

1 Introduction
Gene expression in eukaryotes is commonly regulated by elements, enhancers, or silencers, lying far from the gene on the genome. The E. coli arabinose, galactose, deo, lactose, and glp operons are repressed from sites that are clearly separated from the genes, if not far from them. The best-studied example of activation of this kind in E. coli is provided by the genes in charge of nitrogen assimilation (NRI-regulon). In addition to their relevance to E. coli, these regulations offer models for their eukaryotic counterparts, because of the simplicity of the prokaryotic interactions. This review presents recent data concerning the classical systems (galactose operon, NRI-regulon) as well as the latest members of this family (coliphages λ and 186, ula operon, members of the RpoN-regulon).
2 DNA looping for gene repression
2.1 The bacteriophage λ genetic switch
A genetic switch enables the phage to replicate either lytically or lysogenically. Molecular dissection of this process has been achieved in major part by Mark Ptashne's group [1,2]. The key component of the switch is the λCI repressor. Three phage promoters, PR, PL, PRM, are involved in this process. The dimeric repressor binds with a high degree of cooperativity to two of the three naturally adjacent operator sites, OR1, OR2 and OL1, OL2, respectively, to repress transcription of the lytic genes from PR and PL respectively and to activate its own synthesis from PRM (Fig. 1A).
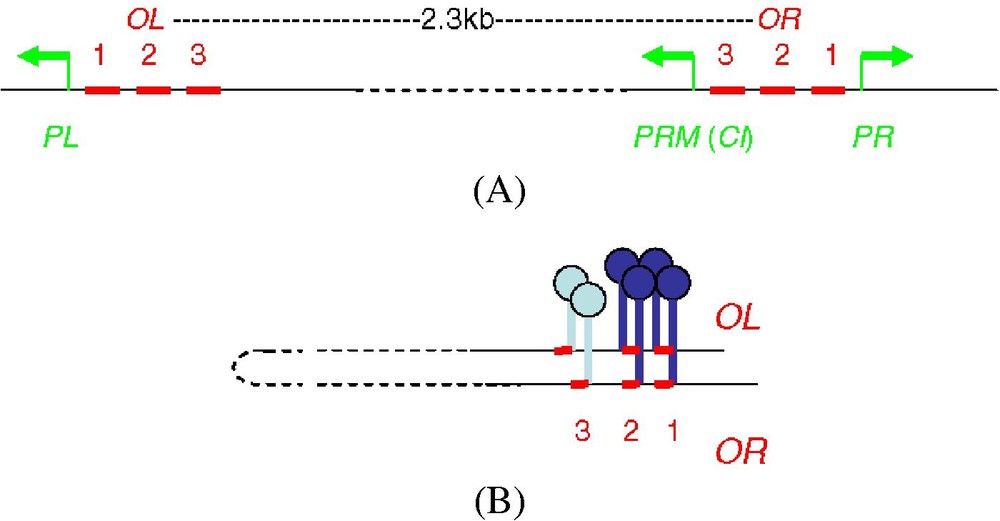
(A) Binding of the dimeric repressor with a high degree of cooperativity to two of the three naturally adjacent operator sites, OR1, OR2 and OL1, OL2, respectively, to repress transcription of the lytic genes from PR and PL respectively and to activate its own synthesis from PRM. (B) Octamerisation of the repressor between the two pairs of high-affinity sites, OL1–OL2, on one side, OR1–OR2, on the other side, inducing looping of the intervening DNA.
These sites can be separated from one another [3], without any biological significance (see [4]). On the contrary, like the three adjacent sites of each operator, OL or OR (Fig. 1A), a first set of biochemical, genetic and functional data seemed to indicate in 1996 that the dimers were closely associated on DNA and that the repressor was preferentially acting as a tetramer or a multimer of higher order. Thus, in addition to their unfitness for long-range action, the dimers have a lateral surface of interaction which is not restricted to the non-DNA-binding C-terminal domain (CTD), their function of activation at PRM is lost when OR1 is separated from OR2, the DNAseI footprint as well as the electron microscopy of the DNA-protein structures they form when their binding sites are close, may simply indicate protein aggregation between the sites, rather than DNA loop formation (see [4]).
In addition, analytical ultracentrifugation indicated that the repressor in solution could tetramerise, and even more easily, octamerise, without further aggregation [5]. Like the tetramers, the octamers bind cooperatively to DNA [6]. However, octamerisation was only detected by ultracentrifugation and this technique could not specify the organization of the subunits.
The simultaneous binding of the protein to four sites in vitro and in vivo indicated that the protein that had tetramerised on two adjacent high-affinity sites could actually octamerise and induce clear DNA loops [7]. Simultaneously, the crystallization of the C-terminal domain (CTD) of the λCI repressor responsible for the dimerisation of the subunits and cooperative binding of the dimers on the DNA, confirmed that the tetramerisation and the octamerisation of the protein were favoured [8,9].
However, the long-range action that the loops induced by the λCI tetramer seemed to uncover was not involved in the repression of the lytic genes from PR and PL, separated by nearly 2300 pb. Indeed, repression of β-galactosidase from PR-lacz fusions is only weakly (4-fold) improved [7], whereas repression of PR (PL respectively) by the repressor bound to OR (OL resp.) is already efficient in the absence of repressor binding to the remote OL (OR resp.) operator [1,2]. In fact, for other E. coli genes, when auxiliary sites are required for full and efficient repression of the genes, they are located at moderate distances from the promoter, and the gain in repression is lost when they are moved away. Thus, whereas the repression of the deoxyriboaldolase gene of the deo operon is 10.5-fold improved when the two operator sites, deoO2 and deoO1, are artificially separated by 1245 bp, it is only 3.3-fold enhanced for 4076 bp under the same conditions, and the natural distance is 600 bp [10]. Similarly, an auxiliary wild type or artificial lac operator site can efficiently improve β-galactosidase repression (up to 35-fold), only when it is relatively close from the first one (100 or 400 bp) [11–13]. The repression of β-galactosidase is no longer enhanced when the sites are separated by 3600 bp, even though these sites have a high affinity for the repressor [13] and are able to induce DNA loops as large as 2 kb [14].
The octamerisation of the non-DNA bound repressor only takes place at high repressor concentration exceeding that necessary for the lysogeny. Thereby, it had been suggested that some of these preformed octamers, once bound to the high-affinity OR1 or OR2 sites of the OR operator, facilitate the binding of the repressor to the weakest affinity site, OR3 (Fig. 1A), in order to stop repressor synthesis from PRM (OR3 overlaps PRM) when the repressor is produced in excess (negative autoregulation of the λCI repressor) [5,15]. In fact, a phage with a mutation in OR3 that eliminates the ability of λCI to repress PRM forms a lysogen that produces elevated λCI levels and is defective in prophage induction by UV [16]. This revealed the importance of OR3 and of the negative autoregulation of the repressor in switching from lysogeny to lytic development. The RecA protein that cleaves the repressor when the cell is irradiated is unable to work correctly when the concentration of repressor exceeds the lysogenic concentration. A 2.5-fold increase is sufficient to achieve this situation [16].
Repression of PRM also requires the third site of the remote OL operator, OL3, [16], as well as each of the four OL1, OL2, OR1, OR2 sites [17]. For this reason, it has been suggested that the repressor is octamerised between the two pairs of high-affinity sites, OL1–OL2, on one side, OR1–OR2, on the other side, inducing looping of the intervening DNA (Fig. 1B). This loop brings closer the two weakest affinity sites of the OL and OR operators, OL3 and OR3. The repressor is then able to bind to OR3 and OL3, without any contact with the repressor bound to the four other sites (Fig. 1B). The cooperative binding of the dimer at the OR3 and OL3 sites for repression of PRM is essentially similar to that of the dimers at the two other pairs of sites, OR1–OR2 or OL1–OL2, for repression of PR and PL, respectively.
This is a new function of the loop: the structural loop induced by the interaction of two proteins at distant sites, brings closer two other sites in their vicinity and allows protein binding to these sites.
2.2 The bacteriophage 186 genetic switch
Coliphage 186, a member of the P2 family of phage, shows essentially no similarity with λ at the protein or DNA level. Nevertheless, the lysis–lysogeny switches of each phage show superficial similarity. Like for λ, UV irradiation induces the lytic cycle. However, at this signal, the CI immunity repressor is not cleaved by the RecA protein, like the λCI repressor. Instead, it is reversibly inactivated by a SOS-induced phage protein [18]. Thereby, the two proteins display some structural differences. Furthermore, the lytic genes are transcribed from only one promoter (instead of two promoters, PR and PL, for the λ phage). The synthesis of the 186CI repressor is maintained during lysogeny by transcription from the PL promoter.
In spite of their genomic and functional differences, the two phages and their CI repressor maintain lysogeny in a related molecular way, remarkably adjusted to their differences.
- A In the absence of repressor (Fig. 2A), transcription from the lytic promoter, PR, is 60 fold stronger than that from PL. It interferes with transcription from PL, and this is sufficient to repress it. This transcriptional interference is a specific feature of bacteriophage 186 [19,20].
- B Like λCI, the repressor can octamerise in solution and on the DNA. At the intermediate concentration necessary for maintenance of the lysogeny (Fig. 2B), the 186CI repressor binds highly cooperatively in an all-or-none manner to the three adjacent sites of the operator overlapping the PR promoter (λCI was bound to only two of these sites, which brings out another structural difference between the two proteins). PR is strongly repressed (400-fold), whereas PL is activated (2.2-fold). Thus, 186CI positively regulates its own transcription, indirectly, without contacting the RNA polymerase at PL like λCI at PRM [21]. More precisely, the repressor is octamerised between the three operator sites at PR and a FL (alternatively FR) site, located some 300 bp away (Fig. 2B). The FL and FR sites are important for the lysogeny and are another specific feature of phage 186. By strengthening PR repression, they increase the convergent transcription from PL. In this process, the difference in expression between PR and PL is 10-fold increased.
- C When the CI concentration exceeds the lysogenic concentration (Fig. 2C), by only 2-fold, the PL promoter is partially repressed (negative autoregulation). For this, the repressor binds simultaneously to the three operator sites at PR and to a fourth operator site located at PL, 62 bp from PR [21]. This is possible because FL and FR are independently occupied by the repressor. Therefore, another role of FL and FR is to prevent repressor binding to PL at the lysogenic concentration.
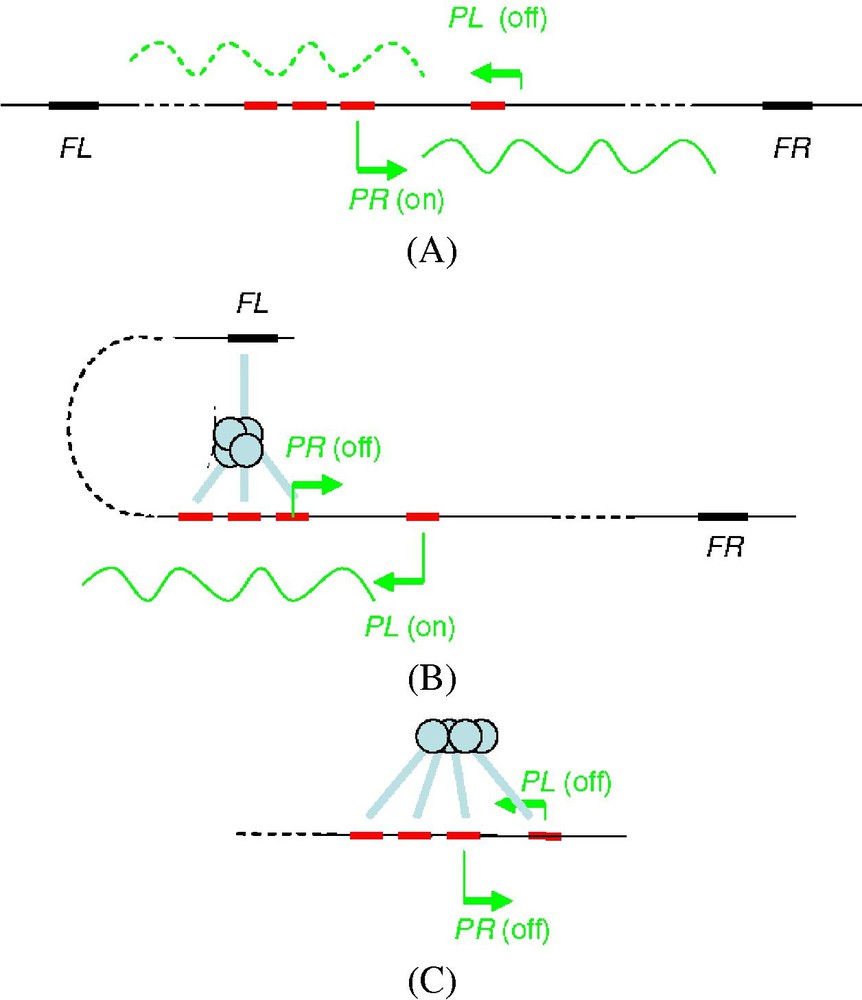
(A) Transcription from the lytic promoter, PR, in the absence of repressor. (B) Binding of the 186CI repressor in an all-or-none manner to the three adjacent sites of the operator overlapping the PR promoter at the intermediate concentration necessary for maintenance of the lysogeny. (C) Partial repression of the PL promoter when the CI concentration exceeds the lysogenic concentration by only 2-fold (negative autoregulation).
2.3 Tetramerisation of the GalR repressor
The gal operon is responsible for galactose metabolism in E. coli. It is the first E. coli operon, together with the arabinose operon, that was found regulated from multiple genomic sites. Its repression requires the GalR protein and two sites with a good affinity for this protein, an interior site within the operon, Oi, and an exterior site, Oe [22], as shown in Fig. 3A.
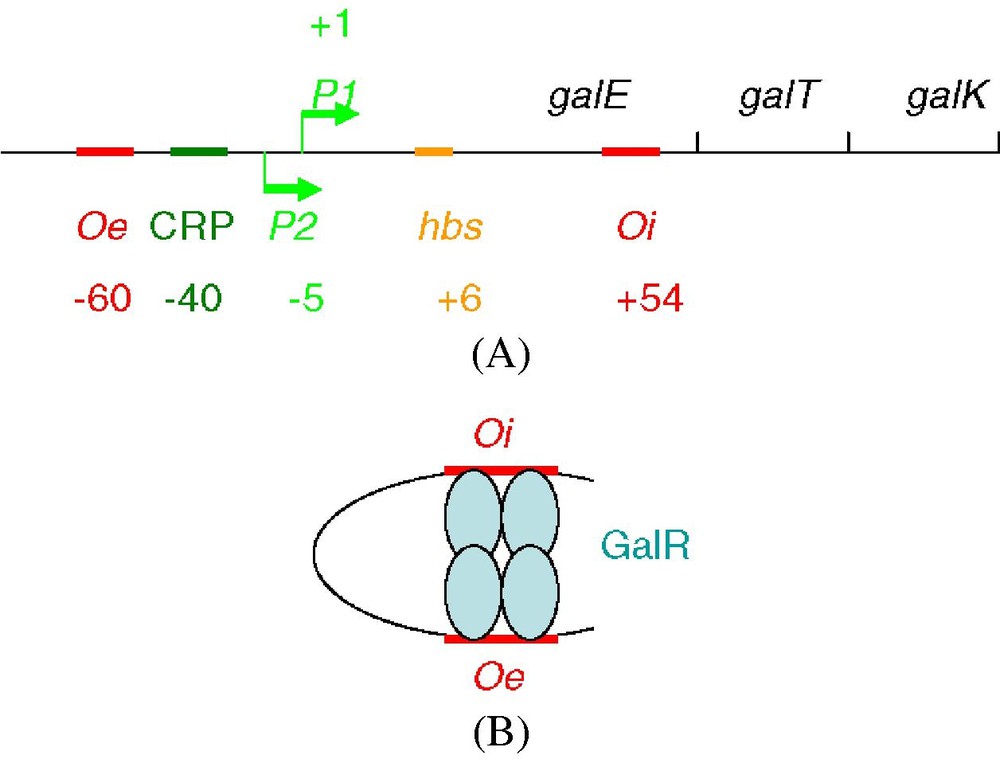
(A) Repression of the gal operon requiring the GalR protein and two sites with a good affinity for this protein, an interior site within the operon, Oi, and an exterior site, Oe [22]. (B) Tetramerisation of GalR at Oi and Oe.
Contrary to several other repressors, the dimeric GalR repressor does not tetramerise in solution or on the DNA in vitro. This suggests that the two dimers have a weak affinity for each other. Moreover, GalR and LacI belong to the same family of repressors. The lack for a tetramerisation domain present in LacI, has long been thought to be responsible for this apparent impossibility to tetramerise (see [23]).
In fact, repression seems to require various auxiliary proteins, in addition to GalR: the HU nucleoid protein, which specifically interacts with GalR [24], from an hbs site between Oi and Oe, as well as the RNA polymerase or the activating CRP protein (see [23] and Fig. 3A).
However, the repression of the gal operon does not seem to be fundamentally different from that of the arabinose, deo, or lac operons. A functional assay designed to detect protein–protein interactions directly in situ in E. coli indicated that the two GalR dimers could directly interact in vivo from artificial constructs excluding the binding of the assumed auxiliary proteins between Oi and Oe and that the effect of HU deduced from the use of HU-deficient strains was overestimated [23]. This artificial situation is not essentially different from the wt one. One consequence of short-range DNA looping is to require that the sites lie on the same face of the DNA helix. In the artificial constructs, the angular orientation between Oi and Oe was nearly the same as in the wt ones [23]. As to HU, it does not act to improve this angular orientation, as shown by transcriptional assays with the wild type promoter regions in vitro [25], nor is an adaptor inserted between the two dimers [26].
The gal repressor has never been crystallized. Genetic assays such as the one described in [23] allow us to detect protein–protein contacts and to specify them by screening the protein mutants deficient in these contacts. A genetic map of the interface between the two GalR dimers was determined by this genetic approach, in conjunction with site-directed mutagenesis at the inferred sites of interaction [27,28]. The tetramerisation of GalR at Oi and Oe [23] (Fig. 3B) has been confirmed by mutations that compensate for the lack of tetramerisation as well as by mutations that strengthen the tetramerisation [26,28].
Finally, GalR is able to repress efficiently RNA synthesis from the wt promoters in vitro in the absence of HU. However, DNA loop formation is only observed in the presence of HU by AFM microscopy, even though HU cannot be detected on these loops. This again suggests a transient role of HU [29]. Thus, HU is not essential to the repression of the gal operon. However, it improves repression by stabilizing the weak interaction between the two GalR dimers.
Another recent aspect of repression of the gal operon is noteworthy, though it is not related to DNA looping. The physiological role of E. coli Spot 42 RNA, encoded by the spf gene, has long remained obscure. In fact, if GalR represses the three genes of the gal operon at the transcriptional level, the last gene of the operon, galk, is specifically repressed by Spot42, functioning as an anti-sense RNA, when the cell derives its energy from sources other than galactose [30].
2.4 The ula regulon
The ula regulon, responsible for the utilization of l-ascorbate in E. coli, is formed by two divergently transcribed operons, ulaG, and ula ABCDEF. The regulon is negatively regulated by a repressor of the DeoR family, which is encoded by the ulaR gene. Full repression of the ula regulon requires simultaneous interaction of the repressor with both divergent promoters, at sites separated by 187 pb. This process is helped by the integration host factor [31].
3 The lac repressor–operator loop as a gene insulator
The question of whether lac operator–repressor loops can insulate a gene in the loop from its genomic environment is an intriguing possibility (M. Amouyal, B. Pineau, D. Bienvenu, unpublished results).
A DNA loop might have intuitively this function, since it forms a closed topological domain.
Known insulator elements vary greatly in their DNA sequences and the specificity of proteins that bind to them. However, they share at least one of the two following properties: (i) they have the ability to act as an enhancer blocker when placed between an enhancer and the promoter, (ii) they have the ability to protect against position effects due to the chromosomal environment (for a review, see [32]).
Additionally, like the lac operator sequences, known insulators do not decondense chromatin and do not have any enhancer effect by themselves. In that sense, they constitute a neutral barrier for the domain that they delimit. Furthermore, like the lac operator, the gypsy insulator of Drosophila, Su(Hw), is likely to operate through DNA looping when it flanks a portion of genome on each side [33,34].
Recently, Bondarenko et al. [35] have used the lac operator sequences as a model for eukaryotic insulators. They have shown that the E. coli glnAp2 promoter is no longer activated by NRI in vitro, when the promoter is inserted within a lac repressor–operator DNA loop. DNA loop formation is essential since only one lac repressor–operator element inserted between the NRI enhancer and the gene does not block the action of the enhancer. This does not reproduce the effect of an insulator, but may contribute to its understanding.
4 DNA looping for gene activation: the RpoN regulon
In E. coli, there are seven different sigma factors (listed in [36]). Core RNA polymerase has the capacity to bind each of them to form seven different holoenzymes recognising different sets of promoters. These sigma subunits are all homologous to sigma70, except one, sigma54 transcribed by the rpoN gene. Contrary to the sigma70-holoenzyme, the sigma54-holoenzyme cannot make an open complex in the absence of activator. DNA loop formation between the transcription machinery (sigma54-holoenzyme) and the activator bound to the upstream enhancer sites, 100 bp or more from the promoter, mediates the activation of all sigma54-dependent promoters of the RpoN regulon. In E. coli, there are 12 known activators of this family (reviewed in [36,37]). At their interface with RNA polymerase, they all contain a stretch of amino acids first described for the AAA+ proteins, a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. However, each of them responds to a specific environmental signal or stress.
4.1 The NRI regulon
The expression of these genes is modulated by the deprivation or excess of nitrogen compounds issued from a variety of sources [38]. Ammonium salts are the preferred source, but this source can be replaced by other sources (amino acids, nucleosides, nucleobases…), as well as by the scavenging of some cellular peptides [39] in case of ammonia deprivation. This substitution is controlled and constitutes the nitrogen (Ntr) response. In view of the number of nitrogen sources and genes involved, the assimilation of nitrogen compounds must adjust itself to a level of complexity fairly greater than the other regulations reviewed here.
At least six operons are regulated by the same protein, NRI (or NtrC), in correlation with nitrogen variations and nitrogen metabolism (NRI regulon). NRI activates the expression of glnALG (glutamine synthetase and nitrogen response regulators), astCADBE (arginine catabolism), glnK-amtB (an alternate PII enzyme for adenylation of glutamine synthetase and an ammonia transporter), nac (a sigma70-dependent transcriptional activator), and glnHPQ (glutamine transport) in E. coli. Several lines of evidence also suggest that it controls the expression of gltIJKL (glutamate–aspartate transport). In addition to these genes, microarray analysis suggests that NRI might also activate ddpXABCDE (d-alanine-d-alanine metabolism), potFGHI (putrescine transport), yeaGH (unknown function), ygjG (a transaminase), and yhdWXYZ (amino acid transport) [39].
In response to ammonia deprivation, the intracellular concentration of NRI rises dramatically. The protein is phosphorylated. The unphosphorylated NRI protein forms a dimer in solution and on the DNA. Upon phosphorylation, NRI undergoes conformational changes that lead to the multimerisation of the protein on the DNA and to assembly of a DNA-bound complex with ATPase activity [40,41]. This energy is conveyed to the transcriptional complex to activate transcription. To this end, the core RNA polymerase must associate with the sigma54 factor [42], in place of the six alternative sigma subunits that the core RNA polymerase has the capacity to bind in order to recognize the different sets of promoters.
Furthermore, the transition to nitrogen limitation induces the genes of the NRI regulon, sequentially, in a cascade. The low concentration of phosphorylated NRI initially present during the transition is sufficient for expression of the glnALG operon. However, a higher NRI concentration is required in vitro and in vivo for nac, glnk, the astCADBE operon and probably for other operons [43]. In correlation with this finding, the cells that have been genetically manipulated such that the NRI concentration is always low retain the ability to fully activate the glnAp2 promoter of the glnALG operon, but are unable to grow on arginine as a nitrogen source, for activation of the astC promoter, or to activate the glnK promoter (see [43]).
The primary response to nitrogen limitation is indeed to express the three genes of the glnALG operon (Fig. 4): (i) the increase in the level of the glnA product, the enzyme glutamine synthetase, compensates in part for the reduced availability of ammonia (ammonia deprivation results in a decline in the intracellular concentration of glutamine, precursor molecule, with glutamate, for all cellular nitrogen compounds); (ii) the kinase NtrB, which catalyses NRI phosphorylation, is the product of glnL; (iii) NRI is the product of glnG and its synthesis needs to be increased.
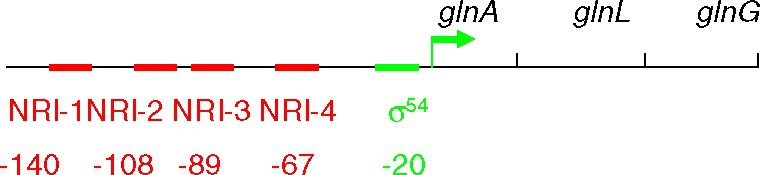
Primary response to nitrogen limitation expressing the three genes of the glnALG operon: (i) the increase in the level of the glnA product, the enzyme glutamine synthetase, compensates in part for the reduced availability of ammonia (ammonia deprivation results in a decline in the intracellular concentration of glutamine, precursor molecule, with glutamate, for all cellular nitrogen compounds); (ii) the kinase NtrB, which catalyzes NRI phosphorylation, is the product of glnL; (iii) NRI is the product of glnG and its synthesis needs to be increased.
In a second step, with the increase in phosphorylated NRI, the genes and operons, whose products have the potential to increase the intracellular concentration of glutamine or are otherwise useful under starvation conditions, are activated.
The glnAp2 promoter is activated at low NRI concentration from two high-affinity sites 1 and 2, centred at −108 and −140 upstream from the transcription start (Fig. 4). These sites are able to activate transcription efficiently up to 1400 bp [44]. The protein binds highly cooperatively to these sites. In addition, there are two low affinity sites, 3 and 4, located between the first sites and the start of transcription, at −89 and −67 (Fig. 4). These sites are only occupied at high NRI concentration. When this is the case, the activity of the glnAp2 promoter is reduced [45]. DNA loop formation between the distant sites and the transcription start, is thought to be disturbed by NRI binding to sites 3 and 4.
On the contrary, for the same high concentrations, other promoters of the gene cascade and of the NRI regulon, using nitrogen sources other than ammonia, can be activated. The arrangement of the NRI binding sites, the association of at least two adjacent sites of variable affinity, is different at the corresponding promoters: for example, there are two overlapping high-affinity sites at the glnHp2 promoter, but a weak affinity site and a strong one at the nac promoter, and these tandem sites are occupied for different NRI concentrations (see [43]).
The involvement of auxiliary proteins (Nac, IHF, ArgR…) at intermediate sites between NRI and sigma54 to modulate the level of activation seems to be a current feature of sigma54-controlled promoters [36]. However, their presence has been found only accessory in some instances [46].
Thus, the Nac protein at the nac promoter of Klebsiella aerogenes is another transcription factor specific to the genes regulated by nitrogen deprivation. When the Nac protein binds between the NRI enhancer and the transcription start at this sigma54 dependent-promoter, it reduces its activity, probably like NRI at the 3 and 4 sites of glnAp2 [47].
Thereby, like for phage λ and contrary to what is commonly admitted, the homologous sites of a series of adjacent, tandemly arranged sequences, are not necessarily functionally equivalent.
4.2 Sigma54-dependent genes that are not involved in nitrogen metabolism
Eleven such activators are known (reviewed in [36,37]). Their architecture depend on how they respond to the signal of activation [37]. Some need to be phosphorylated by a separate kinase in a two-component response, like the NtrB/NRI pair. For others, for example PspF, interaction with another protein is the cue for activation. For others, such as FhlA, this signal is given by the binding of an inducer directly or indirectly connected to the primary agent responsible for the environmental changes or stresses.
ZraR (or HydG) controls the heavy-metal (Zn++, Pb++) tolerance system expressed by the zraP and zraSR genes [48]. The products of zraSR (hydHG) are a membrane-associated sensor kinase, ZraS, and the response regulator, ZraR.
AtoC regulates expression of the atoDAEB operon. This operon is involved in acetoacetate and short-chain fatty acid catabolism. The gene located just upstream of atoC encodes the AtoS sensor kinase that modulates AtoC activity. AtoC also plays a central role in the regulation of polyamine biosynthesis by binding to ornithine decarboxylase and inhibiting it [49].
The formate-sensing transcription regulator, FlhA, controls the formation of the formate hydrogen lyase complex required for formate metabolism. Activation of the hyp, hyc, fdhF, and hydN-hypF operons is induced by direct formate binding to FlhA (reviewed in [36]).
The products of the prpBCDE operon degrade propionate. Most of the genetics of propionate catabolism and analysis of gene expression has been studied with S. enterica serovar Typhimurium. In addition to sigma54, expression requires IHF and PrpR, which is homologous to NRI. 2-Methylcitrate or a product of its metabolism has been proposed to bind PrpR and induce the operon. It is assumed that regulation in E. coli is similar [36].
The PspF protein that controls the phage shock response of the pspABCDE operon does not contain any known regulatory input domains. Its activity is controlled by formation of a repressive complex with another protein, PspA [50].
NorR (also called YgaA) controls the nitric oxide detoxification system, expressed by the norVW operon. The genetics and biochemistry of this system has been recently specified. norV (YgaK) encodes a flavorubredoxin and norW (YgbD), an NADH:(flavo)rubredoxin oxydoreductase [51]. The norVW genes also have a role in the protection against reactive nitrogen intermediates [52]. It is not yet clear how the signal of activation is transduced to NorR [51].
The hyf locus (hyfABCDEFGHIJ–hyfR–focB) of E. coli encodes a 10-subunit hydrogenase complex (hydrogenase-4 [Hyf]); a potential sigma54-dependent transcriptional activator, HyfR (related to FhlA); and a putative formate transporter, FocB (related to FocA). Since FhlA activates the Hyf operon under aerobic conditions, the hyf operon belongs to the formate/FhlA regulon of E. coli. Then, it is expected that the Hyf complex has a role similar to that of the Hyc complex in fermentative formate metabolism. However, HyfR only activates the hyf operon under anaerobic conditions. Thus, hyf seems to be a vestigial, unexpressed operon and its physiological purpose remains obscure [53,54].
Last, little is known about four other sigma54-dependent transcriptional activators, YfhA, YgeV, DhaR, and RtcR [37].