la vipérine, qui modifie le CTP en une forme toxique pour le développement du virus, le 3′-désoxy-3′,4′-didéshydro-CTP (ddhCTP) [19]. Une conséquence intéressante de cette conversion métabolique est que la diminution de la teneur en C du génome va permettre au processus de réplication du virus d’être moins sensible à la présence de cette nucléobase. Il s’ensuit que, lors du passage d’un virus relativement riche en C d’un hôte animal à l’homme, l’évolution vers la perte de C peut être transitoirement concomitante d’une augmentation de sa pathogénicité. À long terme, cependant, la perte de C restreint fortement le paysage évolutif du virus et tendra très probablement à son atténuation [20].
5. Quelques exemples de corrélations permettant de proposer une fonction pour les protéines virales
Des milliers de mutations ont été identifiées à ce jour. Il est possible de suivre leur émergence le long de l’arbre de l’évolution phylogénétique du virus et de mettre ensuite en évidence quelques caractéristiques intéressantes qui pourraient nous permettre d’anticiper son futur.
5.1. Mutations conduisant à une fin de traduction prématurée
L’existence de mutations conduisant à terminer prématurément la synthèse de protéines du virus est attendue avec une fréquence élevée. C’est d’autant plus vraisemblable ici que les codons de fin de traduction UAA, UAG et UGA ne contiennent pas de C, et sont donc favorisés par la disparition de ce nucléotide. Comme la plupart de ces mutations conduisent à des polypeptides non-fonctionnels il est généralement probable que les virus en question ne donnent pas naissance à une descendance significative. Il s’ensuit que lorsque ces mutations sont observées — et qu’elles ne résultent pas d’erreurs de séquençage — elles indiquent que le rôle de la protéine tronquée correspond à une fonction peu sollicitée, ou que la protéine est restée fonctionnelle à un niveau suffisant pour permettre la reproduction du virus. Quelques observations nous permettent cependant de proposer une explication au fait que les virus en question aient pu survivre. Voici trois exemples qui révèlent des caractères intéressants du virus.
Dans une souche du virus isolée en Islande, la succession des mutations G1440A (Gly392Asp, protéine Nsp2) et G2891A (Ala876Thr, domaine ubiquitine de la protéine Nsp3) est ensuite présente en de multiples pays [21]. Cette séquence se termine par C27661U (qui modifie l’acide aminé Gln90 en une fin de traduction prématurée, près de l’extrémité carboxy-terminale de la protéine Orf7a). Cette protéine virale est présente dans le réticulum endoplasmique, l’appareil de Golgi et l’espace péri-nucléaire [22]. Plusieurs variantes ont été identifiées au cours de l’épidémie [23]. Fait remarquable, plusieurs délétions ont été isolées dans le gène, ce qui suggère que la fonction de cette région n’est pas essentielle [24]. Cependant, beaucoup de ces mutations, comme celle dont il est question ici, maintiennent intact le petit gène de la protéine hydrophobe Orf7b en aval de l’Orf7a. Cette très petite protéine est présente dans l’appareil de Golgi et se retrouve aussi dans le virus purifié [25]. Il faut noter qu’elle est synthétisée in vivo via un changement de cadre de lecture qui recouvre le codon de fin de traduction du gène Orf7a (…GAA TGA TT…devient …GA ATG ATT…). Cela peut être interprété comme la concurrence dans cette région entre la traduction de l’Orf7a et de l’Orf7b, créant un conflit coût / bénéfice pour l’expression de l’une ou l’autre de ces protéines. Il sera donc important de surveiller la descendance future du virus dans cette région car elle pourrait donner lieu à des formes atténuées intéressantes.Exemple 1.
Une autre succession des mutations qui conduit à l’arrêt prématuré de la traduction d’une protéine virale commence avec G11083U (protéine Nsp6, Leu37Phe). Cette mutation est désormais largement répandue dans le monde entier. Elle est susceptible de favoriser une liaison plus stable de la protéine au réticulum endoplasmique (RE), ce qui pourrait augmenter l’infection par le coronavirus en compromettant le transfert des composants viraux aux lysosomes où ils sont dégradés [26] ; ensuite, nous avons G1397A (Nsp2, Val378Ile), susceptible là encore de favoriser la propagation du virus [27], suivi par G29742U (3′UTR du virus), et U28688C (synonyme) ; puis nous avons le couple de mutations C884U (Nsp2 à nouveau, Arg207Cys [27]) et G8653U (Nsp4, essentiel pour l’assemblage de l’enveloppe [28]). La mutation Met2796Ile correspondante est située à la frontière du domaine de la protéine situé dans le lumen du RE. On sait que, pour fonctionner correctement, le RE nécessite la présence d’oxygène [29], et les dérivés réactifs de l’oxygène (DRO) sont associés à un repliement anormal des protéines dans ce compartiment. La protéine Nsp4 contient un certain nombre de résidus cystéine, susceptibles d’être oxydés. Le rôle de la méthionine dans la protéine originelle pourrait être d’agir comme un tampon contre les DRO, de sorte que le mutant serait sans doute atténué ; cette mutation est suivie par A19073G (dans le domaine méthylase de la protéine Nsp14, Asp1869Gly, position qui a déjà évolué depuis le virus SARS-CoV-1 [30], et donc probablement plus ou moins neutre), puis par un couple comprenant la mutation entraînant la fin de la traduction : G27915U, Gly8 vers fin de traduction de l’extrémité N-terminale de l’Orf8 et C29077U (synonyme) ; la succession se termine par un couple de mutations conduisant aux changements synonymes C19186U et G23608U. Cette région des coronavirus liés au SRAS est hypervariable. Elle change au cours des épidémies, ce qui montre qu’elle est soumise à une pression de sélection permanente produisant parfois deux peptides Orf8a et Orf8b [31]. Elle correspond à des protéines exprimées à la fin du cycle d’infection. Il sera important de les suivre en fonction de l’évolution de la virulence du virus. Il s’agit d’une ramification de l’arbre évolutif apparue dans quatre pays différents et dans sept échantillons, sur une période de six semaines entre la première et la dernière mutation.Exemple 2.
Nous avons ici une succession de mutations qui commence à l’extrémité 5’ du génome du virus, C241U, suivie de la mutation C14408U (Pro314Leu) à l’extrémité d’un doigt de zinc dans la réplicase Nsp12, et qui apparaît dans de nombreuses branches de l’arbre d’évolution du virus. Elle est examinée en détail plus bas (origine des efflorescences). Cette mutation est suivie par A23403G (Asp614Gly), mutation largement répandue de la protéine de la spicule de la capside virale (à nouveau discutée plus loin), C3037U (synonyme), la mutation G25563U (Gln57His) dans Orf3a formant des canaux potassiques est supposée interférer négativement avec la fonction de la protéine [32], C1059U (Thr265Ile) dans la protéine Nsp2, discutée plus haut, et le triplet G4181A (Ala1306Thr) dans le domaine SUD-N de la protéase Nsp3, puis les mutations G4285U (Glu1340Asp), et G28209U qui entraîne une fin de traduction après le glutamate 106 de la protéine Orf8. Comme discuté précédemment, de nombreuses mutations, y compris des délétions dans la protéine Orf8, ont souvent été observées. Cela indique une fois de plus que l’évolution de ces régions doit être suivie avec attention pour rechercher des formes atténuées du virus. Cette mutation spécifique induisant une fin de traduction est très significative car elle a été trouvée dans un échantillon de Croatie, un autre de Thaïlande, sur deux branches largement séparées et avec un mois de différence. La séquence de mutations discutées ici correspond ici à l’échantillon de Thaïlande.Exemple 3.
5.2. Inversion de la tendance du génome viral à perdre ses résidus cytosine
Nous avons ici retenu deux exemples d’une situation où, à partir d’un point de branchement amont, il apparaît que la descendance du virus perd moins ses cytosines, et peut même tendre à en regagner. Ces exemples sont les suivants (Figure 4).
Dans le jeu de données numéro 1, il s’agit de deux sous-arbres, le premier concernant plutôt des pays d’Asie et ayant pour racine le nœud associé aux mutations M2 et M3. Le second contient des échantillons d’Amérique du Nord et d’Océanie, et son nœud racine est lié aux mutations M6 et M7. Le premier arbre naît de la succession de mutations C8782U (synonyme), U28144C (Leu84Ser) dans la protéine Orf8, dont la fonction a été discutée plus haut et définit un clade important de variants du virus [23], C24034U (synonyme), et enfin du doublet U26729C (synonyme), G28077C (Val62Leu), dans la protéine Orf8 à nouveau. Comme il s’agit de l’origine du phénomène observé on est amené à penser que c’est l’altération du rôle de l’Orf8 (8a ou 8b) qui en est responsable. La région de l’Orf8 est particulièrement variable et a été clairement impliquée dans la transmission entre espèces [33]. Une hypothèse répandue est que l’altération du gène correspond à une perte de fonction active chez les ancêtres chiroptères [34]. Comme ceux-ci sont généralement plus riches en cytosine que les formes humaines [20], on peut se demander si l’une des fonctions de cette protéine ne serait pas de moduler l’activité de la CTP synthase.
De fait, le second provient de la même filiation, à laquelle s’ajoutent les mutations U490A (Asp75Glu) dans la protéine Nsp1, qui contrôle la traduction spécifique de l’ARN viral [35], systématiquement associée à la mutation, C3177U (Pro971Leu) dans le domaine acide, sans fonction bien identifiée, de la protéase multifonctionnelle Nsp3 [36], puis pour finir le doublet U18736C (Phe1757Leu) de l’exonucléase, N7-méthyltransférase Nsp14, et A29700G dans la région 3′UTR du virus. La modification Phe1757Leu se trouve au milieu d’un site de liaison au zinc à l’interface entre les deux domaines de la protéine Nsp3. On peut donc penser que cette mutation pourrait changer de façon subtile le mécanisme de correction des erreurs de réplication, d’une façon qui accommoderait moins bien l’entrée de l’UTP en face d’un A dans la matrice virale négative. Ensuite, 3 des 5 échantillons ayant acquis le plus de C l’ont fait à travers une transition de U vers C. Le premier, HongKong/HKPU1_2101, met en évidence deux transitions simultanées en positions 12877 et 23857. Ces mutations étant synonymes, elles sont peu susceptibles de changer le mécanisme de réplication correction. Le second, USA_SC_3571, et le troisième, Australia/VIC209, montrent des transitions du même type, elles-aussi synonymes, respectivement en positions 11635 et 19713. Enfin, les deux derniers échantillons, USA/WI-33 et USA/WI-28, sont issus de la transversion de A en C en position 29567, mutation à la fin de l’ORF9b.
Deux sous-arbres du premier jeu de données où la tendance du génome à perdre ses résidus cytosine est inversée. Partie supérieure de la figure. Premier sous-arbre. Les sous-échantillons affichés sont ceux ayant acquis le plus de C, en dehors de quelques échantillons esseulés sur d’autres branches. Le nœud siège de la mutation M1 suit directement ceux respectivement associés aux mutations C8782U et U28144C. Partie inférieure de la figure. Second sous-arbre. On retrouve dans cet arbre une majorité de souches ayant un solde neutre de C (autant gagnés que perdus), ainsi que 3 souches en ayant plus acquis que perdus.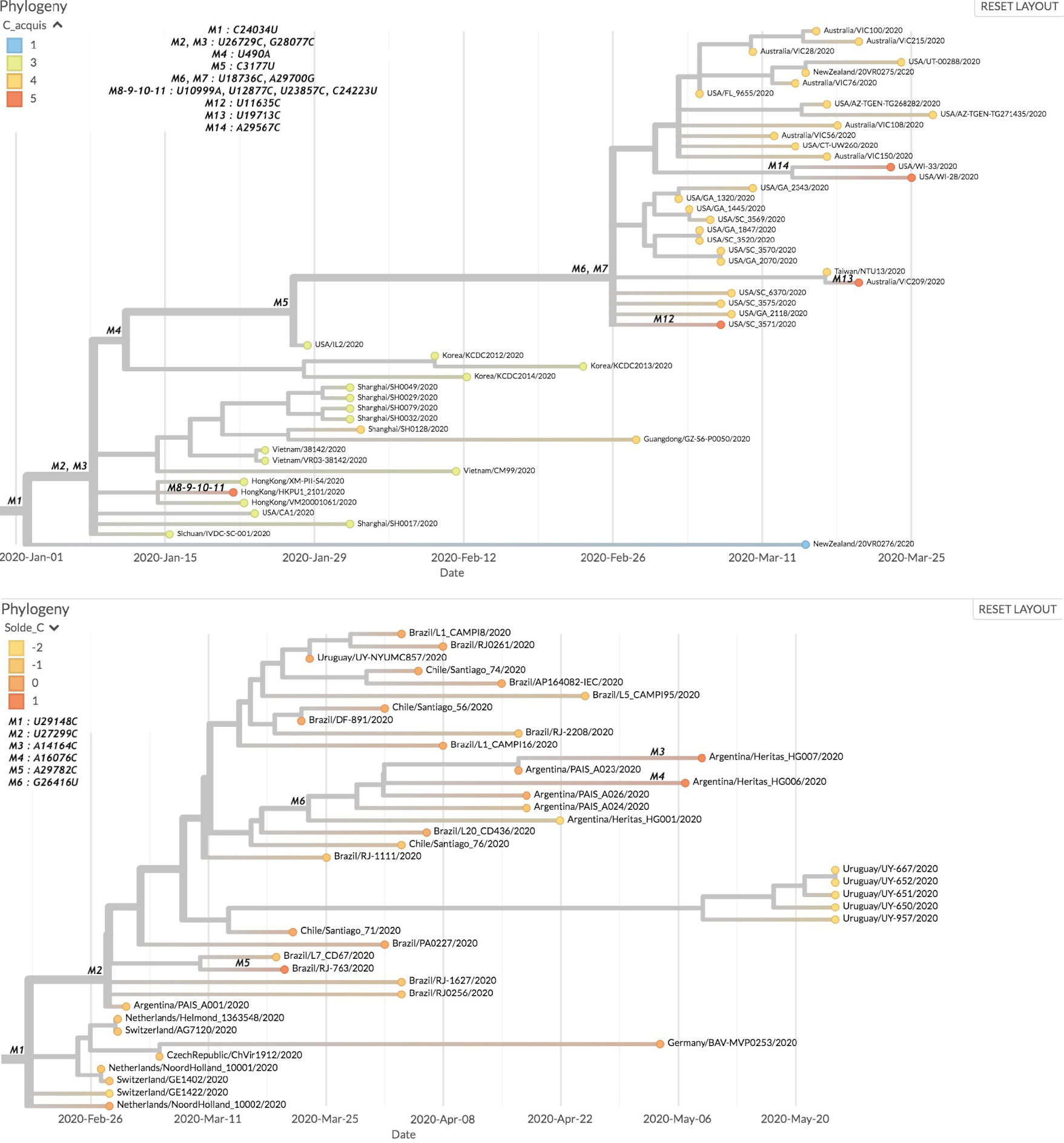
Pour le jeu de données numéro 2, cette inversion de tendance concerne majoritairement des souches d’Amérique latine. La succession de mutations C241U, C14408U, puis A23403G discutées à propos de la genèse de codons de fin de traduction dans les gènes du virus, est suivie de C3037U (synonyme), et du triplet G28881A, G28882A, G28883C, chevauchant les codons en position 203-204 du gène de la nucléocapside N, ce qui transforme un dipeptide arginine-glycine en lysine-arginine. Cela modifie la charge positive de la protéine et peut contribuer à améliorer son rôle dans l’assemblage du génome du virus dans la capside, comme discuté plus bas à propos de l’apparition d’efflorescences (36). Après cette triple modification on constate plusieurs inversions de la tendance à perdre le C dans le génome. On trouve U29148C (Ile292Thr) à nouveau dans le gène de la nucléocapside N, puis U27299C (Ile33Thr) dans le gène de l’Orf6, ce qui débouche sur un ensemble d’échantillons ayant au pire autant gagné de C qu’ils n’en ont perdu (Brazil/RJ-763, Argentina/Heritas_HG007, Argentina/Heritas_HG006). On trouve aussi 3 échantillons parmi les 39 du sous-arbre qui ont gagné un C de plus qu’ils n’en ont perdu. A chaque fois, la dernière acquisition de C provient d’une transversion à partir d’une adenine (en positions 14164 (Met233Leu), 16076 (Asp870Ala), et 29782, dans la fin 3′UTR du génome viral. Dans l’ensemble c’est le changement dans la nucléocapside qui paraît le plus propice à l’inversion de tendance à perdre le C. En effet, cette protéine, exprimée à un niveau élevé au cours de l’infection, régule le processus de réplication / transcription du virus et cela pourrait expliquer cette remarquable observation [38].
Exemple d’efflorescences. Le sous-arbre illustré contient 10 des 20 efflorescences les plus significatives au sens de la méthode que nous avons utilisée. Le nœud N0 est le siège de la mutation C14408U. 50 pays différents sont représentés sur le sous-arbre descendant de N0. La sous-branche supérieure est marquée par une prédominance marquée d’échantillons nord-américains (USA et Canada, points rouges).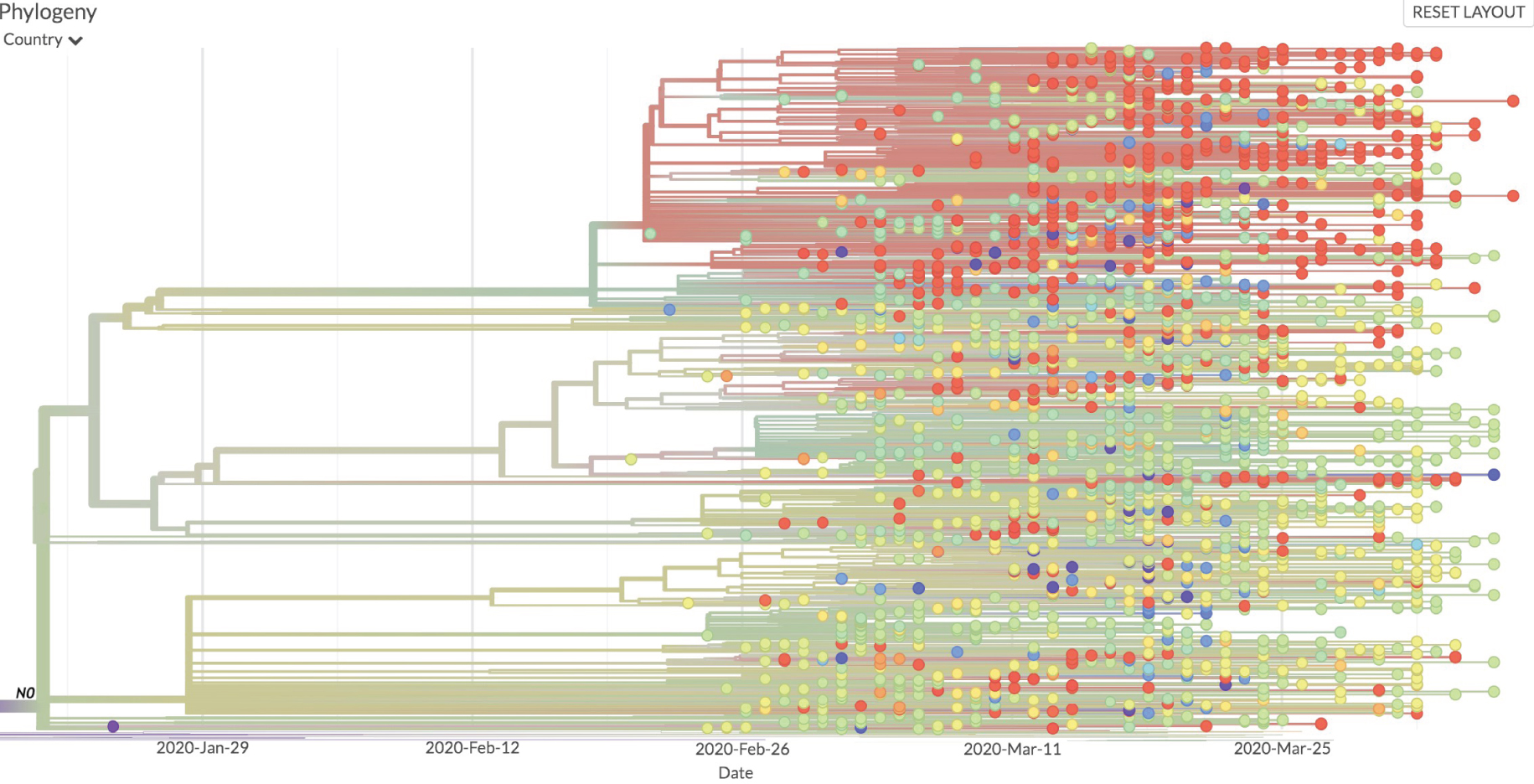
5.3. Apparition d’efflorescences
La succession C3037U, (C241U, A23403G), C14408U est présente en amont de 12 sous-arbres, que nous avons considérés comme significatifs (voir Figure 5 et Matériels et Méthodes).
La mutation synonyme C3037U est située à l’extrémité du domaine 1 de type ubiquitine de la protéine Nsp3. Cela conduit à une séquence (UUUUUU) qui favorise les changements dans le cadre de lecture et pourrait diminuer l’efficacité de traduction des protéines de la région ORF1a. La mutation C241U est très souvent observée [39]. Elle se trouve dans la région qui démarre la réplication du virus. On peut donc supposer que cela peut modifier la fréquence de réplication. A23403G est une mutation non synonyme largement répandue qui conduit au remplacement d’un aspartate par une glycine à la position 614 de la spicule qui sert à la liaison du virus à son récepteur cellulaire. Pour cette raison, plusieurs analyses ont suggéré que cette mutation a un rôle important dans la propagation du virus [40, 41]. Ici, le fait qu’elle fasse partie d’une efflorescence majeure peut être considéré comme un argument supplémentaire en faveur de cette interprétation. Le changement C14408U modifie une proline en une leucine (Pro314Leu) juste après la fin du domaine NiRAN (domaine nucléotidyl transférase de la réplicase RdRp associée aux nidovirus) de la protéine Nsp12 se terminant par un “doigt de zinc”. Le domaine NiRAN, essentiel pour la réplication du virus, agit comme une nucléotidyltransférase, préférant l’UTP comme substrat pour une fonction qui n’a pas encore été clarifiée [42]. La proline modifiée dans le mutant fait partie d’un dipeptide diproline qui joue le rôle de charnière de séparation entre le domaine NiRAN et le domaine suivant.
Une deuxième efflorescence, qui partage plusieurs éléments avec la précédente, commence avec la même séquence C3037U, (C241U, A23403G) et C14408U. Cependant, elle se poursuit par une série de mutations contiguës entraînant une modification (G28881A, G28882A, G28883C) de la nucléocapside N, comme nous l’avons vu. Il peut être intéressant de remarquer que ce changement pourrait être impliqué dans l’assemblage du génome du virus dans la capside par séparation de phases [37]. Cela pourrait accroître l’efficacité de la transmission du virus et ainsi contribuer à la formation d’efflorescences. Le fait qu’il s’agisse d’un groupe de mutations impliquant G est intriguant. Il peut résulter du fait qu’il recouvre une séquence GGGG.
Nous avons vu précédemment que la mutation G11083U (protéine Nsp6, Leu37Phe) a démarré une autre succession de mutations qui ont conduit à l’arrêt prématuré de la traduction d’une protéine virale. Ici, cette mutation largement répandue est à l’origine d’efflorescences. Comme on l’a vu, elle favorise peut-être l’infection par le coronavirus en compromettant le transfert des composants viraux aux lysosomes pour leur dégradation. Cela favoriserait certainement les efflorescences. Cette mutation est suivie, dans une première succession génératrice d’efflorescences, par G26144U (Gly251Val) dans la protéine Orf3a, qui forme des canaux potassiques importants pour la réponse de l’immunité innée - mais la fonction exacte de la protéine reste encore à déterminer [43]. Les mutations ultérieures sont C14805U (synonyme) et U17247C (synonyme). Cette succession suggère que la première mutation de la protéine Nsp6 et peut-être la seconde sont les causes principales de l’efflorescence [26]. Le rôle de la première mutation est confirmé par une deuxième succession génératrice d’efflorescences, où elle est suivie d’un quadruplet : C6312A (Thr2016Lys), dans une région inter-domaine qui précède le domaine G2M de la protéase multi-domaine Nsp3, puis associée à trois mutations C → U, qui devraient donc être plus fréquentes : C13730U (Ala88Val) dans le domaine NiRAN de la protéine Nsp12, C23929U (synonyme), et enfin C28311U (dans une suite de quatre C, Pro13Leu) au début de la protéine de nucléocapside, N.
Une deuxième suite de mutations qui aboutit à des efflorescences est C8782U (synonyme), U28144C (Leu84Ser) dans la protéine Nsp8 déjà notée à deux reprises et définissant un clade spécifique du virus [23], aboutissant à C26088U (synonyme). La mutation Leu84Ser co-évolue de manière significative avec les mutations Asp614Gly de la protéine de pic dont il a été question plus haut [36], ce qui en fait un autre candidat probable pour une sélection positive conduisant à une propagation accrue du virus, donc aux efflorescences.
5.4. Changement dans la fréquence des transitions / transversions
Parmi les mutations en amont de branches montrant des changements significatifs dans le flux de transition / transversion, on trouve la mutation C17747U, qui modifie un résidu proline en un résidu leucine dans la protéine Nsp13 (voir Figure 6 et Matériels et Méthodes).
Une des lignées considérée comme significative concernant le changement du processus d’évolution moléculaire. La lignée issue de la mutation C17747U est représentée en jaune, et son processus d’évolution est modélisée par un modèle TN93 à 6 paramètres (processus 1). Le reste de l’arbre (feuilles bleues) est modélisé par un modèle TN93 à 3 paramètres.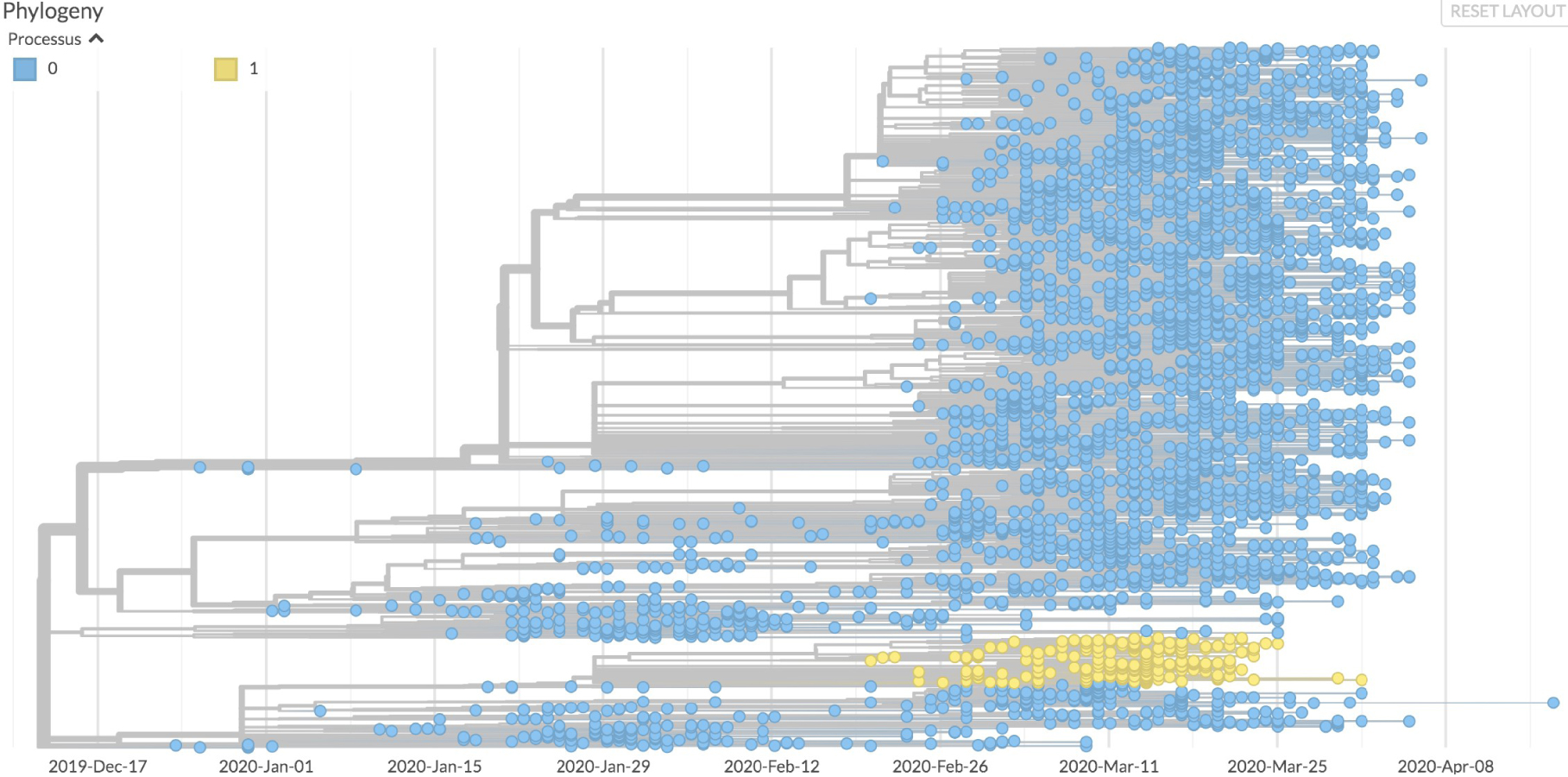
Cette mutation affecte le domaine de la protéine qui a une activité nucléoside triphosphatase, dont le rôle exact n’est pas connu mais en accord avec une activité de correction [44]. On peut imaginer qu’elle est impliquée dans le contrôle de qualité du produit de la réplication du virus via la stabilisation de la forme « anti » des nucléotides, évitant ainsi le mésappariement conduisant aux transversions. De fait, cette protéine a été identifiée parmi celles qui conduisent à une altération significative de la diversité du génome viral [15]. L’existence d’un changement notable dans le type de mutations en aval de l’arbre est donc un argument fort pour le rôle discriminateur de la région correspondante de la protéine. Par ailleurs, dans la mesure où cette mutation augmente la fréquence de mutations d’une façon biaisée, on peut s’attendre à ce que la lignée qui s’en suit conduise à une atténuation du virus. Cependant, comme cela change le paysage évolutif cette évolution pourrait conduire à des mutations “innovantes” modifiant la pathogénicité du virus, et cela surtout dans des conditions où la recombinaison due à des co-infections serait favorisée. Il s’agit d’un argument de plus pour choisir une politique de santé publique qui tend à éviter la formation de grappes d’infection.
6. Conclusions et perspectives
L’épidémie de COVID-19 est une expérience grandeur nature sur l’évolution des virus. Il est remarquable que nous ne connaissions pas la véritable origine du virus [45], ni où il nous mènera. Cela explique pourquoi la grande majorité des études sur le virus SARS-CoV-2 et son évolution sont essentiellement descriptives. Ici, nous avons essayé d’utiliser l’évolution continue du virus pour étudier certaines de ses contraintes connexes en utilisant une approche de modélisation probabiliste de l’évolution moléculaire du virus basée sur l’hypothèse que le virus est assujetti à son hôte. En nous fondant sur la structure métabolique des cellules hôtes, qui agit comme cadre matériel obligatoire pour la multiplication des particules virales, nous avons mis en évidence des changements spécifiques dans le schéma d’évolution de la descendance du virus, dont témoignent les modifications de la composition du génome viral au fil du temps. En utilisant comme ligne de base le changement de C vers U largement répandu dans la composition de ce génome, nous avons identifié des nœuds où le changement est déplacé de cette direction à une autre, favorisant les transversions plutôt que les transitions, inversant la tendance de l’enrichissement pour aller de U vers C, ou faisant naître des efflorescences avec l’apparition soudaine de branches multiples dans l’arbre d’évolution. Cela nous a permis de mettre en évidence une série de fonctions qui évoluent vers une propagation plus efficace du virus (par exemple, la mutation Asp214Gly de la protéine de la spicule virale précédemment identifiée, mais aussi la mutation Gln57His du canal potassique Orf3a). Nous avons encore remarqué que l’Orf8 est le site probable d’une compétition permanente pour l’expression de deux protéines chevauchantes Orf8a et Orf8b formées aux dépens du décalage du cadre de lecture au cours de la traduction. De même la région instable de l’Orf7 pourrait favoriser la synthèse de la très petite protéine membranaire Orf7b, dont la fonction reste inconnue à ce jour. Enfin, l’inversion de la tendance à favoriser U par rapport à C indique que la protéine N pourrait être impliquée dans le contrôle de la synthèse de CTP chez l’hôte, ce qui suggère une cible intéressante pour le contrôle futur du développement du virus. Nous espérons que cette combinaison de connaissances mathématiques et biochimiques nous aidera à concevoir d’autres entreprises contre les conséquences désastreuses de COVID-19. Nous avons remarqué que l’un des moyens possibles pour le virus d’échapper au contrôle dépendant du CTP dans les cellules serait d’infecter des cellules qui ne sont pas censées se multiplier, comme les neurones. Cela pourrait expliquer les sites de développement viral inattendus observés dans l’épidémie actuelle.
7. Matériels et méthodes
7.1. Traitement des données
Un total de 4792 séquences du virus SARS-CoV-2 ont été récupérées à partir de la banque GISAID [46] à la date du 17 Avril 2020 pour le premier jeu de données. Seuls les génomes des virus d’hôtes humains du SARS-CoV-2 d’une longueur supérieure à 25 000 pb ont été retenus. Les séquences dont la date d’échantillonnage était insuffisamment renseignée (absence du jour de collecte, parfois du mois) ont également été écartées. Pour les séquences présentes de multiples fois, seul le premier isolat a été retenu. Nous avons aussi réutilisé le travail des équipes de Nextstrain et écarté les échantillons trop divergents ou instables qu’ils avaient eux-mêmes laissés de côté (github.com/nextstrain/ncov/blob/master/defaults/exclude.txt). La séquence de 26 régions codantes (Nsp1, Nsp2, Nsp3, Nsp4, Nsp5, Nsp6, Nsp7, Nsp8, Nsp9, Nsp10, Nsp11, Nsp12, Nsp13, Nsp14, Nsp15, Nsp16, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N et ORF10) a été caractérisée en utilisant NC_045512 comme référence. Le total des séquences retenues à l’issue du traitement est de 4088 séquences. Un second jeu de données de 3246 séquences, dont 510 sont communes avec le premier jeu de données, a été récupéré le 6 Juillet 2020 en utilisant directement l’API de Nextstrain [47].
Nous notons ici qu’au fil du temps, la disponibilité des données n’a cessé d’être altérée, certaines séquences étant retirées des échantillons, tandis que d’autres entraient dans la base de données. En outre, il était généralement difficile d’extraire de grands échantillons de séquences, de sorte qu’il était extrêmement difficile de constituer un dépôt de données cohérent où des approches statistiques robustes pouvaient être mises en œuvre. Il semble très malheureux que la majeure partie des séquences d’un virus d’importance mondiale n’ait pas été mise à disposition dans la base de données internationale des séquences, malgré les recommandations des principales institutions de recherche [48, 49].
7.2. Reconstruction phylogénétique
Le processus de reconstruction commence par un alignement de l’ensemble des séquences sur la séquence de référence. Il n’était pas question ici de prendre en compte les insertions et suppressions de nucléotides, seules les potentielles substitutions ont été étudiées. Nous avons utilisé le programme MAFFT [50] pour créer ces alignements. Certaines positions ambigües peuvent être mises en évidence lors du processus d’alignement. Par exemple, certaines régions du génome peuvent présenter une forte instabilité et une grande variabilité en fonction des paramètres de l’algorithme utilisé pour l’alignement. Pour pallier ce problème, nous avons utilisé les mêmes masques que ceux utilisés par l’équipe de Nextstrain. Les sites 18529, 29849, 29851, 29853, ainsi que les 130 premiers et 50 derniers sites du génome ne sont ainsi pas considérés dans le processus de substitution. Nous avons ensuite eu recours à un modèle General Time Reversible (GTR) pour inférer le processus de substitution en jeu à l’aide du logiciel IQTREE [51]. Ce premier arbre est une version assez brute qui ne prend pas en compte l’aspect temporel de l’évolution. Le logiciel Treetime [52] permet de raffiner cet arbre en prenant aussi en compte les dates d’échantillonnage des séquences. Il reconstruit alors l’arbre ayant le maximum de vraisemblance par rapport aux séquences échantillonnées. Il infère aussi par maximum de vraisemblance les compositions des séquences ancestrales des échantillons, ainsi qu’un intervalle de confiance à 90% autour de la date la plus vraisemblable de ces ancêtres communs. Une fois l’arbre constitué, il nous était alors possible de reconstituer l’ordre d’apparition des mutations de chaque échantillon au sens du maximum de vraisemblance. Pour la visualisation de l’arbre et la production des Figures 2 à 6, nous avons utilisé le programme Auspice développé par Nextstrain, auquel nous avons apporté quelques modifications pour afficher les grandeurs auxquelles nous nous sommes intéressés. Nous avons pour cela développé un script Python permettant de modifier le fichier JSON utilisé en entrée par le programme Auspice (accessible sur demande). Cela nous a permis d’enrichir les capacités de visualisation du logiciel en y ajoutant des grandeurs comme le nombre de C acquis ou perdus par un échantillon par rapport à la référence et de générer des présentations de l’arbre originales.
7.3. Identification des efflorescences
Le principal écueil auquel nous avons dû faire face lors de la sélection des efflorescences était le biais de sélection des échantillons de l’arbre phylogénétique. Ainsi, certains hôpitaux étaient susceptibles d’échantillonner plus que d’autres du fait des politiques sanitaires et des moyens mis en œuvre différents selon les pays. Afin d’éviter de sélectionner des nœuds susceptibles de présenter une efflorescence en raison d’un suréchantillonnage, nous avons choisi de développer une méthode statistique sur mesure.
On appellera sous-arbre tout ensemble de nœuds et de feuilles prenant pour racine l’un des nœuds de l’arbre principal. L’idée est d’exploiter l’information fournie par l’identité des pays représentés dans chaque sous-arbre : plus une souche se transmet facilement, plus le nombre de pays dans lequel on s’attend à l’observer est élevé. Pour mettre en oeuvre cette heuristique, il faut contrôler deux facteurs : la taille de l’arbre (deux arbres de profondeurs inégales, c’est-à-dire enracinés à des dates différentes, montrent naturellement des diversités de pays différentes) et l’hétérogénéité d’échantillonnage (des pays où l’échantillonnage et le séquençage sont effectuées avec des intensités différentes ont des probabilités différentes d’apparaître dans un sous-arbre donné).
Ces deux facteurs interagissent, car la taille d’un arbre (le nombre de ses feuilles par exemple) varie évidemment avec l’intensité d’échantillonnage. Une manière de contrôler cette interaction est de mesurer la taille d’un arbre par sa longueur totale, ou somme des longueurs de branches, en unités de temps. En effet, cette observable est peu sensible aux effets de suréchantillonnage car la présence de nombreuses séquences échantillonnées au même endroit à peu près au même moment engendre un sous-arbre dont la longueur est proche de zéro.
Pour contrôler l’effet du facteur longueur L sur le nombre de pays représentés, N, nous cherchons à apprendre la relation N = f(L) dans un arbre typique afin de pouvoir ensuite identifier les sous-arbres dont le nombre de pays représentés, pour une longueur connue L, excède l’attendu f(L). Un modèle statistique simple consiste à supposer que le nombre d’apparitions du pays i dans un arbre de longueur L est poissonnien de paramètre 𝜃i L et que ces nombres sont indépendants. Si K est le nombre total de pays référencés par Nextstrain, le nombre de pays N représentés dans un arbre de longueur L est donc la somme de K variables de Bernoulli indépendantes de paramètres 1 −exp(−𝜃i L). Si l’on suppose par exemple que les pays se divisent en deux groupes, les k1 “fréquents” d’intensité 𝜃1, et les k2 “rares” d’intensité 𝜃2≪𝜃1, N a pour moyenne K − k1 exp(−𝜃1 L) − k2 exp(−𝜃2 L), qui se comporte lorsque L est grand comme K − k2 exp(−𝜃2 L).
De plus, lorsque L est grand, en supposant que 𝜃2 L = O(1), la distribution de N est approximativement égale à k1 + N2, où N2 est poissonnien de paramètre k2(1 −exp(−𝜃2 L)).
Ainsi, nous avons utilisé la paramétrisation :
N = a − b exp(−cL), en interprétant les paramètres comme suit : a est le nombre maximal de pays, b est le nombre de pays dont les intensités d’échantillonnage/séquençage sont faibles et c est une densité de présence de ces pays par unité de longueur de l’arbre. Sous l’hypothèse nulle, N est distribuée comme a − b + N1, où N1 suit la loi de Poisson de paramètre b(1 −exp(−cL)). Nous avons enfin sélectionné les 20 efflorescences les plus significatives, c’est-à-dire celles dont le comportement déviait le plus de celui attendu par notre estimateur. Cela nous a alors permis de reconstituer les lignées ainsi que les mutations apparues successivement en amont de chaque nœud au niveau duquel une efflorescence avait eu lieu. Par ce biais, nous avons pu mettre en évidence les successions de mutations communes à certains de ces nœuds et donc celles donnant lieu à une majorité d’efflorescences statistiquement significatives. Par ailleurs, nous avons restreint la sélection automatique des nœuds de sorte qu’aucun nœud sélectionné ne soit présent dans la lignée d’un autre. Les efflorescences sélectionnées sont donc mutuellement indépendantes, même si elles peuvent bien évidemment avoir des ancêtres communs. Pour arbitrer le choix entre deux nœuds présents dans une même lignée, nous avons systématiquement conservé le nœud le plus ancien, et donc l’arbre le plus fourni.
Évolution moléculaire et cliquet de Muller
La biologie repose sur les lois de la physique. Parce que les processus biologiques se déroulent autour de 300 K, la vie est soumise au stress universel du bruit thermique, impliquant une énergie qui ne diffère pas beaucoup de celle qui régit les liaisons chimiques de la chimie biologique. Il s’ensuit que les réactions qui se produisent et organisent les êtres vivants ne peuvent pas se développer avec une stricte reproductibilité. Des erreurs inévitables font que le produit d’une réaction diffère de ce qu’il est censé être. La réplication du génome ne peut échapper à cette contrainte. Il s’ensuit que, dans la descendance d’un virus, il y a toujours un certain nombre de variants, appelées mutants lorsqu’ils portent des altérations du génome. Dans la plupart des cas, ces mutants correspondent au passage d’un des quatre nucléotides à un autre. Ce processus, en gros, est aléatoire — la position de la mutation peut être n’importe où dans le génome, et le remplacement d’un nucléotide par l’un des trois autres. Au fil du temps, tous les nucléotides du génome sont susceptibles de se transformer en d’autres nucléotides. Cela affectera les fonctions nécessaires à la multiplication du virus, et certains changements continueront à se propager (fixation), tandis que d’autres finiront sans descendance. Une mutation suivie dune fixation est appelée substitution. La substitution d’une purine par une pyrimidine (ou vice versa) est appelée transversion; les autres substitutions sont appelées transitions. La probabilité qu’une mutation particulière revienne à l’état ancestral est très faible. Cela oblige l’évolution à toujours aller de l’avant, sans possibilité de revenir en arrière. Ce processus a été remarqué en 1932 par Hermann Muller dans le cas particulier des effets de l’irradiation sur la mutagenèse. Sa réflexion a depuis été simplifiée et popularisée. Elle est connue sous le nom de “cliquet de Muller” [54]. Il est évidemment très probable que la majorité des mutations conduisent à la perte partielle ou totale des fonctions codées par les régions altérées du génome. Il s’ensuit que cela conduit généralement, à long terme — mais pas à court terme — à l’atténuation des fonctions permettant la multiplication et la virulence des espèces pathogènes. C’est pourquoi Louis Pasteur et ses successeurs ont eu la chance d’isoler des organismes atténués qui, dans certains cas — rares —, ont pu être utilisés pour la vaccination des personnes infectées [55]. Cependant, ce processus devient improductif dès que la co-infection avec différents mutants se produit dans des circonstances où la recombinaison est possible. Deux mutants différents peuvent se recombiner dans la forme ancestrale de l’agent pathogène et effacer tout le bénéfice de l’atténuation. Cela est d’autant plus dommageable que les anciennes formes sont aussi, très souvent, celles qui se propagent le plus facilement.
Légende de la figure. Cliquet de Muller et recombinaison. La figure est extraite de la référence [56]. Les gènes (majuscules) sont mutés au hasard sous une forme différente (minuscules). Les mutations s’accumulent comme un cliquet car la probabilité de retour à la forme parentale est négligeable. Cela se produit indépendamment pour les virus de descendance différente. Cependant, si des virus de descendance différente se trouvent dans la même cellule, ils peuvent se recombiner. Cela leur permet de recréer la forme ancestrale du virus.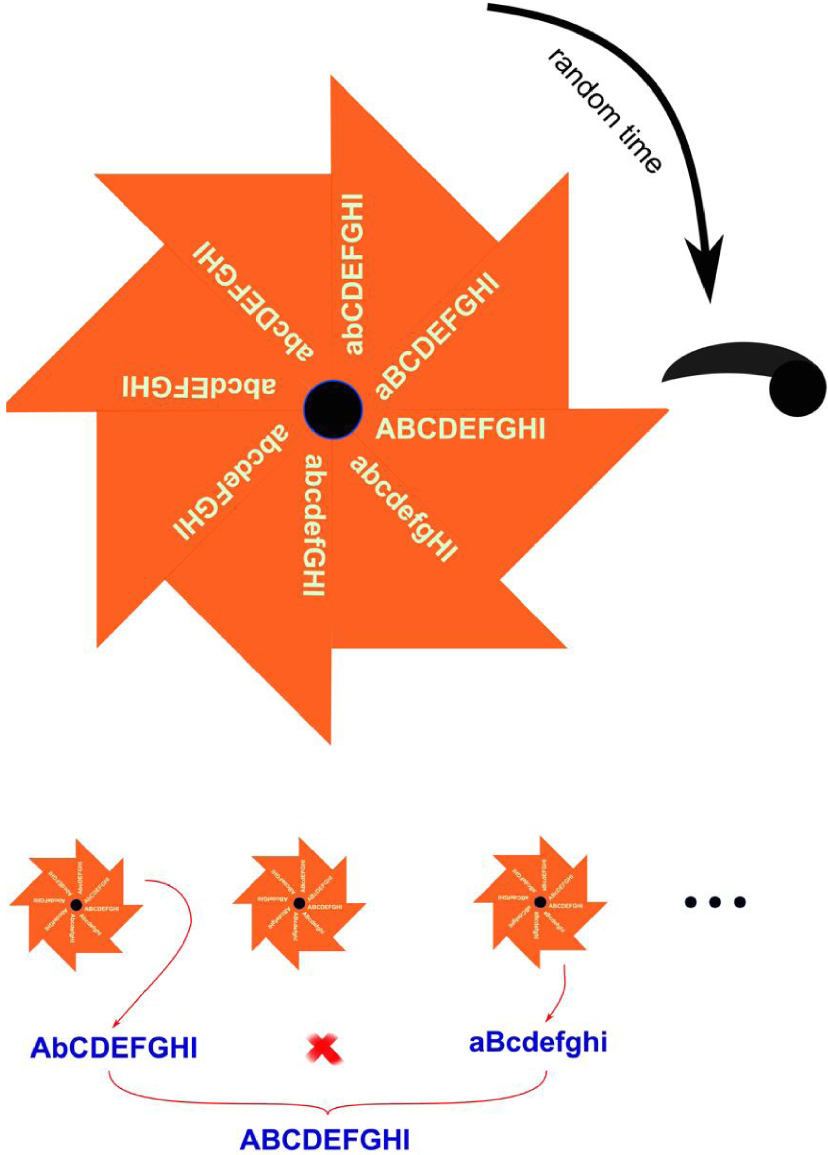
7.4. Détection de changements dans le processus d’évolution moléculaire
Nous avons cherché à déterminer si, dans certains sous-arbres, le processus de substitution se comportait de manière statistiquement différente de ce qui est observé dans le reste de l’arbre. Pour cela, nous avons utilisé le modèle classique TN93 de Tamura et Nei [53] à 3 paramètres (taux de transition des purines, taux de transition des pyrimidines et taux de transversions), et avons permis à ces trois taux de prendre, en aval d’un nœud candidat Ni, des valeurs différentes de celles qu’ils prennent dans le reste de l’arbre. Nous avons ensuite utilisé un second modèle (à 6 paramètres) imbriqué dans le premier (à 3 paramètres), avec comme statistique de test le ratio de vraisemblance 2Δl = 2(l1 − l0), où l0 est la log-vraisemblance sous l’hypothèse H0 (modèle TN93 à 3 paramètres estimant l’ensemble des éléments de l’arbre) et l1 est la log-vraisemblance sous l’hypothèse H1 (modèle TN93 à 6 paramètres avec une différenciation locale des paramètres en aval de chaque nœud retenu). Nous comparons alors le ratio de vraisemblance à une distribution du 𝜒2 à 3 degrés de liberté, dont le seuil de significativité à 5% est de 7.81. Nous pouvons dès lors identifier les nœuds à partir desquels le processus d’évolution varie significativement et quantifier les variations des différents taux de substitution, c’est-à-dire les nœuds pour lesquels nous pouvons rejeter l’hypothèse H0 selon laquelle le modèle TN93 à 3 paramètres produit de meilleures estimations des taux de substitution de l’arbre que le modèle TN93 à 6 paramètres. Nous avons choisi d’implémenter nous-mêmes ces modèles en Python, de manière à disposer de cette flexibilité de paramétrage. Le script permet de déterminer l’ensemble des nœuds et feuilles présents en aval d’un nœud d’intérêt et de réaliser le test d’hypothèse en calculant le ratio de vraisemblance et les différents taux de substitution.
Remerciements
AL remercie le Centre Interdisciplinaire de Recherche en Biologie (CIRB, Collège de France) pour son financement, ainsi que les membres de l’équipe SMILE (Stochastic Models for the Inference of Life Evolution) du CIRB pour de nombreuses discussions fructueuses au sujet de la modélisation de l’épidémie de COVID-19. AD remercie Stellate Therapeutics pour le soutien de son laboratoire.
English version
1. Introduction
The development of the COVID-19 pandemic is being explored in a myriad of articles. Despite this abundance, and because of our anthropocentrism, it is exceptional that these studies focus on the virus’ standpoint. Of course, much work is looking into the details of the composition and structure of the SARS-CoV-2 virus genome, the proteins it codes for and its animal-infecting relatives. However, there are very few major studies on how the virus exploits the metabolism of its host’s cells. The urgent necessity to contain the disease led investigators to emphasize vaccination or, more generally, the involvement of the host’s immune system. It is well known, alas, that while it has sometimes been relatively easy to generate a vaccine that is both effective and harmless against a widespread disease, the opposite is also true. There are still very serious and very common diseases for which there is no vaccination. Vaccinating effectively assumes, in particular, that the progeny of a pathogen remains the same long enough to prevent escape of the immune response triggered by the vaccine. Coronaviruses are viruses made up of a long genome and an envelope. The length of the genome could have led to a very high mutation rate, but these viruses, thus avoiding the universal constraint of Muller’s ratchet—see Box, p. 2—have recruited a specific function that proofreads and corrects replication errors [1]. This means that, while coronaviruses do indeed tend to produce genetic variants over time, the number of these variants remains quite low. This mutation rate may appear very limited, but the sheer number of viral particles generated during an infection is enormous, while the human population currently recognized as infected exceeds twenty million people. It follows that the mutation rate per nucleotide—of course very heterogeneous due to the selection pressure on certain locations in the genome—is around 8 × 10−4 changes per site per year [2].
Here, this situation was placed in the perspective of the fundamental theorem of natural selection proposed by Fisher, which links the evolution of environmental fitness and genetic variance [3]. We wished to use the marks left by the evolution of the virus’ fitness—observed in the form of genomic sequences—in the presence of the biochemical constraints that bias the choices available for evolution. We had to take into account, however, that the terms of the problem are not as explicit as one might have wished: fitness is not known, nor are the time markers (estimated from phylogenetic trees or simply taken as physical time) and the frequency of certain strains in the phylogenetic trees may be less due to natural selection than to heterogeneity in sampling and sequencing depth. This motivated our use of procedures that are robust enough to cope with these uncertainties. Nevertheless, the advantage of such an analysis is that it allowed us to propose anticipations for the evolution of the virus. It is therefore an explicit means of feeding epidemiological or clinical models with relevant observations.
In this context, it seemed to us of great interest to explore the details of how SARS-CoV-2 mutated over time, in the various places where COVID-19 has spread, highlighting relevant descents in relation with the host metabolism. This should allow us to anticipate some of the future of the virus’ progeny, with important consequences for control of the disease. The analysis of the constraints that govern access to the metabolism of the nucleotides that make up the virus genome has shown us that the content of cytosine (C) in its genome is subjected to strong negative pressure, leading to systematic depletion, over time, in cytosine monophosphate [4]. This bias has long been believed to result from a major causal effect of the “editing” of the C content of the genome by the family of APOBEC deaminating enzymes [5, 6]. We now know that it is the organization of the metabolism of pyrimidines in animal cells, and more particularly of cytosine triphosphate [7], which drives the corresponding pressure on evolution (Figure 1).
CTP controls all crucial metabolic steps required to build up a functional SARS-CoV-2 virus. (1) CTP is a precursor of the virus genome; (2) the lipids of its envelope derive from cytosine-based liponucleotide precursors; (3) all transfer RNA molecules produced by the host must be maturated to a form ending in a CCA triplet at their 3′OH end; and (4) post-translational glycosylation of viral proteins, in particular its spike protein require a dolichyl-phosphate anchor in the endoplasmic reticulum (ER) and dolichol kinase is specifically dependent on CTP. See text and Ref. [7] for details.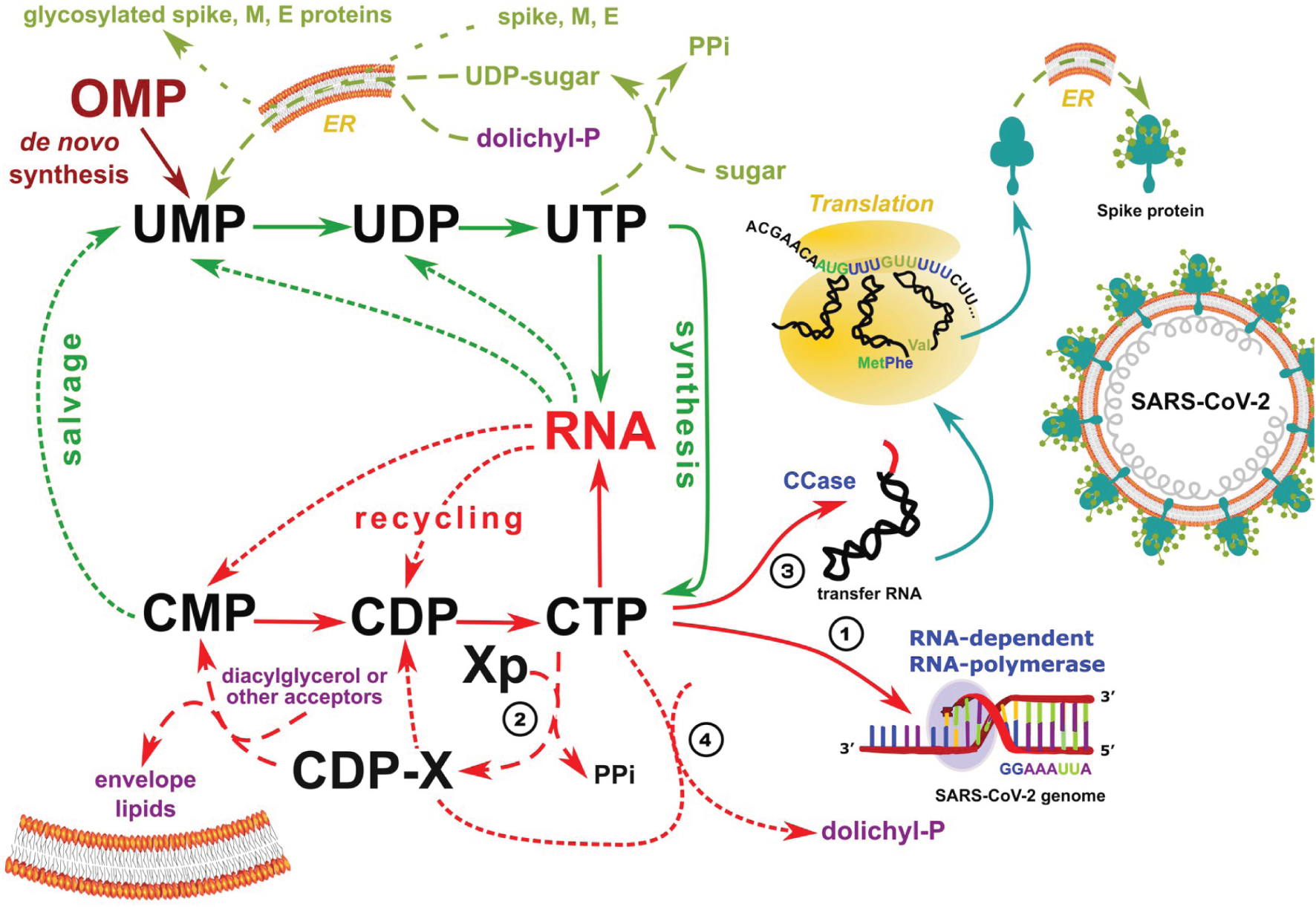
Indeed, due to the extreme asymmetry of the replication of the virus—which replicates 50 to 100 times from its complementary template [8]—a genome editing effect of these highly context-dependent enzymes would only be significant when a C into U is modified on the negative RNA template, which would lead to a major enrichment in A of the viral genome, or possibly from a U → C transition due to another class of deaminating enzymes acting on double stranded RNA, ADAR, that deaminates adenine into inosine [9]. Furthermore, both APOBEC and ADAR are highly specific enzymes and this hardly fits with the widespread C → U transitions that we keep observing as the virus evolves. Here, we have focused on the dynamics of the loss of C in the genome, and sought for the locations and the causes of changes in this driving force. In the first paragraph, we summarized the metabolic reasons accounting for this remarkable phenomenon. Subsequently, in the body of the article, we showed that the constraint on the C content of the genome leads to specific descents which can be used to reveal the existence of important functions of the virus as well as the role of the host’s response.
2. A universal metabolic requisite, the biosynthesis of cytidine triphosphate (CTP), guides the evolution of the virus
What do we know about the synthesis of the building blocks that allow the generation of a viral particle (a virion)? During a viral infection cells usually stop multiplying. All their resources are quickly diverted in favour of the multiplication of the virus. Yet, growth is a universal property of life. This means that, almost always—differentiated neurons are an exception—the cell’s metabolism that the virus faces is organized to allow cell growth as soon as the opportunity to multiply arises. The moment it infects a cell—again, with the exception of those that do not multiply—any virus will therefore have to manage the metabolic pressure that organizes the availability of the building blocks necessary for its construction. In our usual physical space (three-dimensional), growing introduces an inevitable constraint. The cell must put together the growth of its cytoplasm (three-dimensional, therefore), that of the membrane that encloses it (two-dimensional) and that of its genome (one-dimensional, because nucleic acids are linear polymers). However, it is a common metabolism, developed mainly in the cytoplasm, which produces the building materials needed to build up these three major compartments. So, here we have a question similar to the one asked by economists when they raise the question of “non-homothetic” growth [10]. Unfortunately, because life developed from a primitive metabolism in several stages over 3.5 billion years, we might fear that many organisms had found an idiosyncratic solution to this constraint, as often witnessed in the huge diversity of life forms. Unexpectedly, it appears that the solution to this quandary is universal: a single metabolite, the nucleotide cytidine triphosphate (CTP), has been recruited to this purpose [4, 7].
The key role of CTP appears in four essential places in cellular metabolism, and these places are essential for the formation of new virions. (1) It is the immediate precursor of one of the four nucleotides forming the genome of the virus; (2) CTP is required for the synthesis of liponucleotide precursors of the viral envelope; (3) human transfer RNAs are synthesized from 415 genes which do not encode their 3′OH-CCA terminal end—this sequence is synthesized from CTP by a specific nucleotidyltransferase [11]; and finally (4) the “decoration” of proteins by complex glycosylations is performed in parallel with their translation in the endoplasmic reticulum (ER) via the anchoring of substrates by dolichyl-phosphate, produced by a kinase which uses CTP, not ATP, as its phosphate donor [12]. In addition, intermediate metabolism is based on an original organization of the metabolism of pyrimidines, which systematically recycles and salvages them via uridine triphosphate (UTP) which makes CTP a pivot metabolite and limits considerably its availability (Figure 1). As a result, accidental replication errors will tend to replace cytosine with uracil in the genome.
3. General evolution of the SARS-CoV-2 virus
Using the available sequence data gathered in the SARS-CoV-2 GISAID database (https://www.gisaid.org) we have, like others [13, 14], reconstituted a phylogenetic tree of the evolution of the virus. As the sequences of each viral genome, as well as the date of identification of these sequences are known with fairly great precision, this tree makes it possible to explore the orderly lineage of the mutations which appear over time. In particular, unless we can suspect a recombination event due to the infection of the same patient by two or more viruses, when two identical mutations appear in separate branches of the tree, we can assume that this is the result of evolutionary convergence [15]. The reasons for this convergence are discussed on a case by case basis when analysing each relevant mutation. A second observation, which needs to be put in perspective (see below), is that the shape of the tree is not at all homogeneous. We noticed indeed the presence of “blooms” where, at a particular node of the tree, a large number of branches appear, demonstrating an “explosive” appearance of new mutations (Figure 2). We have therefore devised a statistical approach that allowed us to characterize them explicitly.
An example of bloom detected by our statistical approach. At node N0, there are 25 different states in the 40 samples of the subtree and a high number of branches. This behaviour differs significantly from that of the other sub-trees. Each country is represented by a distinct color at the level of the tree leaves.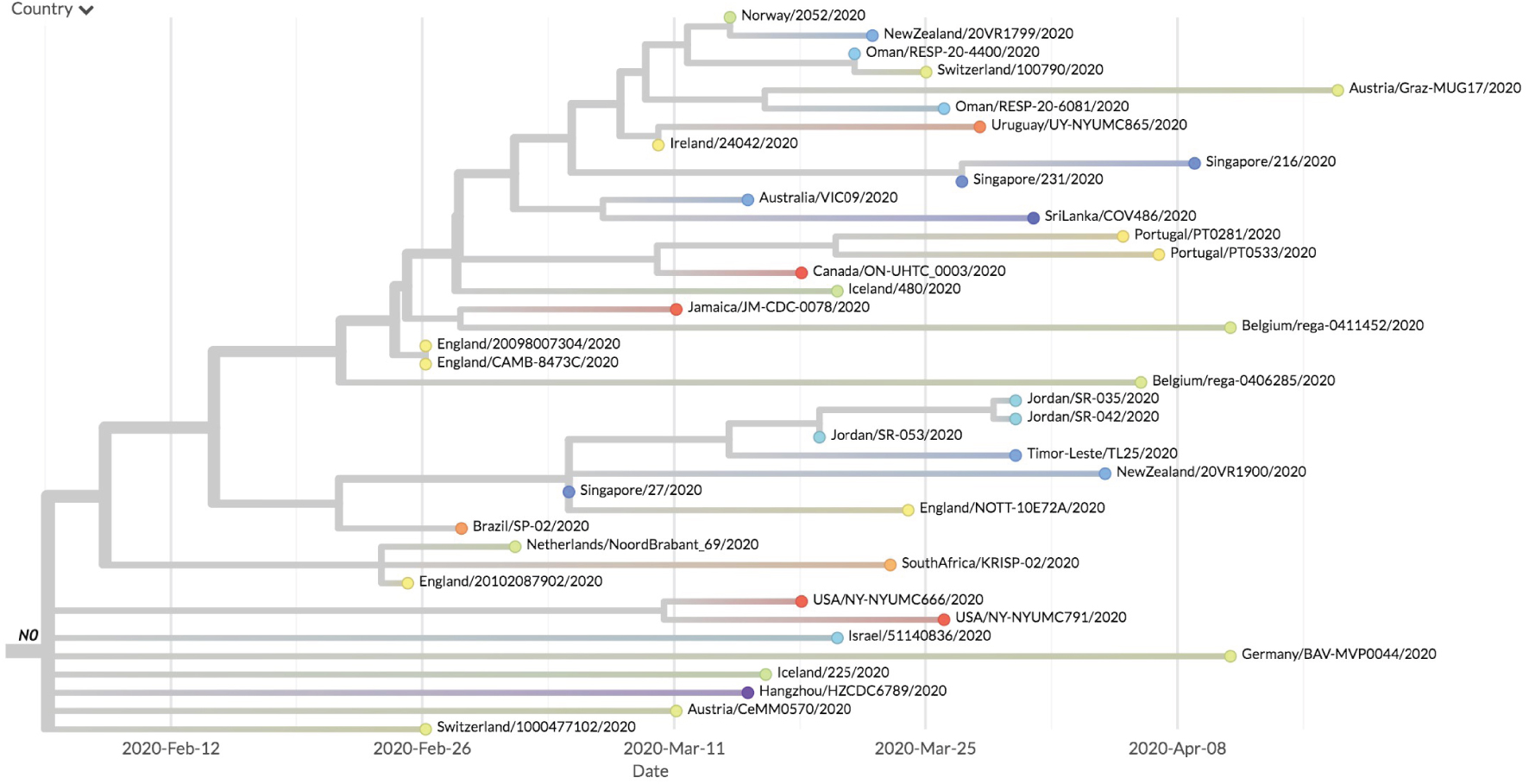
The causes of these blooms are multiple, but the adaptation of important viral functions can be at their origin, and we retained a few cases of this kind for further discussion (see Materials and Methods for the statistical definition of blooms).
4. Description and analysis of the evolution of the C content of the genome
Generally speaking, the coronavirus genome tends to evolve by adapting its C content to the metabolism of its host. More specifically SARS-CoV-2 evolves towards forms less rich in C as the epidemic develops [7]. However, this development is not homogeneous.
In the two data sets of interest, 77% of the transitions between pyrimidines are represented by transitions from cytosine to uracil. These transitions represent 48% of all substitutions identified in the first set (respectively, 49% in the second). An important imbalance can also be noted at the level of the transversions, knowing that more than 73% of those pertain to a substitution from purine to pyrimidine in the first set (respectively, 74%). However, only 20% of these 73% lead to the occurrnce of cytosine (respectively, 17%), indicating once again a tendency to favour the generation of uracil, thus demonstrating that the major constraint of the mutagenic process is the availability of each one of the nucleoside triphosphates in the cell. This inhomogeneity is also salient at the tree level. At the level of branch B4 (20% of the samples), the tendency is strongly marked to lose less C as compared to the rest of the tree (Figure 3).
Heat map of C losses from the original sequence. Branches 1 and 4 can be readily discriminated by their extreme values.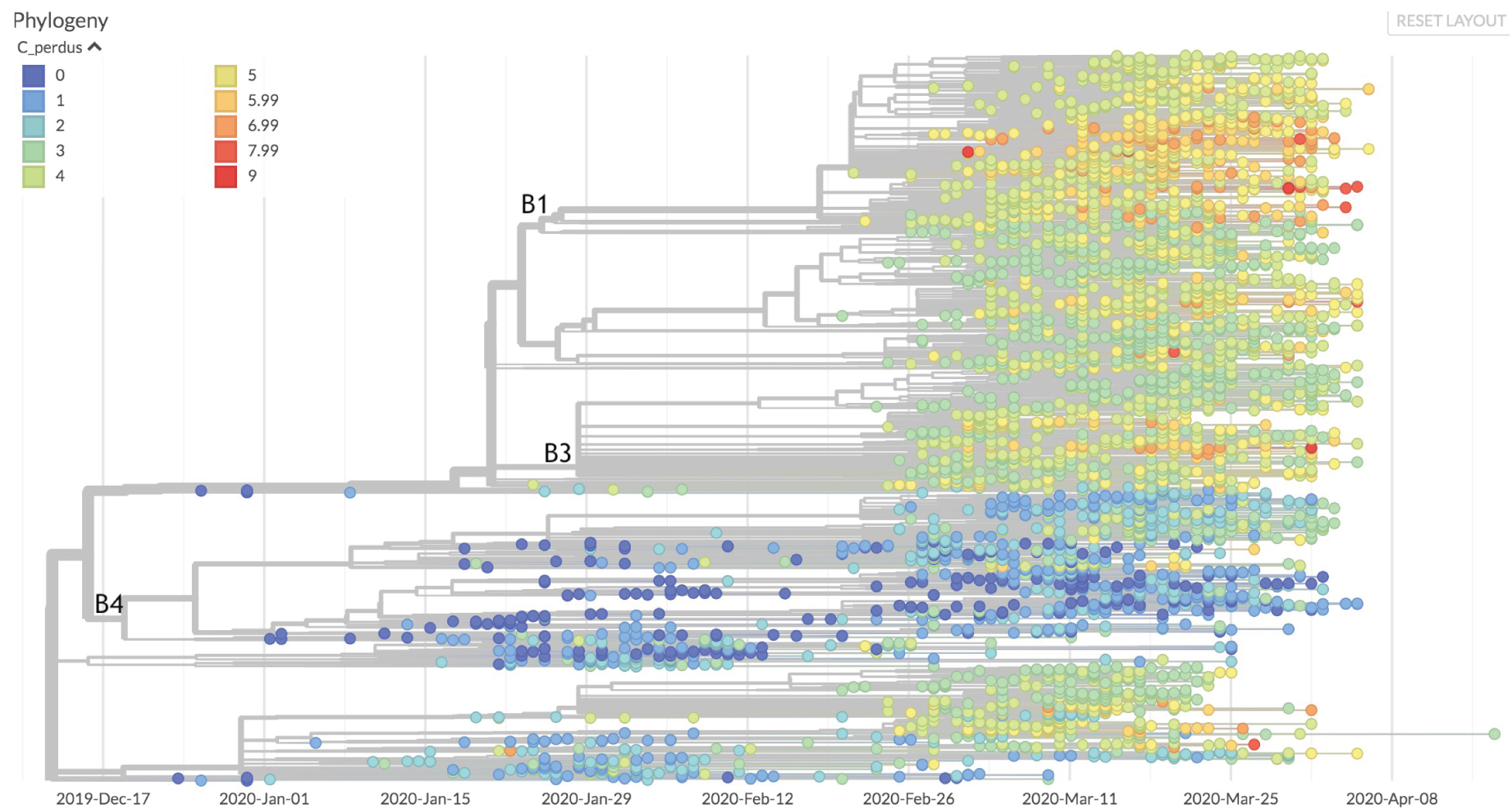
Interestingly, this branch is also the one that comprises on average the strains with the least divergence from the original strain of the virus. By contrast, in branch B1, the loss of C looks larger. The rate of virus mutation also seems to be accelerating in this branch, with a rate of transversions 20% higher than the rest of the tree (and also higher transition rates, but in more anecdotal proportions). Finally, for branch B3, the main site of blooms, a 29% decrease in the transition rate of pyrimidines and a 30% decrease in the rate of purines compared to the rest of the tree is noteworthy.
This inhomogeneity can be the consequence of many constraints:
(1) The very structure of the genome, which must fold into a compact capsid envelope requires certain regions to maintain the presence of specific C residues. This is the case of the regions which control the origin of replication [8] or transcription, AACGAAC, for example [16]. In the case of the translated regions, the pressure on the presence of C varies depending on its position in the codon trinucleotides. When C is located at the first position of a codon, it is used to input arginine, glutamine, histidine, leucine or proline into proteins. Histidine and glutamine are coded in two codon families, discussed below. For arginine, the selection pressure is lower because the CGN codons can be replaced by AGR codons—we used here the IUPAC convention for labelling nucleotides or aminoacids, e.g. N is for aNy, R for puRine, etc. (https://www.bioinformatics.org/sms/iupac.html). The selection pressure on the leucine content is also lower, since in addition to the CUN codons, this amino acid can be input using the UUR codons. In the second codon position, C is again used to code for proline, but also threonine (ACN), alanine (GCN) and serine (UCN). Again, the latter amino acid escapes a large part of the constraint imposed by the availability of C because it can also use the AGY codons. Finally, the third position of the codons is much less constrained because it can be replaced by U but also by A or G in the families with four codons (alanine, proline, threonine, valine). The two codon families UGY, AGY and NAY are discriminated along a pyrimidine / purine axis. A pyrimidine is used to maintain the same nature of the coded residue, as the codon uses a U or C as the 3′ end (aspartate, asparagine, cysteine, histidine and tyrosine). Finally, isoleucine is coded by three codons (AUH), and ending in U or C is taken into account by relevant tRNAs [17];
(2) The function of the virus proteins can impose the presence of certain amino acids in their sequence. For example, the proline residue encoded by the CCN codons is not strictly an amino acid, but is essential for the folding of key domains of viral proteins [18];
(3) Further stressing the importance of CTP, during evolution, innate antiviral immunity recruited the activity of an enzyme, viperin, which modifies CTP into a form toxic to the development of the virus, 3-deoxy-3,4 didehydro-CTP (ddhCTP) [19]. An interesting consequence of this pathway is that decreasing the C content of the genome will allow the virus replication process to be less sensitive to the presence of this nucleobase. It follows that, during the transfer of a virus relatively rich in C from an animal host to human beings, the evolution towards the loss of C may be transiently concomitant with an increase in its pathogenicity. In the long term, however, the loss of C severely restricts the evolutionary landscape of the virus and most likely will tend to its attenuation [20].
5. Examples of correlations allowing us to propose a function for viral proteins
Thousands of mutations have been identified at this date. It is possible to follow their emergence along the tree of its phylogenetic evolution of the virus and then highlight some interesting features that may allow us to anticipate some of its future.
5.1. Mutations leading to an early translation termination
Mutations leading to premature termination of the virus protein synthesis are expected to appear with high frequency. In the present context, this is all the more likely because the translation termination codons UAA, UAG and UGA do not contain C, and are therefore favoured by the disappearance of this nucleotide. Since most of these mutations lead to non-functional polypeptides, it is generally probable that the affected viruses do not give rise to a significant progeny. It follows that when these mutations are observed—and that they do not result from sequencing errors—they indicate that the role of the truncated protein corresponds to a function which is not critical, or that the protein has remained functional at a sufficient level to allow virus reproduction. However, a few observations allowed us to offer an explanation for the fact that the viruses in question may have survived. Here are three examples which reveal interesting features of the virus.
In a strain from Iceland, the succession of mutations G1440A (Gly392Asp, protein Nsp2) and G2891A (Ala876Thr, ubiquitin-like domain of protein Nsp3) is now present in multiple world locations [21]. This sequence ends up with C27661U (which modifies amino acid Gln90 into a premature translation end, near the carboxy-terminal end of protein Orf7a). This viral protein is found in the endoplasmic reticulum, the Golgi apparatus and the perinuclear space [22]. Several variants have been identified in the course of the epidemic [23]. Remarkably, several deletions have been isolated in the gene, which suggests that the function of this region is not essential [24]. However, we noticed that many of these mutations, as the one discussed here, keep the small hydrophobic protein Orf7b gene intact, downstream of Orf7a. This very small protein is present in the Golgi apparatus and is also found in the purified virus [25]. It must be noticed that it is synthesized in vivo via a frameshift that spans the termination codon of the Orf7a frame (…GAA TGA TT…becomes …GA ATG ATT…). This can be interpreted as a conflict in this region between translation of Orf7a and Orf7b, creating a cost / benefit dilemma for the expression of either one of these proteins. Hence it will be important to monitor the future descent of the virus in this region as it may result in interesting attenuated forms.Exemple 1.
Another succession of mutations that leads to premature translation termination of a viral protein begins with G11083U (protein Nsp6, Leu37Phe). This mutation is now widely distributed worldwide. It is likely to induce a more stable binding of the protein to the ER, possibly favouring coronavirus infection by compromising delivery of viral components to lysosomes for degradation [26]; then we have G1397A (Nsp2, Val378Ile), also likely to favour virus propagation [27]; followed by G29742U (3′UTR of the virus), and U28688C (synonymous); subsequently, we have the couple of mutations C884U (Nsp2 again, Arg207Cys [27]) and G8653U (Nsp4, essential for envelope assembly [28]. The corresponding change (Met2796Ile) is located at the border of the ER lumenal domain of the protein. It is known that, in order to function properly, the ER requires the presence of oxygen [29], and reactive oxygen species (ROS) are associated to misfolding of proteins in this compartment. Nsp4 has a number of cysteine residues, prone to be oxidized. The role of methionine in the parent might be to act as a buffer against ROS, so that the mutant would be slightly attenuated). These mutations are followed by A19073G (in the methylase domain of protein Nsp14, Asp1869Gly, a position that already evolved from SARS-CoV-1 [30], hence likely to be more or less neutral), then the couple with the mutation resulting in end of translation: G27915U, Gly8 to end of translation at the N-terminus of Orf8 and C29077U (synonymous); the succession ends with the couple of mutations leading to synonymous changes C19186U and G23608U. This region of SARS-related coronaviruses is hypervariable. It changes during the course of epidemics, showing that it is subject to ongoing selection pressure, sometimes producing two peptides Orf8a and Orf8b [31]. It corresponds to proteins expressed at the end of the infection cycle. It will be important to monitor the way they function in the course of the evolution of virulence of the virus. This displays a branching that appeared in four different countries and in seven samples, spanning six weeks between the first and the last mutation.Exemple 2.
Here we have a succession of mutations that begin within the 5’end of the virus genome, C241U, followed by mutation C14408U (Pro314Leu) at the end of a zinc finger in replicase Nsp12, which appears in many branches of the evolution tree of the virus. It is discussed in details below (origin of blooms). This mutation is followed by A23403G (Asp614Gly) a widely spread mutation of the spike protein (also discussed below), C3037U (synonymous), mutation G25563U (Gln57His) in Orf3a forming potassium channels is supposed to negatively interfere with the function of the protein [32], C1059U (Thr265Ile) in protein Nsp2, discussed previously, and the triplet G4181A (Ala1305Thr) in the SUD-N domain of protease Nsp3, then mutations G4285U (Glu1340Asp), and G28209U which results in an end of translation at glutamate 106 of protein Orf8. As discussed previously, many mutations, including deletions in Orf8, were frequently observed. This is again an indication that evolution of this regions should be carefully monitored to look for attenuated forms of the virus. This particular mutation to an end of translation is significant as it was found in a sample from Croatia, another one from Thailand, on two significantly separated branches and with one month difference. The sequence of mutations here corresponds to the Thailand sample.Exemple 3.
5.2. Reversal of the tendency of the viral genome to lose its cytosine residues
We have here retained two examples of a situation where, from an upstream branching point in the evolution tree, it appears that the descendants of the virus stop losing their cytosines, and may even tend to regain them. These examples are as follows (Figure 4).
Two sub-trees in the first dataset where the tendency of the genome to lose its cytosine residues is reversed. Upper panel. First sub-tree. The sub-samples displayed are those that have acquired the most C, apart from a few isolated samples on other branches. The node with the M1 mutation directly follows those respectively associated with the C8782U and U28144C mutations. Lower panel. Second sub-tree. This tree contains a majority of strains with a neutral C balance (both gained and lost), as well as 3 strains with more C gained than lost.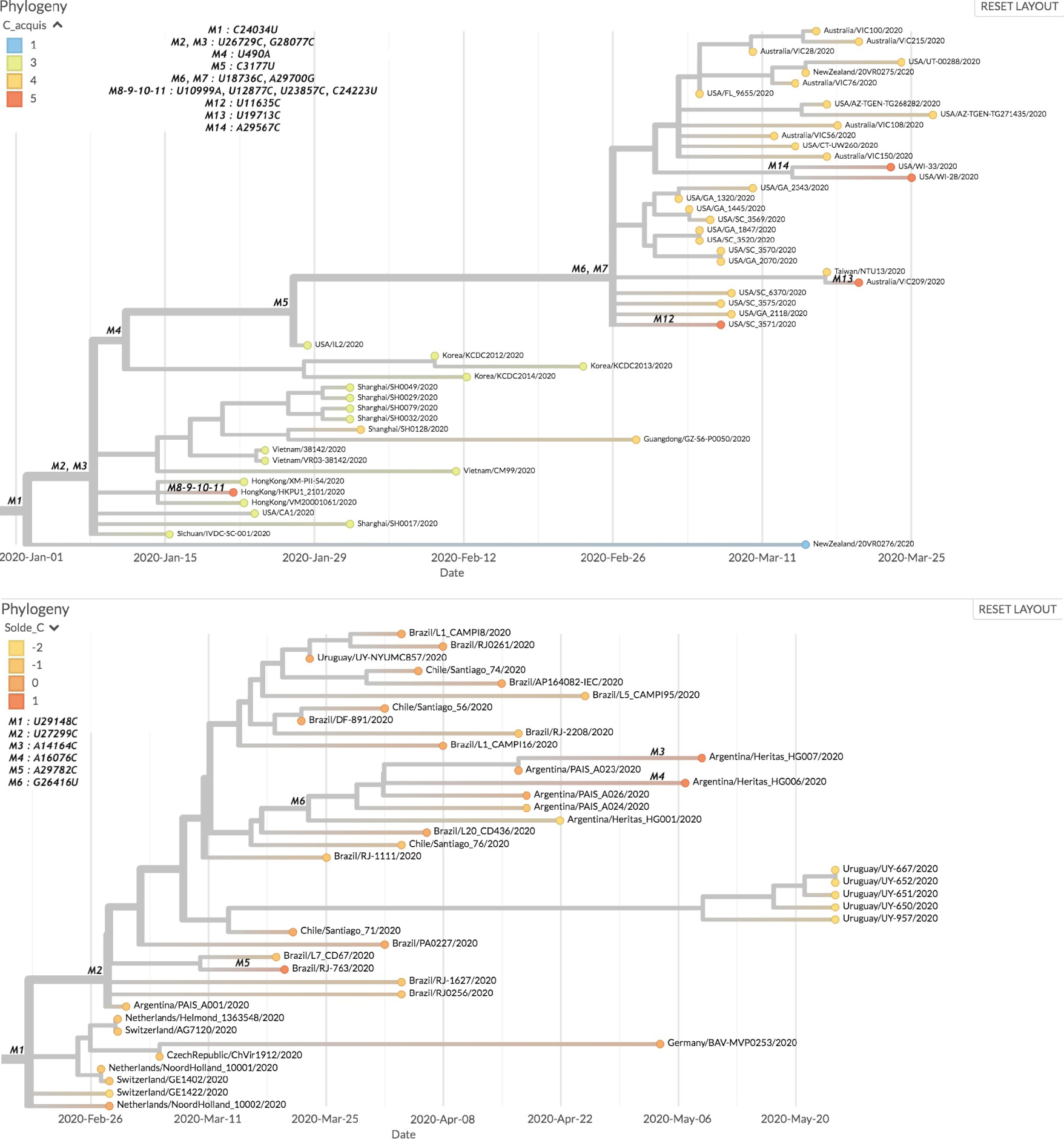
In dataset 1, there are two sub-trees, the first of which is more of an Asian sub-tree with the root of the node associated with the M2 and M3 mutations. The second contains samples from North America and Oceania, and its root node is related to the M6 and M7 mutations. The first tree arises from the succession of C8782U (synonymous), U28144C (Leu84Ser) mutations in the Orf8 protein, whose function was discussed above. It defines a major clade of variants of the virus [23], C24034U (synonymous), and finally the doublet U26729C (synonymous), G28077C (Val62Leu), in the Orf8 protein again. As this is the origin of the observed phenomenon, we are led to believe that it is the alteration of the role of Orf8 (8a or 8b) that is responsible. The Orf8 region is particularly variable and has been clearly implicated in interspecies transmission [33]. A common hypothesis is that the alteration of this gene corresponds to a loss of active function in chiropteran ancestors [34]. Since these are generally richer in cytosine than the human forms [20], one might ask whether one of the functions of this protein is to modulate the activity of CTP synthase.
In fact, the second branch comes from the same descent, to which is added the U490A (Asp75Glu) mutation in the Nsp1 protein, which controls the specific translation of viral RNA [35], systematically associated with the mutation C3177U (Pro971Leu) in the acidic domain, without any clearly identified function, of the multifunctional protease Nsp3 [36], and finally the U18736C (Phe1757Leu) doublet of the exonuclease, N7-methyltransferase Nsp14, and A29700G in the 3′UTR region of the virus. The Phe1757Leu modification is located in the middle of a zinc binding site at the interface between the two domains of the Nsp3 protein. It can therefore be surmised that this mutation could subtly change the proofreading process correcting replication errors in a way that would be less amenable to the entry of UTP opposite an A in the negative viral template. We noted that 3 out of the 5 samples that acquired the most C did so through a transition from U to C. The first one, HongKong/HKPU1_2101, shows two simultaneous transitions at positions 12877 and 23857. These mutations being synonymous, they are unlikely to change the replication-correction mechanism. The second one, USA_SC_3571, and the third one, Australia/VIC209, show transitions of the same type, also synonymous, at positions 11635 and 19713 respectively. Finally, the last two samples, USA/WI-33 and USA/WI-28, were derived from the transversion from A to C at position 29567, a mutation at the end of ORF9b.
For dataset number 2, this reversal of the trend concerns mostly Latino-American strains. The succession of mutations C241U, C14408U, then A23403G discussed in relation to the generation of end of translation codons in the virus genes, is followed by C3037U (synonymous), and the triplet G28881A, G28882A, G28883C, overlapping the codons at position 203-204 of the N nucleocapsid N gene. They mutate an arginine-glycine dipeptide into a lysine-arginine dipeptide. This alters the positive charge of the protein and may help improve its role in the assembly of the virus genome in the capsid, as discussed below in relation to the appearance of blooms (36). After this triple modification, we see several reversals of the tendency to lose C in the genome. U29148C (Ile292Thr) is found again in the nucleocapsid N gene, then U27299C (Ile33Thr) in the Orf6 gene, resulting in a set of samples that have at worst gained as much C as they have lost. There are also 3 samples among the 39 in the subtree that gained one more C than they lost (Brazil/RJ-763, Argentina/Heritas_HG007, Argentina/Heritas_HG006). Each time, the last C acquisition comes from a transversion from an adenine (in positions 14164 (Met233Leu), 16076 (Asp870Ala), and 29782, in the late 3′UTR of the viral genome. Overall, it is the change in the nucleocapsid that appears to be most conducive to reversing the tendency to lose C. Indeed, this protein, expressed at a high level during the infection, regulates the process of replication / transcription of the virus and this may account for this remarkable observation [38].
5.3. Emergence of blooms
The succession C3037U, (C241U, A23403G), C14408U is present upstream of 10 sub-trees, which we considered to be significant (see Figure 5 and Materials and Methods).
Example of blooms. The subtree shown here contains 10 of the 20 most significant blooms in the sense of the method we used. Node N0 is the place where mutation C14408U emerges. 50 different countries are represented on the sub-tree descending from N0. The upper sub-tree is marked by a predominance of North American samples (USA and Canada, red dots).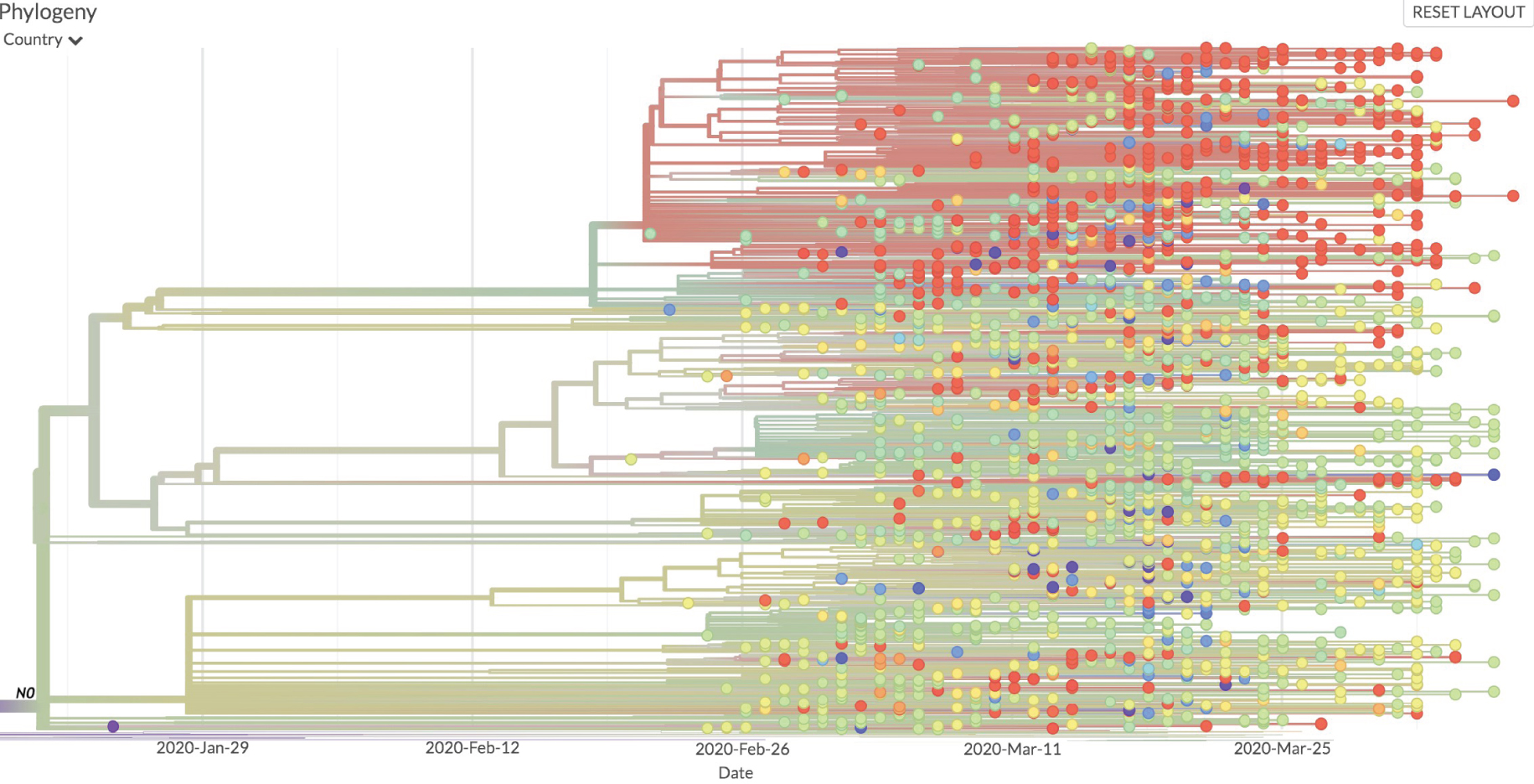
The synonymous mutation C3037U is located at the end of the ubiquitin-like domain 1 of protein Nsp3. This leads to a sequence (UUUUUU) that promotes changes in the reading frame and could decrease the translation efficiency of the proteins of the ORF1a region. The C241U mutation is observed very often [39]. It is found in the region that initiates replication of the virus. We can therefore assume that this may alter the frequency of replication. A23403G is a widely spread non-synonymous mutation which leads to the replacement of an aspartate by a glycine at position 614 of the spike protein, which is used by the virus to bind its host cell’s receptor. For this reason, several previous analyses have suggested that this mutation has an important role in the spread of the virus [40, 41]. Here, the fact that it is part of a major bloom can be considered as an additional argument favouring this interpretation. The C14408U changes an amino acid from proline to leucine (Pro314Leu) just after the end of the NiRAN domain (nidovirus RdRp-associated nucleotidyl transferase) of the protein Nsp12 ending in a “zinc finger”. The NiRAN domain, essential for the replication of the virus, acts as a nucleotidyltransferase, preferring UTP as a substrate for a function which has not yet been clarified [42]. The proline modified in the mutant is part of a dipeptide diproline which plays the role of hinge of separation between the NiRAN domain and the following domain.
A second bloom, which shares several elements with the preceding one begins with the same sequence C3037U, (C241U, A23403G) and C14408U. However it continues with a series of contiguous mutations resulting in a change (G28881A, G28882A, G28883C) in nucleocapsid N, as we saw previously. It is worth noticing that this change might have a role in assembling the virus genome in the capsid by phase separation [37]. This might increase the efficiency of virus transmission and thus contribute to the formation of blooms. The fact that it is a cluster of mutations involving G is intriguing. It may result from the fact that it spans a GGGG sequence.
We have previously seen that mutation G11083U (protein Nsp6, Leu37Phe) has initiated another succession of mutations that led to premature translation termination of a viral protein. Here, this widely distributed mutation is at the root of blooms. As discussed, it is possibly favouring coronavirus infection by compromising delivery of viral components to lysosomes for degradation. This would certainly favour blooms. The mutation is followed, in a first bloom-generating succession, by G26144U (Gly251Val) in protein Orf3a, that forms potassium channels important for innate immunity response—but the exact function of the protein still remains open to question [43]. Subsequent mutations are C14805U (synonymous) and U17247C (synonymous). This succession suggests that the first mutation in protein Nsp6 and perhaps the second one are the primary causes of the bloom [26]. The role of the fist mutation is further substantiated by the second bloom-generating succession where it is followed by a quadruplet: C6312A (Thr2016Lys) in the inter-domain region that precedes domain G2M of multi-domain protease Nsp3, then associated with three C → U mutations, hence expected to be more frequent: C13730U (Ala88Val) in the NiRAN domain of protein Nsp12, C23929U (synonymous), and finally C28311U (in a sequence of four C, Pro13Leu) at the beginning of the nucleocapsid protein, N.
A second succession of mutations that ends up in blooms is C8782U (synonymous), U28144C (Leu84Ser) in protein Orf8, the function of which has been discussed previously and defines a significant clade of the virus variants [23], ending up with C26088U (synonymous). The Leu84Ser mutation co-evolves significantly with the Asp614Gly mutations of the spike protein discussed above [36], which makes it another likely candidate for positive selection leading to increased spreading of the virus, hence blooms.
5.4. Change in frequency of transitions / transversions
Among the mutations upstream of branches showing significant changes in the transition / transversion flow is the mutation C17747U, which modifies a proline residue into a leucine residue in the protein Nsp13 (Figure 6 and Materials and Methods).
One of the descents considered to be significant for the change in the process of molecular evolution. The progeny resulting from the C17747U mutation is shown in yellow, and its evolutionary process is modelled by a 6-parameter TN93 model (process 1). The rest of the tree (blue leaves) is modelled by a 3-parameter TN93 model.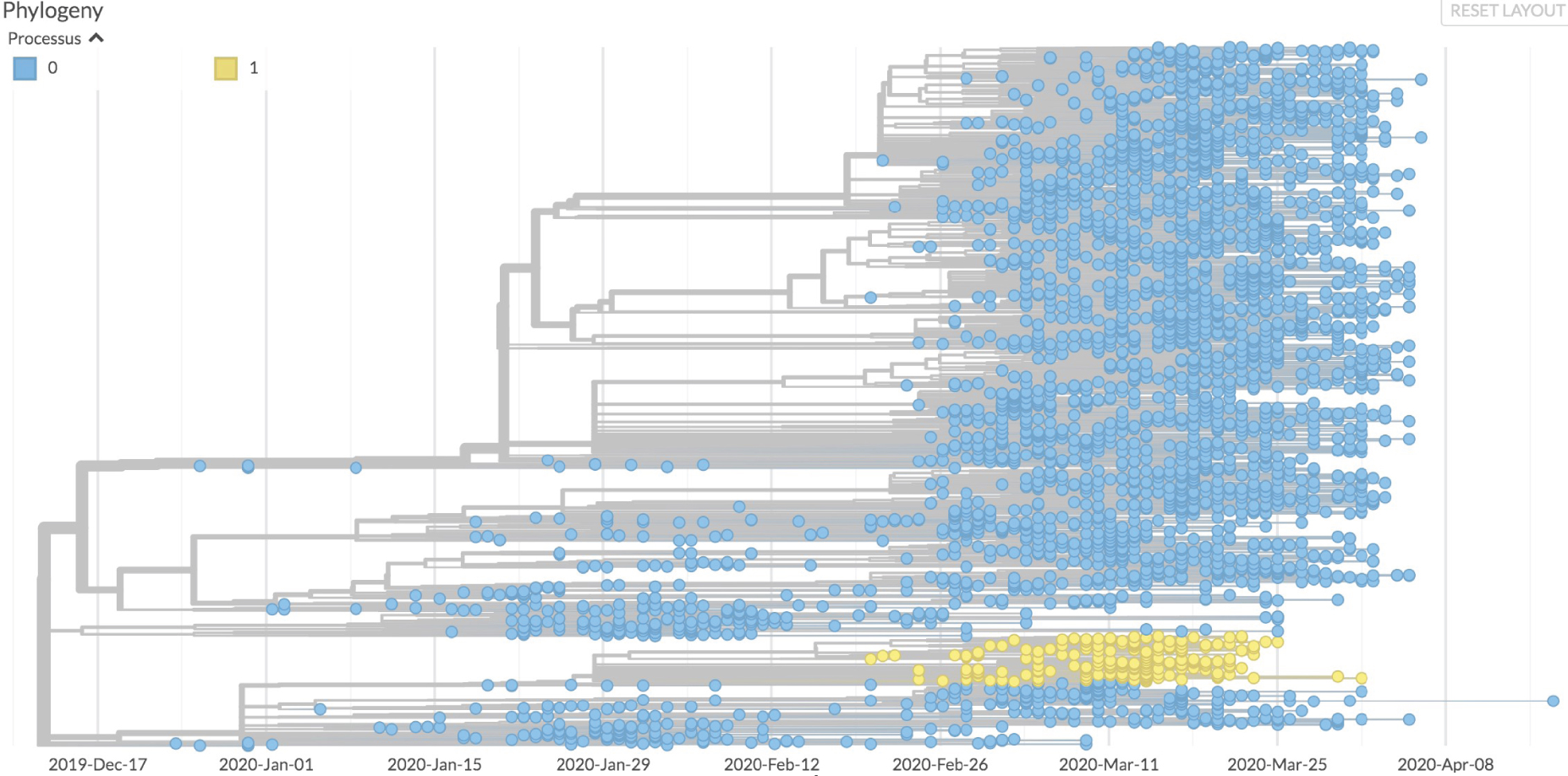
This mutation affects the protein domain which has nucleoside triphosphatase activity, the exact role of which is unknown but consistent with a proofreading activity [44]. We might propose that it is involved in the quality control of the product of the replication of the virus for example via stabilizing the “anti” form of nucleotides, thus avoiding the mismatching leading to transversions. In fact, this protein has been identified among those which lead to a significant alteration in the diversity of the viral genome [15]. The existence of a notable change in the type of mutations located downstream of the tree is therefore a strong argument for the discriminating role of the corresponding region of the protein. Furthermore, to the extent that this mutation increases the frequency of mutations in a biased manner, we can expect the ensuing descent to lead to an attenuation of the virus. However, as this changes the evolutionary landscape, this evolution could lead to “innovative” mutations modifying the pathogenicity of the virus, and this especially under conditions where recombination due to co-infections would be favoured. This is yet another argument for choosing a strong public health policy which tends to avoid the formation of clusters of infection.
6. Conclusions and perspectives
The COVID-19 epidemic is a life-size experiment in virus evolution. Remarkably, we neither know the real origin of the virus [45], nor where it will lead us. This explains why the vast majority of studies of the SARS-CoV-2 virus and its evolution are essentially descriptive. Here, we tried to make use of the ongoing evolution of the virus to investigate some of its related constraints using a hypothesis-driven probabilistic modelling approach to the molecular evolution of the virus. Based on the assumption that the virus’metabolism is ruled by its host. Based on the metabolic set up of the host cells, acting as a compulsory material framework for the multiplication of viral particles, we pointed out specific changes in the evolution pattern of the virus descent, witnessed by changes in the virus genome composition as time passes. Using the widely spread C to U change in this genome’s composition as a base line, we identified nodes where the change is shifted from this direction to another one, favouring transversions rather than transitions, reversing the C to U trend towards U to C enrichment or generating blooms with sudden appearance of multiple branches in the evolution tree. This allowed us to point out a series of functions that are evolving towards a more efficient spread of the virus (e.g. the previously identified Asp214Gly mutation of the spike protein, but also the Gln57His mutation of the Orf3a potassium channel). We also noticed that Orf8 is the likely site of an ongoing competition for expression of two frameshift-dependent overlapping proteins Orf8a and Orf8b. Similarly, the unstable region of Orf7 could promote the synthesis of the very small membrane protein Orf7b, whose function remains unknown to date. Finally, the reversion of the tendency to favour U over C indicates that nucleocapsid protein N may be involved in the control of CTP synthesis in the host, suggesting an interesting target for future control of the virus development. We hope that this combination of mathematical and biochemical knowledge will help us devise further enterprises against the dire consequences of COVID-19. We noticed that among the possible way for the virus to escape CTP-dependent control in cells would be to infect cells that are not expected to grow, such as neurons. This may account for unexpected body sites of viral development observed in the present epidemic.
7. Materials and methods
7.1. Data processing
A total of 4,792 sequences of the SARS-CoV-2 virus were recovered from the GISAID databank [46] on April 21, 2020 for the first dataset. Only the genomes of viruses from the human hosts of SARS-CoV-2 of a length greater than 25,000 bp were retained. Sequences for which the sampling date was insufficiently informed (absence of the harvest day, sometimes of the month) were also excluded. For sequences present multiple times, only the first isolate was retained. We also reused the work of the Nextstrain teams and discarded the too divergent or unstable samples that they themselves had left out (github.com/nextstrain/ncov/blob/master/defaults/exclude.txt). The sequence of 26 coding regions (Nsp1, Nsp2, Nsp3, Nsp4, Nsp5, Nsp6, Nsp7, Nsp8, Nsp9, Nsp10, Nsp11, Nsp12, Nsp13, Nsp14, Nsp15, Nsp16, S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N and ORF10) was characterized using NC_045512 as a reference. The total number of sequences retained at the end of the treatment is 4,088 sequences. A second dataset of 3,246 sequences, 510 of which are common with the first dataset, was retrieved on July 6, 2020 using directly the Nextstrain API [47].
We note here that, over time, data availability kept being altered, with some sequences deleted from the samples, while other ones entered the database. Furthermore, it was generally difficult to extract large samples of sequences so that it was extremely difficult to build up a consistent data repository where correct statistical approaches could be implemented. It seems very awkward that the bulk of the sequences of a virus of worldwide importance has not been made available at the International Nucleotide Sequence Database despite recommendations of the major research institutions [48, 49].
7.2. Phylogenetic reconstruction
The reconstruction process begins with aligning all of the sequences to the reference sequence. Insertions and deletions of genome regions were not taken into account. It was out of the question here to take into account nucleotide insertions and deletions. We retained only the potential one to one substitutions. We used program MAFFT [50] to generate these alignments. Some ambiguous positions were highlighted during the alignment process. For example, some regions of the genome may display high instability and wide variability depending on the parameters of the algorithm used to perform the alignment. To overcome this problem, we used the same masks as those used by the Nextstrain team. Sites 18529, 29849, 29851, 29853, as well as the first 130 and last 50 sites of the genome were therefore omitted from the substitution analysis. We used a General Time Reversible (GTR) model to infer the substitution process at work using the IQTREE software [51]. This first tree is a fairly raw version which does not take into account the temporal aspect of evolution. The Treetime software [52] allows you to refine this tree by also taking into account the sampling dates of the sequences. It then reconstructs the tree with maximum likelihood compared to the sampled sequences. Using maximum likelihood approaches it also infers the compositions of the ancestral sequences of the samples, as well as a 90% confidence interval around the most likely date of these common ancestors. Once the tree has been created, we could then reconstruct the order in which the mutations in each sample appeared, in the sense of maximum likelihood. For the visualization of the tree and the production of Figures 2 to 6, we used the Auspice program developed by Nextstrain, to which we made some modifications to display the quantities we were interested in. To this purpose, we developed a Python script to modify the JSON file used as input by the Auspice program (available on request). This allowed us to enrich the visualization capabilities of the software by adding quantities such as the number of C acquired or lost by a sample compared to the reference and to generate original tree presentations.
7.3. Identification of blooms
The main pitfall we had to face when identifying blooms was the bias introduced when selecting samples from the phylogenetic tree. In particular, some hospitals were likely to provide more samples than others, due to the different health policies and means implemented depending on the country. In order to avoid selecting nodes likely to generate a bloom due to oversampling, we chose to develop a custom-made statistical method meant to cope with this difficulty.
A subtree is any set of nodes and leaves rooted in one of the nodes of the main tree. The idea is to use the information provided by the identity of the countries represented in each sub-tree: the easier a strain is spread, the higher the number of countries in which it is expected to be observed. To implement this heuristic, it is necessary to control two factors: the size of the tree (two trees of unequal depth, that is to say rooted on different dates, naturally show diversity as different countries) and the heterogeneity of sampling (countries where sampling and sequencing are carried out with different intensities have different probabilities of appearing in a given sub-tree).
These two factors interact, because the size of a tree (the number of its leaves for example) obviously varies with the sampling intensity. One way to control this interaction is to measure the size of a tree by its total length, or sum of branch lengths, in time units. Indeed, this observable is not very sensitive to the effects of oversampling because the presence of many sequences sampled in the same place at about the same time generates a sub-tree whose length is close to zero.
To control the effect of the length factor L on the number of countries represented, N, we seeked to learn the relation N = f(L) in a typical tree in order to be subsequently able to identify the sub-trees whose number of countries represented, for a known length L, exceeds the expected f(L). A simple statistical model consists in supposing that the number of occurrences of country i in a tree of length L is a Poisson distribution of parameter 𝜃i L and that these numbers are independent. If K is the total number of countries referenced by Nextstrain, the number of countries N represented in a tree of length L is therefore the sum of K Bernoulli variables independent of parameters 1 −exp(−𝜃i L). For example, if countries are divided into two groups, the k1 “frequent” of intensity 𝜃1, and the k2 “rare” of intensity 𝜃2≪𝜃1, N has the mean K − k1 exp(−𝜃1 L) − k2 exp(−𝜃2 L), which behaves when L is large like K − k2 exp(−𝜃2 L).
In addition, when L is large, assuming that 𝜃2 L = O(1), the distribution of N is approximately equal to k1 + N2, where N2 foloows a Poisson law of parameter k2(1 −exp(−𝜃2 L)).
So, we used the parameterization:
N = a − b exp(−cL), interpreting the parameters as follows: a is the maximum number of countries, b is the number of countries with low sampling/sequencing intensities and c is a density of presence of these countries per unit length of tree. Under the null hypothesis, N is distributed as a − b + N1, where N1, follows the Poisson’s law of parameter b(1 −exp(−cL)). Finally, we selected the 20 most significant blooms, i.e., those whose behaviour deviated the most from that expected by our estimator. This then allowed us to reconstruct the lineages and the mutations that appeared successively upstream of each node at which a bloom had occurred. This allowed us to identify the succession of mutations common to some of these nodes and thus those giving rise to the majority of statistically significant blooms. Furthermore, we restricted the automatic selection of nodes so that no selected node was present in the lineage of another one. The selected blooms are therefore mutually independent, even though they may obviously have common ancestors. To arbitrate the choice between two nodes present in the same lineage, we have systematically kept the oldest node, and thus the most dense tree.
Molecular evolution and Muller’s Ratchet
Biology rests on the laws of physics. Because it develops at approximately 300 K, it is subject to the universal stress of thermal noise, involving an energy that does not differ considerably from that involved in the chemical bonds of biological chemistry. It follows that the reactions that come through and organize living things cannot develop with strict reproducibility. Inevitable errors cause the product of a reaction to differ from what it is supposed to be. Genome replication cannot escape this constraint. The consequence is that, in the progeny of a virus, there is always a number of variants, named mutants when they carry over alterations of the genome. In most cases, these mutants correspond to the change from one of the four nucleotides to a different one. This process, as a rough approximation, is random—the mutant position can be anywhere in the genome, and the replacement of one nucleotide is by any of the other three. As time goes by, all the nucleotides of the genome are likely to change into others. This will affect the functions necessary for the multiplication of the virus, and some changes will continue to be propagated (be fixated), while others will end up without a progeny. A mutation followed by a fixation is called a substitution. The substitution of a purine for a pyrimidine (or vice versa) is called a transversion; other substitutions are called transitions. The likelihood of a particular mutation returning to the ancestral state is very low. The probability that a particular mutation will return to the ancestral state is very low. This forces evolution to always go forward, without the possibility of going back. This process was noticed in 1932 by Hermann Muller in the special case of the effects of irradiation on mutagenesis. His reflection has since been simplified and popularized. It is now known as the “Muller’s ratchet” [54]. It is obviously highly probable that the majority of mutations leads to the partial or total loss of the functions coded by the altered regions of the genome. It follows that this generally leads, in the long term—but not in the short term—to the attenuation of the functions allowing multiplication and virulence of pathogenic species. This is why Louis Pasteur and his successors could have the luck to isolate attenuated organisms which, in some—rare—cases, could then be used for the vaccination of infected persons [55]. However, this process becomes unproductive as soon as co-infection with different mutants occurs under circumstances where recombination is possible. Two different mutants can recombine into the ancestral form of the pathogen and erase the entire benefit of attenuation. This is all the more harmful since the old forms are also, very often, those which spread most easily.
Figure caption. Muller’s ratchet and recombination. The figure is reprinted from reference [56]. Genes (capitals) are mutated at random in a different form (low case). Mutations accumulate ratchet-like because the probability of reversion to the parent form is negligible. This happens independently for viruses of different descents. However, if viruses from different descent happen to be in the same cell, they can recombine. This allows them to recreate the ancestral form of the virus.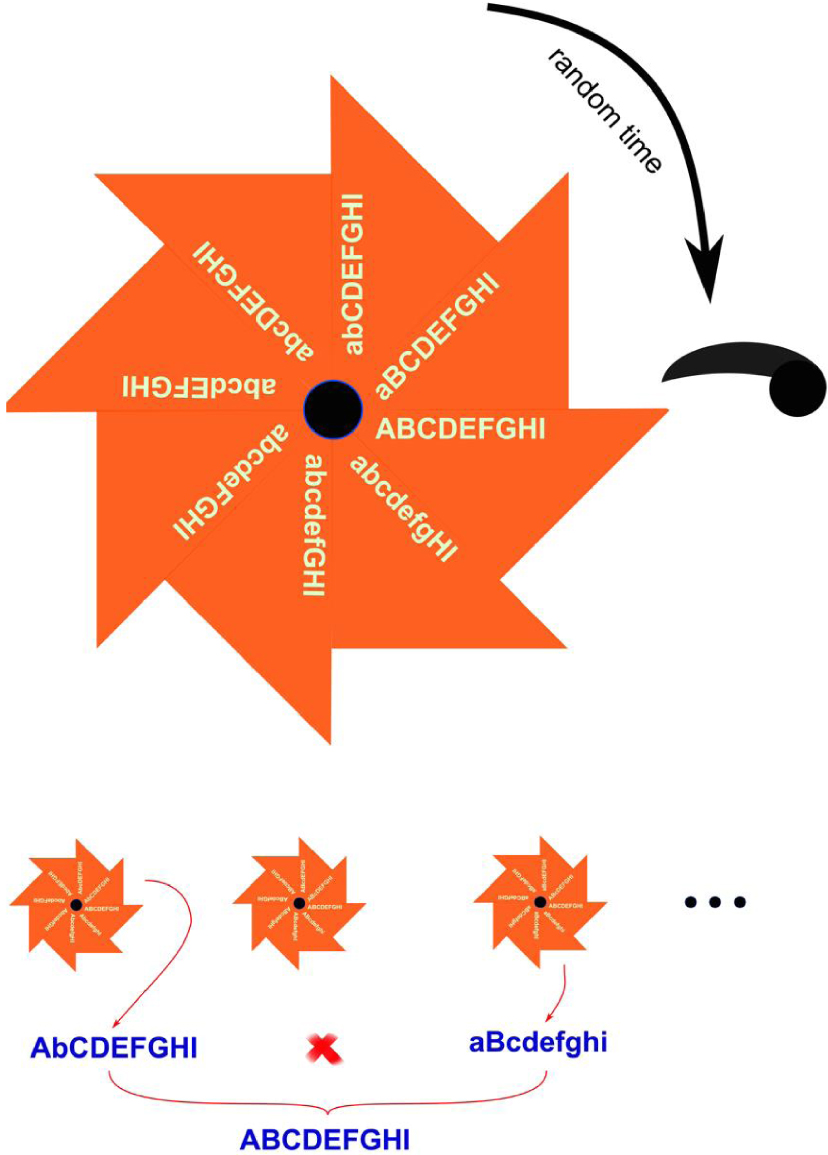
7.4. Detection of changes in the molecular evolutionary process
We investigated whether the substitution process in some sub-trees behaved differently from what was observed in the rest of the tree, statistically speaking. To this aim, we used the classical TN93 model from Tamura and Nei [53] with 3 parameters (purine transition rate, pyrimidine transition rate and transversion rate) and allowed these three rates to take, downstream of a candidate node Ni, values that differed from those they take in the rest of the tree. We then used a second (6-parameter) model. Since this model is nested in the first (3-parameter) model, we used as test statistic the likelihood ratio 2Δl = 2(l1 − l0), where l0 is the log-likelihood under assumption H0 (3-parameter TN93 model estimating all the elements of the tree) and l1 is the log-likelihood under assumption H1 (6-parameter TN93 model with local differentiation of the parameters downstream of a node of interest). We then compared the likelihood ratio to a distribution of the 𝜒2 with 3 degrees of freedom, whose significance threshold at 5% is 7.81. We were then able to identify the nodes from which the evolution process varied significantly and to quantify the variations of the different substitution rates, i.e. the nodes for which we can reject the H0 hypothesis that the 3-parameter model TN93 produces better estimates of tree substitution rates than the 6-parameter model TN93. We have chosen to implement these models ourselves in Python, in order to keep this parametrization flexibility. The program allows us to determine the set of nodes and leaves present downstream of a node of interest and to perform hypothesis testing by calculating the likelihood ratio and the different substitution rates.