

1 Introduction
Monoclonal antibodies have been developed for target cell surface receptor molecules, and they represent the most advanced drugs in clinical investigations. Most kinase inhibitors in clinical development are anticancer therapeutics. Two milestones in drug development have been achieved in this field. In 1998, the monoclonal antibody herceptin (also called trastuzumab, developed by Genentech) directed against the receptor tyrosine kinase (RTK) HER2 has been approved for the treatment of breast cancer by the FDA as the first example of a next-generation anti-cancer therapeutic [1].
ErbB2 is a proto-oncogene located on chromosome 17, whose product, a tyrosine kinase receptor (HER2), is a 1255 amino acid glycoprotein of 185 kDa, which belongs to the family of epidermal growth factor receptors [2]. ErbB2 was found to be overexpressed in several types of human adenocarcinomas, especially in tumours of the breast and the ovary [3–5]. The overexpression was correlated with more aggressive tumours and a poorer prognosis [6–8]. Hence, herceptin (trastuzumab), humanized antibody against the overexpressed ERBB2, is proven to be effective in treating breast cancers with ErbB2 amplification, which is directed against a juxtamembrane epitope in the HER2 ectodomain [9]. High-affinity binding of trastuzumab to HER2 results in attenuation of aberrant HER2 kinase-associated signal transduction, with coordinate changes in cell cycle distribution (e.g., decreases in the fraction of cells undergoing the S-phase), specifically in HER2-overexpressing cells [10].
Single nucleotide polymorphism (SNP) accounts for common form of human genetic variation. About 500,000 SNPs fall in the coding regions of the human genome [11]. Among these, the non-synonymous SNPs (nsSNPs) cause changes in the amino acid residues. These are likely to be an important factor contributing to the functional diversity of the encoded proteins in the human population [12]. Discovering the deleterious nsSNPs is the main task of pharmacogenomics. So, the main aim of this study is to identify deleterious nsSNPs associated with HER2 protein and their nature of stability and binding affinity with herceptin. We here report that the most deleterious nsSNP is rs4252633 on HER2 receptor. We modeled the mutant HER2 based on this nsSNP, which showed more structural deviation from native HER2. In addition to that, herceptin is having higher binding affinity with mutant structure than in native HER2. This shows that herceptin is the best drug for the molecular target of HER2 receptor, even with most deleterious nsSNP.
2 Materials and methods
2.1 Datasets
The SNPs and their related protein sequence for ErbB2 gene were obtained from the dbSNP [13] available at http://www.ncbi.nlm.nih.gov/SNP/ for our computational analysis.
2.2 Predicting stability change on mutation by coding nsSNPs based on support vector machine (I-Mutant 2.0)
Single-base changes in protein coding regions of DNA, which lead to changes in amino acids, have an effect on protein structure and function. However, most of the deleterious non-synonymous single nucleotide polymorphisms (nsSNPs)-associated proteins show less stability [14]. We used the program I-Mutant 2.0 available at http://gpcr.biocomp.unibo.it/cgi/predictors/I-Mutant2.0/I-Mutant2.0.cgi. I-Mutant 2.0 is a support vector machine (SVM)-based tool for the automatic prediction of protein stability changes upon single point mutations. I-Mutant 2.0 predictions are performed starting either from the protein structure or, more importantly, from the protein sequence [15]. This program was trained and tested on a data set derived from ProTherm [16], which is presently the most comprehensive available database of thermodynamic experimental data of free-energy changes of protein stability upon mutation under different conditions. The output file shows the predicted free-energy change value or sign (DDG), which is calculated from the unfolding Gibbs free energy value of the mutated protein minus the unfolding Gibbs free energy value of the native type (kcal/mol). Positive DDG values means that the mutated protein posses high stability and vice versa.
2.3 Analysis of functional consequences of coding nsSNPs by sequence-homology-based method (SIFT)
We used the program SIFT [17], which is specifically aimed to detect the deleterious coding non-synonymous SNPs, available at http://blocks.fhcrc.org/sift/SIFT.html. SIFT is a sequence-homology-based tool, which presumes that important amino acids will be conserved in the protein family. Hence, changes at well-conserved positions tend to be predicted as deleterious [17]. We submitted the query in the form of SNPids or as protein sequences. The underlying principle of this program is that SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence. SIFT is a multistep procedure that, given a protein sequence, (a) searches for similar sequences, (b) chooses closely related sequences that may share similar function, (c) obtains the multiple alignment of these chosen sequences, and (d) calculates normalized probabilities for all possible substitutions at each position from the alignment. Substitutions at each position with normalized probabilities less than a chosen cut-off are predicted to be deleterious, and those greater than or equal to the cut-off are predicted to be tolerated [18]. The cut-off value in SIFT program is a tolerance index of ⩾0.05. Higher the tolerance index, less functional impact a particular amino acid substitution is likely to have.
2.4 Simulation for functional change in coding nsSNPs by structure-homology-based method (PolyPhen)
Analyzing the damaged coding non-synonymous SNPs at the structural level is considered to be very important to understand the functional activity of the concerned protein. We used the server PolyPhen [19], which is available at http://coot.embl.de/PolyPhen/ for this purpose. Input options for PolyPhen server is protein sequence or SWALL database ID or accession number together with sequence position with two amino acid variants. We submitted the query in the form of protein sequence with mutational position and two amino acid variants. Sequence-based characterization of the substitution site, profile analysis of homologous sequences and mapping of substitution site to a known protein's 3-dimensional structures are the parameters taken into account by PolyPhen server to calculate the score. It calculates position-specific independent counts (PSIC) scores for each of the two variants, and then computes the PSIC score difference between them. Higher the PSIC score difference, higher is the functional impact a particular amino acid substitution is likely to have.
2.5 Modeling nsSNP location on protein structure and the nature of its stability
Structure analysis was performed for evaluating the structural stability of native and mutant proteins. We used the web resource SAAPdb [20] to identify the protein (PDBid: 1n8z) coded by ERBB2 gene. It is to be noted that 1n8z contain three chains, in which chains A and B belong to herceptin and chain C belong to HER2 protein (ERBB2). We also confirmed the mutation position and the mutation residue in PDB id 1n8z of chain C from this server. The mutation was performed by using SWISSPDB viewer and energy minimization for 3D structures was performed by NOMAD-Ref server [21]. This server uses Gromacs as default force field for energy minimization based on the methods of steepest descent, conjugate gradient and L-BFGS methods [22]. We used the conjugate gradient method for optimizing the 3D structure in the C chain of HER2 protein. Divergence in mutant structure with native structure is due to mutation, deletions, and insertions [23], and the deviation between the two structures could affect the stability and alter the functional activity [24], which is evaluated by their RMSD value. In order to check the stability of the native and mutant modeled structures, identification of the stabilizing residues will be useful. We used the server SRide [25] for identifying the stabilizing residues in the native protein and in the mutant model. Stabilizing residues were computed using parameters such as surrounding hydrophobicity, long-range order, stabilization centre, and conservation score, as described by Magyar et al. [25].
2.6 Computation and interaction analysis of binding affinity for native and mutant HER2 with herceptin
Interaction free energies are crucial for analyzing binding propensities in proteins. We used the program FastContact that calculates the binding free energy between protein complexes. It is based on a statistically determined desolvation contact potential and Coulomb electrostatics with a distance-dependent dielectric constant [26]. In order to calculate the binding affinity for native and mutant HER2 protein with herceptin, we once again used SWISSPDB viewer for performing mutation at a specific position in the C chain of 1n8z. NOMAD-Ref server [25] was used for energy minimization for entire protein complex (1n8z), which includes HER2 protein complexed with herceptin. Then, we submitted both the native protein complex and modeled mutant complex independently to this server, which in turn computes the binding energy between herceptin and HER2 protein for both native and mutant proteins. The major interface residues involved in the interaction between herceptin and HER2 protein of native and mutant type were identified by the interface tool provided by iMolTalk [27].
A quantitative measure of the atomic motions in proteins can be obtained from the mean square fluctuations of the atoms relative to their average positions. These can be related to the B-factor [28,29]. Analysis of B-factors, therefore, is likely to provide newer insights into protein dynamics, flexibility of amino acids, and protein stability [30]. Protein flexibility is important for protein function and for rational drug design [31]. Therefore, flexibility of certain amino acids in protein is useful for various types of interactions that can be analyzed by the B-factor, which are computed from the mean-square displacement of the lowest-frequency normal mode using ElNémo server [32].
3 Results and discussion
3.1 SNP dataset from dbSNP
The ERBB2 gene investigated in this work was retrieved from dbSNP database [13]. It contained a total of 135 SNPs. Out of which, 10 were non-synonymous SNPs (nsSNPs), 10 were synonymous SNPs, and five were in non-coding regions. The rest were in the intron region. We selected non-synonymous coding SNPs for our investigation.
3.2 Identification of functional nsSNP by I-Mutant 2.0
More negative is the DDG value, less stable the given point mutation is likely to be, as predicted by I-Mutant 2.0 server [15]. Out of 10 nsSNP, we obtained the 8 nsSNPs that were found to be less stable by this server as shown in Table 1. Out of 8 nsSNPs, 5 nsSNPs, namely rs1801201, rs28933369, rs2172826, rs1058808 and rs36085723, showed a DDG value of >−1.0. The remaining three nsSNPs, namely rs4252633, rs1801200 and rs28933370 showed a DDG value of <−1.0, as depicted in Table 1. Out of eight nsSNPs that showed negative DDG, the four nsSNPs, namely rs4252633, rs28933369, rs1058808 and rs2172826, changed their amino acid from aromatic to polar amino acid, non-polar to polar amino acid, polar to non-polar amino acid and from polar amino acid to positively charged amino acid, respectively. In the other four nsSNPs, three nsSNPs, namely rs1801201, rs1801200 and rs36085723, changed their amino acid from non-polar to non-polar mutation and the remaining one with rs28933370 changed from polar to polar mutation. Since the amino acids mutations in the latter four nsSNPs share the same physiochemical properties, we considered the former four nsSNPs to be less stable and deleterious by this analysis.
List of nsSNPs that were predicted to be functionally significant by I-Mutant 2.0, SIFT and PolyPhen
| SNPs ID | Nucleotide change | AA change | DDG | Tolerance index | PSIC SD |
| rs4252633 | G/T | W452C | −0.92 | 0.04 | 2.420 |
| rs1801201 | A/G | I654V | −1.02 | 0.59 | 0.777 |
| rs1801200 | A/G | I655V | −0.81 | 1.00 | 0.944 |
| rs34602395 | G/T | S703I | 1.65 | 0.00 | 0.879 |
| rs28933369 | G/A | G776S | −1.52 | 1.00 | 1.722 |
| rs28933370 | A/G | N857S | −0.69 | 0.32 | 0.775 |
| rs28933368 | G/A | E914K | 1.37 | 0.00 | 1.470 |
| rs2172826 | C/G | P927R | −1.50 | 0.58 | 2.071 |
| rs1058808 | C/G | P1170A | −1.07 | 0.05 | 0.383 |
| rs36085723 | G/A | V1253M | −1.68 | 0.16 | 0.734 |
3.3 Deleterious nsSNP by SIFT program
The conservation level of a particular position in a protein was determined by using a sequence-homology-based tool, SIFT [17]. Protein sequences of 10 nsSNPs were submitted independently to SIFT program to check its tolerance index. Higher the tolerance index, lesser the functional impact a particular amino acid substitution is likely to have, and vice-versa. Among the 10 nsSNPs, four nsSNPs were found to be deleterious, having the tolerance index score of ⩽0.05. The results are shown in Table 1. We observed that, out of four deleterious nsSNPs, two nsSNPs showed a highly deleterious tolerance index score of 0.00, followed by two nsSNP with tolerance index scores of 0.04 and 0.05, respectively. Among these deleterious 4nsSNPs, two nsSNPs showed a nucleotide change from G → T, one from G → A and the other from C → G (Table 1). Also, according to the SIFT results, the four nsSNPs that showed a deleterious score showed a change in their amino acid from aromatic to polar, polar to non-polar, negatively charged to positively charged and polar amino acid in the native protein to a non-polar amino acid in the mutant protein. We found that two nsSNPs, namely rs4252633 and rs1058808, which that showed less stability by I-Mutant 2.0 server, were also seen to be deleterious according to SIFT. Therefore, these two nsSNPs were found deleterious nsSNPs by this investigation.
3.4 Damaged nsSNP by PolyPhen Server
The structural levels of alteration were determined by applying PolyPhen program [19]. Protein sequence with mutational position and amino acid variants associated with the 10 nsSNPs investigated in this work was submitted as input to PolyPhen server and the results were shown in Table 1. A PSIC score difference of 1.1 and above is considered to be damaging. It can be seen that, out of 10 nsSNPs, four nsSNPs were considered to be damaging. All the four nsSNPs exhibited a PSIC score difference between 1.470 and 2.420.
Three nsSNPs, namely rs4252633, rs28933369 and rs2172826, which were observed to be less stable by I-Mutant 2.0, were damaging according to PolyPhen. Also, two nsSNPs, namely rs4252633 and rs28933368, were deleterious by the SIFT program and were damaging according to PolyPhen. Hence, according to our investigation, only one nsSNP, i.e. rs4252633, was seen to be less stable, deleterious and damaging by all the three programs, namely, I-Mutant 2.0, SIFT and PolyPhen used in this work. Therefore, we considered this nsSNP rs4252633 for our further course of investigations.
3.5 Computing the stability by modeling of mutant structure
Mapping the damaged nsSNP rs4252633 into protein structure information was obtained from Single Amino Acid Polymorphism database (SAAPdb) [20]. The available structure for ERBB2 gene has a PDB id 1n8z that consists of three chains (A, B, and C), in which chains A and B belong to herceptin and chain C belongs to HER2 protein. Based on this nsSNP rs4252633, the mutation was at residue position 452 (W → C) in chain C of 1n8z. So, we performed the tryptophan to cysteine mutation for 1n8z in chain C at position 452 by SWISSPDB viewer to get the modeled structure. Then energy minimizations were performed by NOMAD-Ref server [21] for the native type HER2 protein (C chain of 1n8z) and the mutant type HER2 protein (W452C).
The total energy for the native-type structure and the mutant-type structure were found to be −35,685.387 and −35,523.078 kcal/mol, respectively (Table 2). Higher the total energy, less stable the protein structure will be. The total energy comparison showed that mutant type is little higher in energy than native type. Table 2 also shows that the RMSD value between the native type and the mutant type was 2.81 Å. Higher the RMSD value, larger the deviation between the two structures, which in turn influences the stability and the functional activity. In order to check the stability nature of the mutant type, we used SRide server [25] for identifying the stabilizing residues of native-type structure and mutant modeled structure. It can be seen from Table 2 that six stabilizing residues were identified in native type, whereas only five stabilizing residues were identified in mutant. Three stabilizing residues, namely Tyr(61), Gly(386) and Tyr(387), were found to be common to both the native structure and the mutant model. The remaining stabilizing residues of the native structure were not seen in the mutant model. Therefore, we predict that mutation from tryptophan to cysteine at the residue position 452 in C chain of 1n8z would be more deleterious. The mutation in this position could be important for ERBB2 gene due to slightly higher total energy; it shares only three common stabilizing residues of mutant type compared with the native type. Fig. 1a depicts the superimposed structure of mutant and native HER2 protein and Fig. 1b depicts the superimposed structure of the tryptophan residue in native and cysteine residue in mutant.
RMSD and total energy of native and mutant model of HER2
| Parameters | Native structure (C chain of 1n8z) | Mutant model (C chain of 1n8z at W452C) superimposed with native structure |
| RMSD | 0.00 Å | 2.81 Å |
| Total energy after energy minimization | −35,685.387 kcal/mol | −35,523.078 kcal/mol |
| Stabilizing residues | Tyr61, Leu63, Asn67, Gly386, Tyr387, Arg434 | Tyr61, Leu127, Ser351, Gly386, Tyr387 |
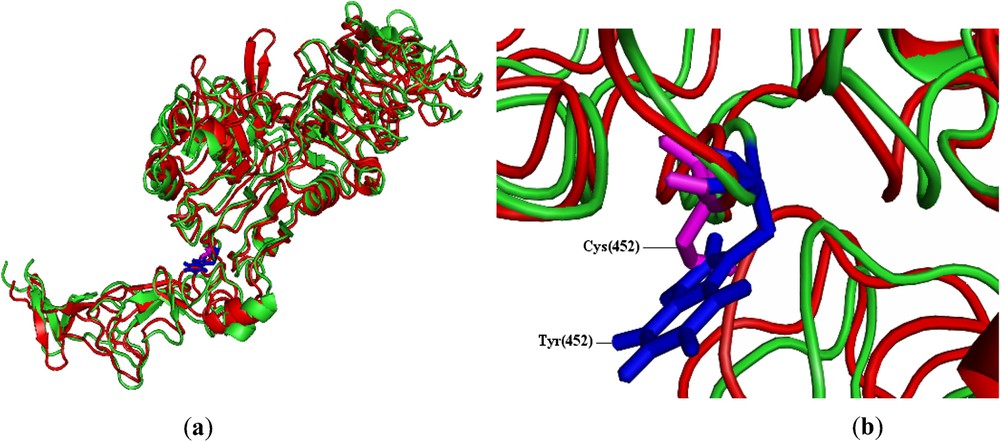
Pymol view of (a) superimposed structures of HER2 protein of native (green) and mutant (red) types. (b) Tryptophan residue (blue) of native HER2 and cysteine residue (magenta) of mutant HER2 at the residue position of 452 (rs4252633). For reference to colours, see the web version of this article.
3.6 Rationale of binding efficiency for native and mutant HER2
FastContact is useful for finding the binding free energy for the given receptor and ligand [26]. We have used this server for our nsSNP analysis with respect to the binding energy between the herceptin and HER2 protein of native and mutant types. In order to understand the binding efficiency of both native and mutant HER2 protein, we selected the same PDB id 1n8z. Then, we performed the mutation at residue position 452 from W → C in chain C by SWISSPDB viewer, and energy minimization were performed by Nomad-Ref [25] for the entire protein complex (PDBid 1n8z is having HER2 protein complex with herceptin) of both native and mutant types to get optimized structures. In this analysis, we found that the binding free energy for herceptin with mutant HER2 protein was −24.40 kcal/mol, whereas with native HER2 protein the binding free energy was −15.26 kcal/mol (Table 3). The higher binding free energy between herceptin and mutant HER2 protein may probably be due to non-covalent interactions. So, we used iMolTalk [27] to find out the various types of interaction between herceptin and HER2 protein of native and mutant types. We found eight interactions that include six hydrogen-bonding and two electrostatic interactions between herceptin and mutant HER2 protein. On the other hand, there were only six interactions, which include four hydrogen-bonding and two electrostatic interactions between herceptin and native HER2 protein. These results are separately shown in Table 3, Figs. 2a and 2b. The complex of herceptin with native HER2 protein is shown in Fig. 2a and the complex of herceptin with mutant HER2 protein is shown in Fig. 2b. The analysis portrays that the higher binding free energy was probably due to additional hydrogen bonding of Asn30 to Asp596 and Asn30 to Glu598 between herceptin and mutant HER2 protein, as shown in Fig. 3. Moreover, we further investigated the reason for this additional hydrogen bonding. We used the ElNémo server [32] to find out the flexibility nature of the two amino acids, namely Asp596 and Glu598 of HER2 protein of native and mutant types. We found that normalized mean-square displacement of amino acids participating in additional hydrogen bonding of HER2 protein of mutant type had slightly higher flexibility as compared to those two amino acids of native type (Table 4). Due to the flexible nature of these two amino acids in mutant HER2 protein, additional hydrogen bonding was formed with herceptin.
Binding free energy and major interactions for native and mutant complexes
| 3D structure | Binding energy (kcal/mol) | Major interaction residue pairs | Interaction between chains | Types of interaction |
| Native type (1n8z) | −15.26 | Asn 30(ND2)⋯Gln602(OE1) | Chain:A–Chain:C | Hydrogen bonding |
| Tyr 92(OH)⋯Lys569(NZ) | Chain:A–Chain:C | Hydrogen bonding | ||
| Thr 94(OG1)⋯Asp560(OD2 | Chain:A–Chain:C | Hydrogen bonding | ||
| Arg 50(NH1)⋯Glu558(OE1) | Chain:B–Chain:C | Salt bridge | ||
| Arg 50(NH2)⋯Asp560(OD2) | Chain:B–Chain:C | Salt bridge | ||
| Gly 103(O)⋯Lys593(NZ) | Chain:B–Chain:C | Hydrogen bonding | ||
| Mutant type (1n8z W452C) | −24.40 | Asn30(ND2)⋯Asp596(OD2) | Chain:A–Chain:C | Hydrogen bonding |
| Asn30(ND2)⋯Glu598(OE1) | Chain:A–Chain:C | Hydrogen bonding | ||
| Asn30(OD1)⋯Gln602(NE2) | Chain:A–Chain:C | Hydrogen bonding | ||
| Tyr92(OH)⋯Lys569(NZ) | Chain:A–Chain:C | Hydrogen bonding | ||
| Thr94(OG1)⋯Asp560(OD2) | Chain:A–Chain:C | Hydrogen bonding | ||
| Arg50(NH1)⋯Glu558(OE2) | Chain:B–Chain:C | Salt bridge | ||
| Arg50(NH2)⋯Asp560(OD2) | Chain:B–Chain:C | Salt bridge | ||
| Gly103(O)⋯Lys593(NZ) | Chain:B–Chain:C | Hydrogen bonding |
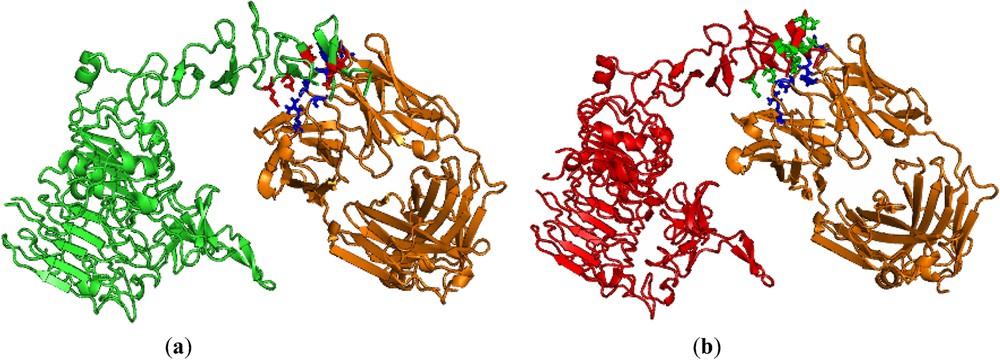
Pymol view of (a) herceptin (orange) with interface residues (blue) complexed with native HER2 protein (green) with interface residues (red). (b) Herceptin (orange) with interface residues (blue) complexed with mutant HER2 protein (red) with interface residues (green). For reference to colours, see the web version of this article.

Pymol view of additional hydrogen bonding between herceptin (Asn30) and mutant HER2 protein (Asp596 and Glu598).
Comparison of normalized mean square displacement of amino acids participating in additional hydrogen bonding of HER2 protein of mutant type with native type
| Position of amino acid | of native type | of mutant type |
| Asp596 | 0.0381 | 0.0415 |
| Glu598 | 0.0307 | 0.0355 |
Since deleterious nsSNPs detection is a major and challenging task in cancer studies, some of the functionally significant nsSNP that are prone to cause breast cancer, including I655V, P1170A, G776S, and W423C, were detected and proved experimentally; they also have been the subject of many clinical studies [33–44]. For instance, it was interesting to note from Table 1 that two nsSNPs, namely rs1801200 and rs1058808, having the mutation from isoleucine to valine at the residue position of 655 and that from proline to alanine at the residue position of 1170, were shown less stable by I-Mutant 2.0; this polymorphism is well known to carry a risk of breast cancer [33–36]. However, several following studies have cast doubt on this association, which remains controversial [37–42]. Also, other functional nsSNPs, including G776S mutation, dramatically increase ErbB2 catalytic activity [43]; they were also shown to be less stable and damaged by I-Mutant 2.0 and PolyPhen, respectively. This could be inferred as an additional significant finding from our analysis.
However, according to our devised criterion, we also identified the most deleterious nsSNP (rs4252633) that cause a mutation from tryptophan to cysteine at the residue position 432 and its binding affinity with herceptin computationally; there was also a good correlation with experiential observation [44]. Our finding could show the optimal path for further clinical and experimental studies to characterize HER2 mutant (W452C) protein in depth.
4 Conclusion
In our analysis, we found out that nsSNP SNPid (rs4252633) showed less stable, deleterious and a damaging score by I-Mutant 2.0, SIFT and PolyPhen, respectively. Structural analysis also revealed that RMSD between native type and mutant type of HER2 receptor was about 2.81 Å. Due to deviation in structure of HER2 receptor by this deleterious nsSNP, stability was decreased by total energy comparison and stabilizing residue's analysis. Further, binding affinity with herceptin increased in mutant type as compared to that of the native type. The reason is attributed to two additional hydrogen bonds between Asn30 to Asp596 and Asn30 to Glu598 of herceptin with mutant HER2 protein, which were not found in native-type HER2. Normal-mode analysis also showed that the two amino acids, namely Asp596 and Glu598 of mutant HER2, forming additional hydrogen bonding with herceptin, had a slightly higher flexibility than the native type. Based on our investigation, we concluded that SNPid rs4252633 could be the most deleterious nsSNP for HER2 receptor. Moreover, herceptin could be the best drug for mutant as compared to native HER2 target. Therefore, the overall analysis shows that herceptin could show a good effect on breast-cancer patients having this type of mutation caused by deleterious nsSNP (rs4252633).
Acknowledgement
The authors thank the management of Vellore Institute of Technology University for providing the facilities for carrying out this work.