

1 Introduction
Cadmium (Cd) is an important environmental pollutant and all Cd compounds have been classified as human carcinogens [1]. Sources of human exposure to this metal include food, cigarette smoke, and alcoholic beverages [2]. It has been shown that long-term exposure of pregnant female rats to Cd results in foetal growth retardation and teratogenic effects. However, despite numerous studies concerning Cd toxicity, the mechanism of its adverse effect still remains unknown. Foetal growth retardation and teratogenic effects induced by Cd exposure could be due to an overproduction of reactive oxygen species produced by pregnant female rats [3]. Piasek et al. [4] showed that parenteral Cd exposure during pregnancy causes perturbations in essential elements in maternal and foetal compartments. Acute Cd exposure in the last trimester of gestation poses a risk for foetal viability, especially when combined with low iron level in maternal diet. However, as far as we know, there are no studies investigating whether Cd could be implicated in oxidative stress during pregnancy in female rat.
The molecular mechanism responsible for the toxic effect of Cd is not well understood. Various studies connect Cd with oxidative stress, since this metal can alter the antioxidant defence system in several animal tissues. This causes depletion in the levels of reduced glutathione, as well as an alteration in the activity of antioxidant enzymes, and a change in the structure of the cellular membrane through a process of lipid peroxidation [5,6]. Studies on mammals showed that Cd stimulated formation of reactive oxygen species, including oxygen free anion radical [7], hydrogen peroxide [8], and probably hydroxyl radical [9]. As a consequence, enhanced lipid peroxidation, DNA damage, altered calcium and sulfhydryl homeostasis, as well as marked disturbances of the antioxidant defence system (ROS), occurred [10–12].
The main target organs for Cd accumulation are the liver and the kidneys [13], where it induces metallothionein (MT) synthesis [14], but almost every other organ of the body can accumulate Cd to a certain extent.
Lipid peroxidation is considered the primary mechanism for Cd-toxicity, despite its inability to directly generate free radicals under physiological condition [15]. The displacement of iron and copper from various intracellular sites by Cd increases the concentration of ionic iron and copper. This causes oxidative stress through the Fenton reaction, producing hydroxyl radical species that are believed to initiate lipid peroxidation [16]. Decomposition products of lipid hydroperoxide such as malondialdehyde and 4-hydroxynoneal can cause chaotic cross-linkage with proteins and nucleic acids, which plays an important role in the process of carcinogenesis. The intracellular concentration of reactive radical species can be increased either by overproduction of ROS or by the inability of the metabolic system to destroy them. ROS may lead to cellular damage when the rate of its generation surpasses the rate of its decomposition by antioxidant defence systems, such as the enzymes superoxide dismutase (SOD), catalase (CAT), or reduced GSH [17,18].
In this study, we investigated the effects of Cd administration on lipid peroxidation levels and oxidative enzymes and DNA damage in tissues of pregnant female rats.
2 Material and methods
2.1 Animals
Female Wistar rats (Pasteur Institute, Tunisia) weighing 150–200 g (12-week-old) at the time of experiments were housed at 25 °C in a cage under a 12 h–12 h light/dark cycle, with free access to food and water. Cd was purchased from Sigma Chemical (St Louis, MO). Vaginal smears were examined and the different stages of the oestrus cycle were followed as described by Snell [19]. The females found in oestrous were selected; each one was mated with one adult male from 8 to 10 p.m. The mid-point of the mating period was considered the beginning of gestation. A total number of 12 pregnant female rats were divided into two groups:
- – group (1) – the animals were subcutaneously injected with 0.1 ml of 0.9% saline solution for 13 successive days (starting from the 6th to the 19th day of gestation). They served as a control group;
- – group (2) – a sublethal dose of Cd (3.0 mg Cd/kg body weight) was subcutaneously injected to pregnant female rats on a daily basis for 13 consecutive days (starting from the 6th to the 19th day of gestation).
The pregnant female rats were allowed to deliver normally. At the end of the experimental period body weight of rats were recorded just after parturition. All animals were sacrificed three days after delivery by decapitation. Liver and kidney were immediately removed.
Animals were cared for in compliance with the Tunisian Code of Practice for the Care and Use of Animals for Scientific Purposes. The experimental protocols were approved by the Faculty's Ethics Committee (‘Faculté des sciences de Bizerte’, Tunisia).
2.2 Tissue preparation
Following Cd treatment, control and treated groups were sacrificed and immediately their livers and kidneys were harvested. The tissues were weighed, rinsed with ice-cold deionized water and dried with filter paper. Fractions of tissues (500 mg) were homogenized in buffer (Tris 10 mmol/l, EDTA 1 mmol/l, PMSF 1 mmol/l; pH = 7.5). The homogenates were centrifuged at 600 g for 10 min and re-centrifuged at 13,000 g for 20 min at +4 °C to obtain a postnuclear homogenate and postmitochondrial supernatant fractions [20].
2.3 Biochemical estimations
Lipid peroxidation was measured as the amount of malondialdehyde (MDA) formed employing thiobarbituric acid using chromatography (HPLC) as described by Richard et al. [21]. CAT activity was assayed by UV spectrophotometry [22]. SOD activity was determined by measuring the inhibition of the auto-oxidant of pyrogallol using a modification of the method of Marklund and Marklund [23]. Se–GPx activity was measured according to Gunzler et al. [24], with a slight modification. GSH and GSSG levels were measured using Akerboom and Sies' method [25]. Protein was determined according to Bradford [26] using bovine serum albumin (BSA) as a standard. All enzyme activities were assayed with a Hitachi UV spectrophotometer and perfused livers and kidneys were taken.
2.4 Determination of MT contents in liver and kidney
The determination of MT was performed according to the technique described by Eaton and Cherian [27]. Quantification of MT was performed by using 109Cd. Briefly, a fraction (about 0.5 g) of the two tissues was homogenized in 1 ml of a 0.25 M sucrose solution. The homogenate were centrifuged at 10,000 g for 10 min at 4 °C, the supernatant was stored at −80 °C for analysis of MT protein. Two hundred microlitres of 109Cd solution (2 μg/ml) were mixed with 200 μl of sample (heat-denatured supernatant) and allowed to incubate at room temperature for 10 min. Then 100 μl of a 2% bovine haemoglobin solution were added to the tubes, mixed and heated in a 100 °C boiling water bath for 2 min. The tubes were placed on ice for several minutes, and centrifuged at 10,000 g for 2 min in a microfuge and another 100-μl aliquot of 2% haemoglobin was added. Heating, cooling and centrifugation were repeated once again. A 500-μl aliquot of the supernatant fraction should be carefully removed. Lastly, aliquots of 500 μl of the supernatant were recovered carefully and transferred into clean tubes for counting their radioactivity.
2.5 DNA extraction from tissues and HPLC-EC analysis
The assay was performed as described by Ravanat et al. [28]. Liver and kidney samples are kept at −80 °C before use. Samples are weighted still frozen and divided into fractions of 150–250 mg. Each portion of tissues is homogenized by using a 2-ml potter glass homogenizer in 1.2 ml of buffer (320 mM sucrose, 10 mM Tris, 5 mM MgCl2, 0.1 mM desferroxamine mesylate, 1% Triton-100, pH 7.5) kept in ice. After homogenization, the sample was transferred into a 15-ml Falcon tube and centrifuged at 1500 g for 5 min at 4 °C. The supernatant was discarded and 0.5 ml of homogenization buffer is added. The sample was vortexed for 10 s and further centrifuged at 1500 g for 5 min at 4 °C. Then, the supernatant was discarded.
8-oxodGuo was quantified in liver and kidney homogenates according to Ravanat et al. [28]. The nuclear pellet obtained previously is suspended in 600 μl of extraction buffer (10 mM Tris, 5 mM EDTA, 0.15 mM desferroxamine mesylate, pH 8). SDS (10% in water, 35 μl) is added. The resulting suspension is homogenized by vortexing. RNase A (3 μl, 100 mg/ml) and RNase T1 (8 μl, U/μl) are then added and the resulting suspension gently vortexed for 10 s. The sample is incubated for 15 min in a water bath at 50 °C. Then, Qiagen proteinase (30 ml) was added and the sample gently vortexed for 10 s prior to be incubated for 60 min at 37 °C. A NaI solution (1.2 ml) was added. The sample was vigorously vortexed for 30 s. Isopropanol (2 ml) was added and the samples gently shaken until complete homogeneity. After centrifugation, the DNA pellet was rinsed by 1 ml of 70% ethanol. The DNA pellet was then solubilized into 100 ml of deionized water containing 0.1 mmol/l of deferroxamine mesylate. The DNA solution was incubated with two units of nuclease P1, the sample was held at 37 °C for 2 h. Then, four units of alkaline phosphatase were added together with palk buffer. After incubation for 1 h at 37 °C, the samples were centrifuged and the aqueous layer collected and analyzed by HPLC-EC.
The HPLC-EC system consisted of a Gilson HPLC system equipped with an Uptisphere ODB octadecylsilyl silica gel column (Interchim, Montluc, France). The retention time of 8-oxodGuo is 21.5 min. The amount of DNA analyzed was determined from the area of the peak of 8-oxodGuo (retention time 15 min) after appropriate calibration.
2.6 Data presentation and statistical analysis
Data presentation and statistical analysis data were analyzed using Stat View 512+ software (Abacus concept, Inc.). Means were given with ± SEM and differences between the controls and treated animals were determined by Student's t-test.
3 Results
In liver, Cd administration during pregnancy have shown depletion (−48%, ) of total GSH and (−71%, ) GSSG (Table 1). No change in the concentration of MDA was noted following the treatment with Cd (Fig. 1). No differences in the activities of Mn-SOD, CuZn-SOD and GPx were observed in treated rats (Table 2). Cd treatment increased MT level () in pregnant female rats.
Effect of cadmium treatment on glutathione levels in liver and kidney from pregnant female rats
| Parameter | Liver control Cd | Kidney control Cd | ||
| GSHT (μmol/g prot) | 50.50 ± 7.46 | 26.18 ± 9.19∗ | 0.76±0.12 | |
| GSSG (μmol/g prot) | 14.01 ± 2.57 | 4.00 ± 1.34∗ | Nd | Nd |
| GSH (μmol/g prot) | 22.38 ± 8.20 | 17.72 ± 7.80 | 0.76±0.12 |
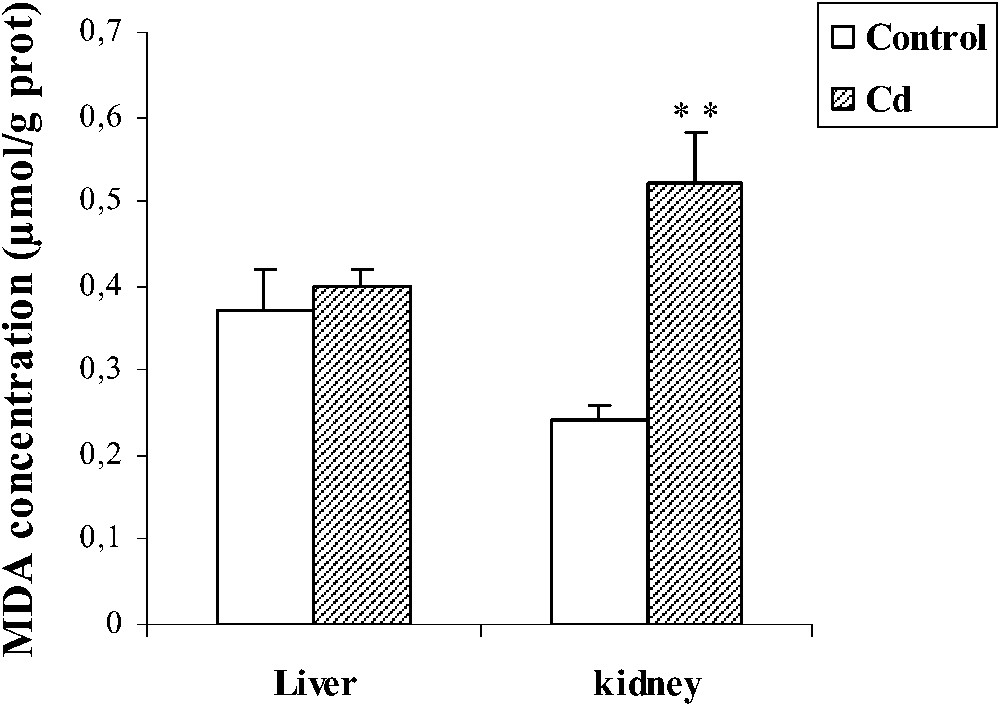
Effect of cadmium treatment on MDA level in liver and kidney of pregnant female rats. Values are means ± SEM calculated from n=6 in each group. **p<0.01 compared to control (Student's t-test). Cd: cadmium; MDA: malondialdehyde.
Effect of cadmium treatment an antioxidant enzymes in pregnant rat liver and kidney
| Parameter | Liver control Cd | Kidney control Cd | ||
| GPX (U/g prot) | 3286.50 ± 311 | 2793.50 ± 257.37 | 1321.66 ± 66.27 | 1162.83 ± 29.91∗ |
| Mn-SOD (U/g prot) | 0.89 ± 0.02 | 0.70 ± 0.05 | 0.95 ± 0.12 | 0.87 ± 0.13 |
| CuZn-SOD (U/g prot) | 30.46 ± 0.43 | 29.68 ± 1.34 | 16.32 ± 0.83 | 14.49 ± 0.70∗∗ |
| CAT (U/g prot) | 376.66 ± 51.22 | 360.66 ± 13.85 | 110.36 ± 13.49 | 90.72 ± 4.97 |
| MT (μg/g prot) | 2.66 ± 1.21 | 72.92 ± 3.54∗∗ | 1.11 ± 0.22 | 16.77 ± 1.05∗ |
In kidney, MDA level increased (+116, ) following Cd treatment, whereas the activities of GPx and CuZn-SOD decreased (−12%, , and −11%, , respectively), while no significant variation in the CAT activity was observed in the Cd group (Table 2). Cd induced depletion (−52%, ) of the total GSH, and (−52%, ) of reduced GSH (Table 1). Moreover, we noted an increase () in the MT level.
Cd treatment did not increase the oxidative DNA damage in liver. By contrast, the level of 8-oxodGuo increased (+51%, ) in the kidney of pregnant female rats (Fig. 2).
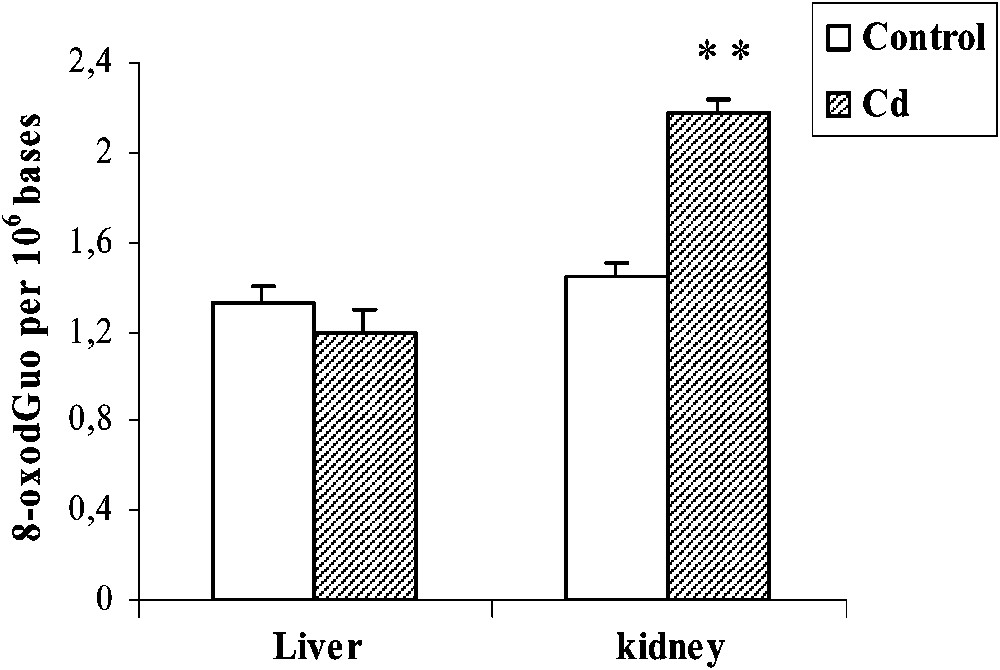
Effects of cadmium treatment on the formation of 8-oxodGuo in liver and kidney of pregnant rats. Values are means ± SEM calculated from n=6 in each group. **p<0.01 compared to control (Student's t-test).
4 Discussion
Cd is perhaps one of the most toxic industrial and environmental metals and it continues to be a health hazard. It is rapidly distributed in the tissues and has a high volume of distribution [29]. The basis of Cd toxicity is its negative influence on enzymatic systems of cells, resulting from substitution of divalent mineral elements (Zn2+, Cu2+, and Ca2+) in metalloenzymes and its very strong affinity to biological structures containing –SH groups, such as proteins, enzymes and nucleic acids [30,31]. Many effects of Cd action result from interactions with necessary micro- and macroelements, especially Ca, Zn, Cu, Fe and Se [29,32].
Our investigation showed that the 8-oxodGuo level increased in kidney of Cd-treated rats, but not in liver. The mechanism of Cd-induced DNA base oxidation is still not fully clarified. Although the molecular mechanism of DNA damage by Cd is not known in detail, it may involve activation of various reactive oxygen species, as suggested by a number of previous studies [7,33], despite the fact that Cd does not catalyse Fenton-type reactions, as do other metal ions [31]. Alternatively, Cd may activate a calcium-dependent endonuclease that induces DNA strand breaks [34]. The absence of the induction of DNA damage by Cd in liver suggests that a protective mechanism may be activated. This may include the synthesis of MT in liver cells, which is higher in liver compared to kidney. As reported by Nordberg et al. [35], Cd is first transported in blood bound to albumin and preferentially taken up by the liver where it induces the synthesis of MT. Then, MT-bound Cd appears in the blood plasma and is efficiently filtered through the glomeruli and thereafter taken up by the tubules, leading to its accumulation in kidney. This can explain the fact that the highest level of MDA was found in kidney. This confirms the high affinity of MTs for Cd, which efficiently reduces free Cd concentrations [36]. In addition, MTs themselves are effective ROS scavengers [37] and block the DNA-damaging activity of hydroxyl radicals [38]. Thus both MT mediated sequestering of free Cd ion and scavenging of Cd induced ROS by MTs could be responsible for the absence of Cd-induced DNA damage in liver of treated rats. Previous studies demonstrated the induction of MT in liver probably implicated in the detoxification of Cd treatment. The high level of Cd (3 mg/kg) used in the present study is partially neutralized by MT in liver, whereas the free fraction could be implicated in the oxidative stress and DNA oxidation observed in kidney.
The present results have clearly demonstrated the ability of Cd to induce oxidative stress in pregnant female rat kidney, as evidenced by increased lipid peroxidation (MDA) after 13 days of Cd treatment. Similar results previously reported by Hussain et al. [39] reported that Cd increased lipid peroxidation by a direct effect or by decreasing the glutathione content. The insignificant MDA increase measured in liver following sub-acute Cd intoxication is probably due to the activation of other defence factors. Several protective agents, including metallothionein, play an important role in detoxification processes [40,41]. An induced metallothionein synthesis has also been observed in various rat tissues after exposure to Cd [42,43]. The enhanced kidney MDA in pregnant female rats exposed to Cd might result from the reduction in the tissues activities of SOD and GSH. The inhibition of SOD activity might be probably a consequence of an interaction between Cd and Zn in SOD molecules. Bauer et al. [44] reported that 111Cd was able to occupy the site of Zn in the CuZn-SOD molecule, creating an inactive form of the enzyme (Cu111CdSOD). Moreover, there is increasing evidence that Cd interacts with Selenium (Se) and disrupts GPx activity. Wasowicz et al. [45] found that Cd exposure decreased Se concentration and GPx activity in both erythrocytes and plasma of exposed workers. Interestingly, Cd is known to interfere with intracellular thiols by direct binding [46]. GSH depletion due to Cd treatment can be either due to direct binding of the metal with GSH or due to leakage of GSH to the extracellular compartment. Hence, GSH is often the first line of defence against oxidative stress. GSH levels can be increased due to an adaptive mechanism to slight oxidative stress through an increase in its synthesis; however, a severe oxidative stress may suppress GSH levels due to the loss of adaptive mechanisms and the oxidation of GSH to GSSG. Similarly, Chin and Templeton [47] proved that exposure to high doses of Cd caused depletion of intracellular GSH due to ROS production. Contrary, Casalino et al. [48] reported an increased GSH level 24 h after Cd administration. The depletion of the glutathione level in the present experiment is thus one of the factors responsible for enhanced lipid peroxidation.
The results suggest that Cd intoxication induces oxidative stress and alters the antioxidant system, resulting in oxidative damage in kidney. Indeed, Fotakis et al. [49] showed that lysosome is one of the main targets for Cd toxicity and that lysosomal-induced damage can be responsible for other cellular events, including DNA damage. Furthermore, a significant increase of lipid peroxidation products and oxidative DNA damage was found in kidney. These oxidative modifications point to pathophysiological alterations of this organ.
In conclusion, sub-acute treatment with cadmium induced an oxidative stress and DNA oxidation in kidney. By contrast, the same investigation reported a protective mechanism of liver by metallothionein against oxidative stress and DNA damage in pregnant female rats.
Acknowledgements
This work was supported by a grant from the Tunisian Ministry of High Education Scientific Research and Technology.