

1 Introduction
L’existence de conformations non planes du macrocycle porphyrinique a été mise en évidence, grâce à la cristallographie, dans un grand nombre d’hémoprotéines et de métalloporphyrines. Les principales distorsions rencontrées sont de type ruffled (froncé), saddled (en selle de cheval), domed (en dôme) ou waved (en vague). Dans les métalloporphyrines synthétiques, les deux premiers types de distorsion sont habituellement la conséquence d’un encombrement stérique important à la périphérie du macrocycle 〚1〛. Nous avons cherché à moduler la distorsion froncée des chiroporphyrines en jouant, non pas sur l’encombrement de leurs substituants méso 〚2〛, mais sur la longueur des brides qui joindraient les groupes esters de deux substituants méso adjacents (Fig. 1). Ces porphyrines bridées sont désignées par l’abréviation H2BnCP, n représentant le nombre de groupes méthylène constituant une bride (B = bride, CP = chiroporphyrine). La présence de ces brides nous semblait a priori être susceptible de limiter la distorsion « naturelle » de la chiroporphyrine due à l’alternance α, β, α, β des substituants méso.
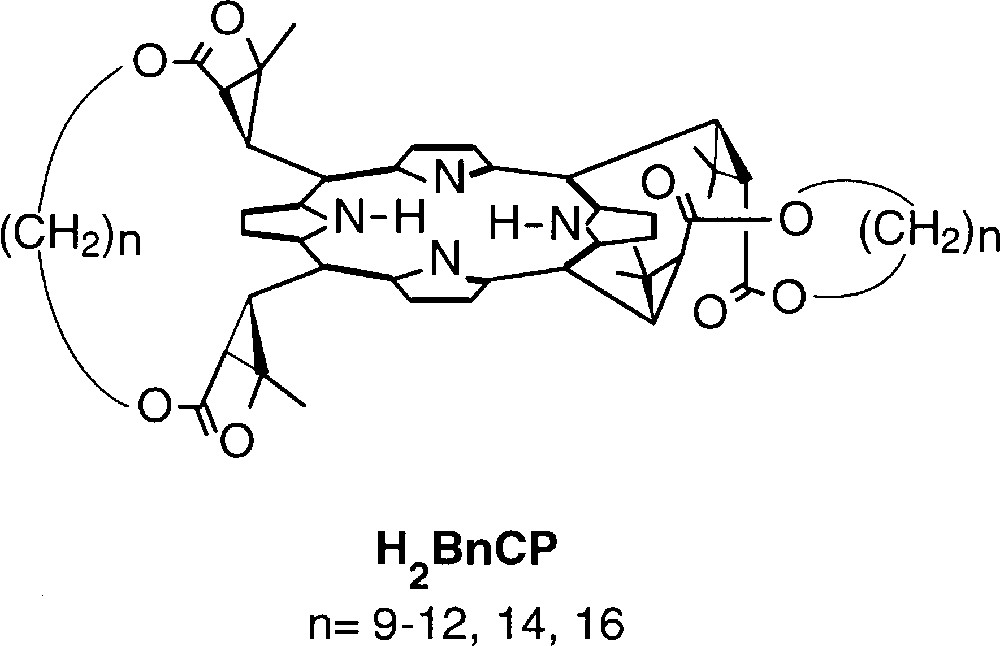
Structure générale des chiroporphyrines bridées.
La distorsion du macrocycle affecte en général un grand nombre de propriétés physiques et spectroscopiques de la porphyrine : les longueurs d’onde des maxima d’absorption UV–visible (qui sont déplacés vers le rouge), les spectres de vibration (Raman), les potentiels d’oxydation, etc. 〚3–6〛. Nous avons donc exploré de façon systématique les propriétés spectroscopiques en solution d’une série de chiroporphyrines bridées H2BnCP (n = 8–12, 14, 16) ainsi celles de leurs complexes avec divers métaux de transition (manganèse, fer, cobalt, nickel, cuivre, zinc). Nous avons également résolu les structures de deux complexes de nickel (n = 8, 10) à l’état cristallin.
2 Spectroscopie UV–visible
Les graphes représentant la longueur d’onde des maxima d’absorption en fonction du nombre de groupes méthylènes constituant une bride pour les bases libres et les complexes métalliques (Fig. 2) montrent que la longueur d’onde des différents maxima d’absorption varie très peu, quelle que soit la longueur des brides. Par souci de clarté, seuls les complexes du cobalt(III) et du nickel(II) sont représentés, mais les mêmes constatations ont été faites avec les autres métaux (MnIII, FeII, CuII, ZnII). Shelnutt et al. 〚1〛 ont montré que la longueur d’onde des maxima d’absorption de porphyrines de nickel, présentant une distorsion froncée, varie en fonction du degré de torsion mesuré par l’angle dièdre entre les plans Cα–N–Cα de deux unités pyrroles opposées. Cependant, lorsque cet angle est compris entre 0 et 40°, la variation de longueur d’onde est faible. Ainsi, la spectroscopie UV–visible serait une sonde peu sensible de la torsion des macrocycles des porphyrines, ne répondant qu’à des déviations très élevées uniquement. Dans le cas de nos chiroporphyrines bridées, il apparaît, d’après la Fig. 1, que la spectroscopie UV–visible ne donne pas d’information sur le degré de distorsion. Cela est compréhensible, puisque les variations de torsion sont faibles au sein de la série : ainsi, les complexes de nickel NiB8CP et NiB10CP présentent à l’état solide des angles dièdres allant de 35 à 45° (voir ci-après), zone où les longueurs d’onde des maxima d’absorption varient très peu, selon Shelnutt et al.
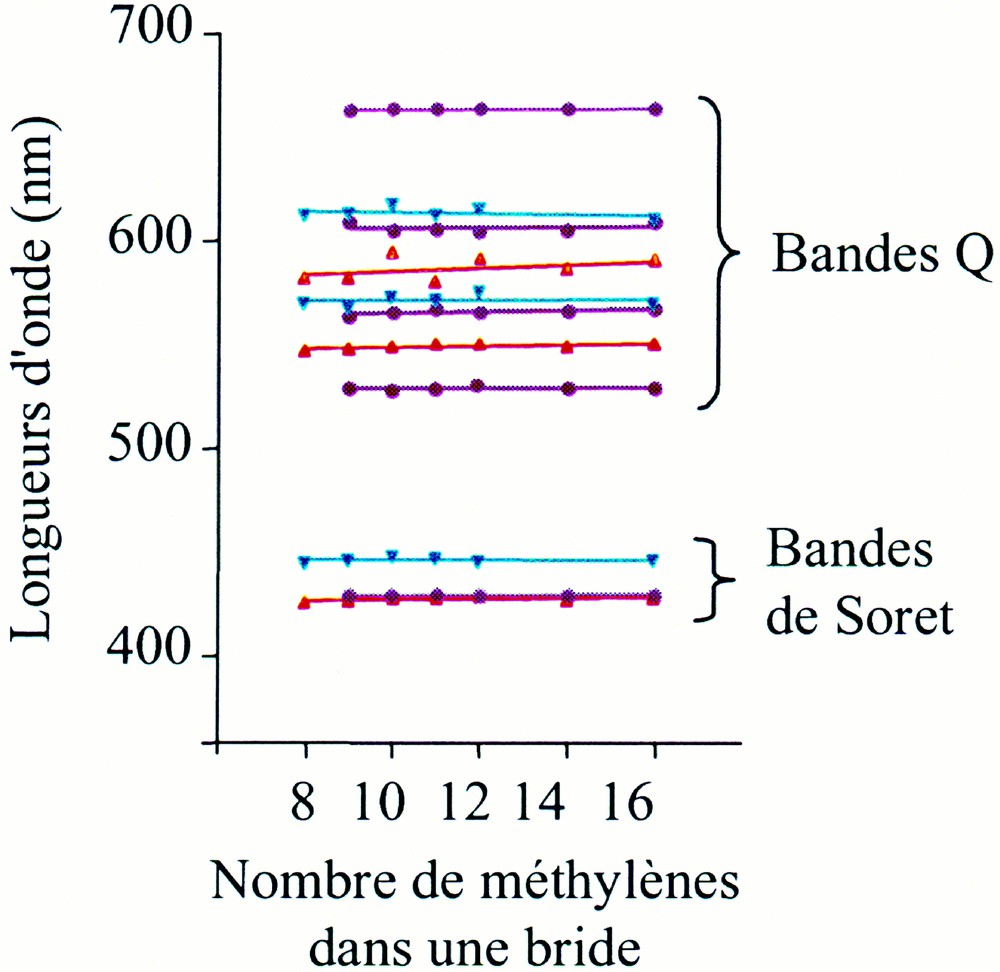
Tracé des valeurs des longueurs d’onde des maxima d’absorption en fonction du nombre de groupes méthylène constituant une bride, pour la série chiroporphyrines bridées (violet) et de leurs complexes avec CoIII (bleu) et NiII (rouge). Les lignes représentent des régressions linéaires obtenues à partir des points expérimentaux.
3 Spectroscopie RMN
Les chiroporphyrines bridées H2BnCP ont été caractérisées par spectroscopie RMN du proton. L’évolution du déplacement chimique des protons NH centraux δNH en fonction de la longueur des brides, ainsi qu’une courbe de régression parabolique, sont illustrées sur la Fig. 3. Un maximum est observé à 13 groupes méthylène. Les variations totales observées sont de l’ordre de 0,2 ppm, valeur bien supérieure à l’incertitude expérimentale, qui est de l’ordre de 0,01 ppm. Deux régimes peuvent être distingués : (1) lorsqu’on allonge les brides de 9 à 12 groupes méthylène, δNH augmente ; (2) si on continue d’allonger les brides au-delà de 13 groupes méthylène, δNH diminue. Ces résultats sont interprétés de la façon suivante. Dans la première zone, l’allongement de la bride permet à la porphyrine de se tordre de plus en plus, ce qui diminue le courant de cycle et induit un déplacement de δNH vers les champs faibles. En revanche, l’allongement de la bride au-delà de 13 groupes méthylène induit l’effet inverse ; tout se passe comme si la porphyrine avait des brides de plus en plus courtes. Ce raccourcissement effectif est probablement une conséquence de la mobilité conformationnelle des groupes esters, que nous avons mise en évidence dans une publication récente 〚2〛. Des conformations où les fonctions carbonyles des esters sont dirigées vers l’extérieur et les groupements alkoxy vers l’intérieur deviennent accessibles lorsque les brides sont suffisamment longues ; l’effet moyen de cette dynamique est un raccourcissement effectif de la bride à l’échelle de temps de la RMN. L’équilibre entre les divers conformères de la fonction ester est rapide et, même à –80 °C, la spectroscopie RMN ne détecte qu’une conformation moyenne.
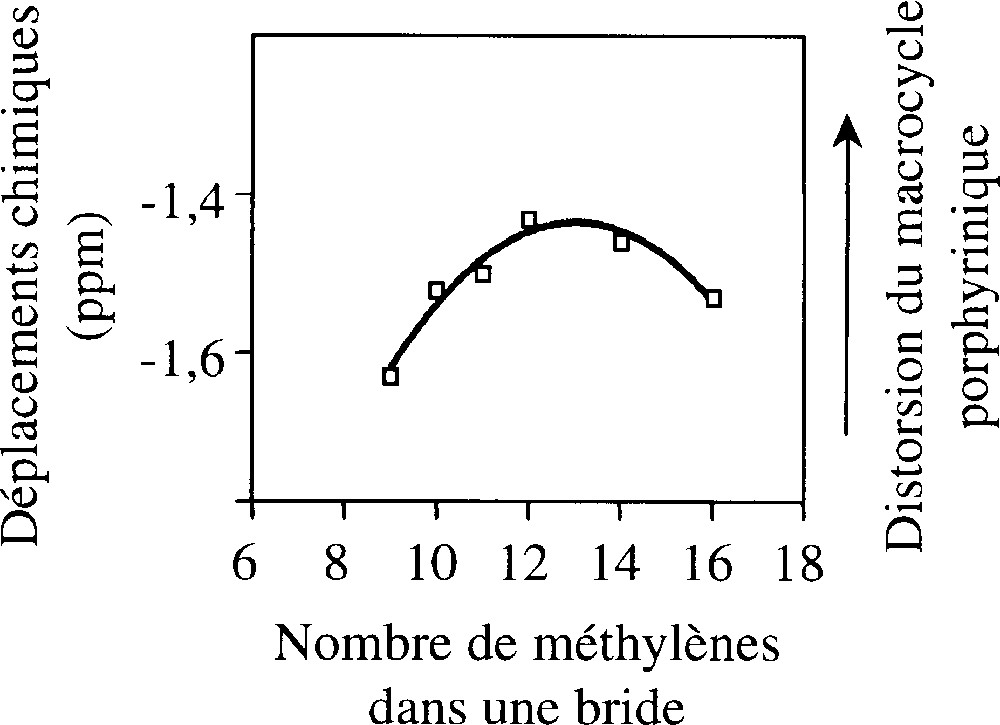
Tracé du déplacement chimique des protons NH centraux en fonction du nombre de groupes méthylènes dans une bride, pour la série des chiroporphyrines bridées (□). La courbe est une régression parabolique obtenue à partir des points expérimentaux.
4 Diffraction des rayons X
Les complexes de nickel des chiroporphyrines bridées à 8 et 10 groupes méthylène, de conformation αβαβ, ont été obtenus à l’état solide, chacun dans deux réseaux cristallins différents : monoclinique et orthorhombique. Le complexe NiB8CP présente deux unités indépendantes dans chacun des deux réseaux. Le complexe NiB10CP présente trois molécules indépendantes dans le réseau monoclinique, et deux dans le réseau orthorhombique. D’une manière générale, quelles que soient la nature du réseau cristallin et la longueur des brides, les complexes de nickel présentent une distorsion froncée, la distance Ni–NPy reste en moyenne de 1,91(2) Å et l’atome de nickel s’inscrit parfaitement dans le macrocycle, avec un déplacement par rapport au plan moyen de la porphyrine voisin de zéro. De plus, les fonctions carbonyles des groupes esters sont orientées vers l’intérieur et les premiers groupes méthylène des brides vers l’extérieur. Ces résultats seront décrits en détail par ailleurs 〚7,8〛.
La caractéristique structurale que nous avons choisie pour décrire la distorsion macrocyclique est l’écart des atomes de carbone méso par rapport au plan moyen du macrocycle. Les valeurs correspondantes sont rassemblées dans le tableau 1, pour chacune des molécules indépendantes des différents réseaux étudiés. Dans le cas du complexe NiB8CP (Fig. 4), l’écart moyen des atomes de carbone méso par rapport au plan macrocyclique est de 0,69(3) Å dans le réseau monoclinique et de 0,64(3) Å dans le réseau orthorhombique. On constate donc que la distorsion est plus ou moins importante, selon l’empilement au sein du réseau cristallin. En ce qui concerne le complexe NiB10CP (Fig. 5), la distorsion du macrocycle est variable à l’intérieur d’une même maille : une des unités indépendantes du réseau cristallin monoclinique présente un écart moyen des atomes de carbone méso par rapport au plan moyen du macrocycle de 0,60(2) Å, alors que les deux autres unités sont considérablement plus tordues, avec des valeurs de 0,71(2) Å et de 0,70(2) Å. Les différences observées entre la molécule peu distordue et les deux autres qui le sont davantage sont largement supérieures à 3 σ (σ est l’écart type), et donc réelles. Cette variation de distorsion est également visible pour le complexe ayant cristallisé dans un réseau orthorhombique, avec une molécule ayant une torsion de 0,59(1) Å et l’autre de 0,70(2) Å. Ainsi, l’ensemble des résultats rassemblés dans le tableau 1 montre que la distorsion du macrocycle est variable pour un même complexe, selon l’environnement cristallin.
Quelques caractéristiques structurales des complexes NiB8CP et NiB10CP à l’état cristallin (réseaux monoclinique et orthorhombique).
| NiB8CP mono | NiB8CP ortho | NiB10CP mono | NiB10CP ortho | |
| d(Ni–NPy) | 1,92(1) Å | 1,92(1) Å | 1,91(1) Å | 1,89(4) Å |
| 1,900(6) Å | 1,917(5) Å | 1,89(1) Å | 1,90(3) Å | |
| 1,92(3) Å | ||||
| Écart des carbones méso au plan moyen du macrocycle | 0,688(6) Å | 0,645(3) Å | 0,596(6) Å | 0,593(8) Å |
| 0,702(7) Å | 0,641(3) Å | 0,713(6) Å | 0,703(7) Å | |
| 0,704(6) Å |
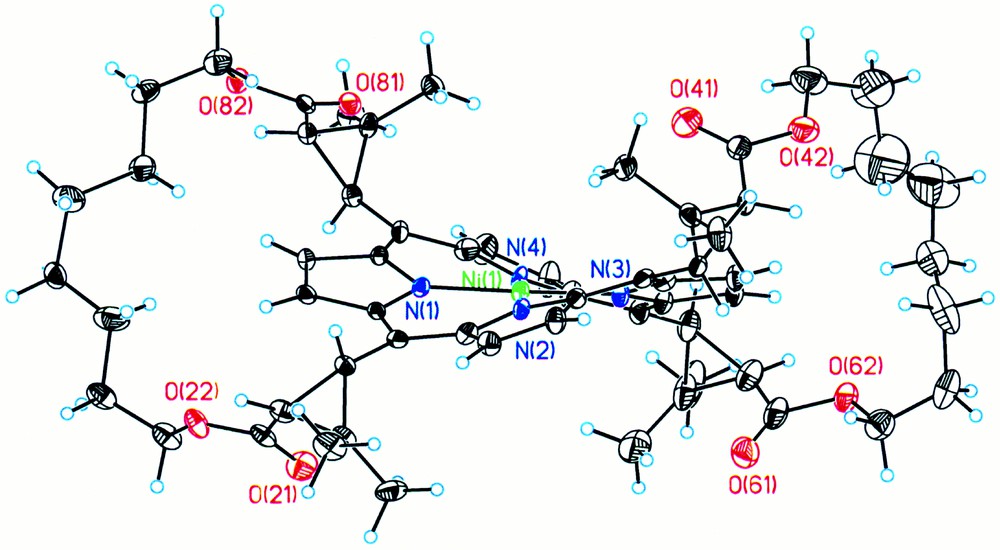
Représentation Ortep d’une unité du complexe NiB8CP dans le réseau monoclinique.
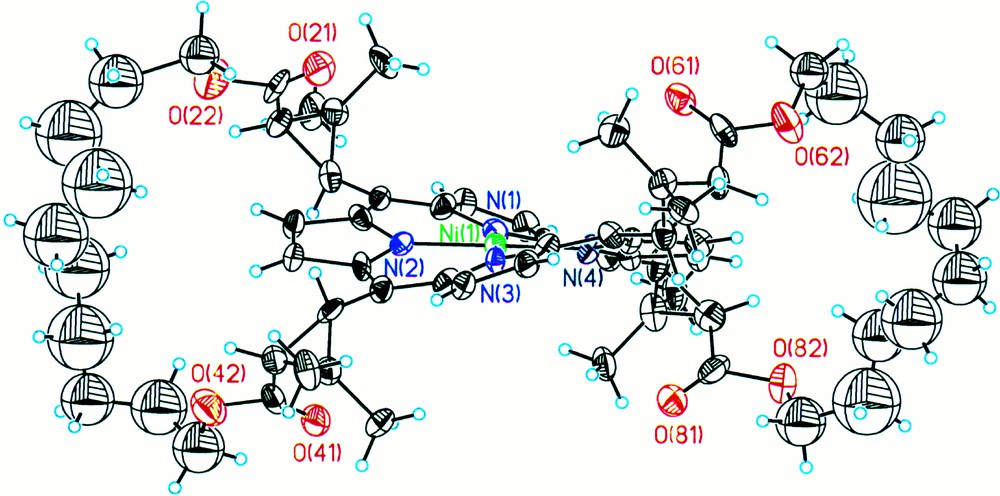
Représentation Ortep d’une unité du complexe NiB10CP dans le réseau monoclinique.
Nous supposons que des conformations très distordues, ou très peu distordues, existent de façon transitoire en solution, et peuvent être stabilisées au contact du réseau cristallin en cours de formation, puis conservées au sein du cristal. Ces espèces transitoires ne sont détectées, ni par la spectroscopie UV–visible, ni par la RMN, ces deux techniques n’observant qu’une conformation moyenne.
5 Conclusion
La distorsion froncée du macrocycle est contrôlée par la longueur des brides qui lient les groupes méso adjacents. La spectroscopie UV–visible n’est pas une sonde suffisamment sensible pour détecter la variation de torsion induite par les brides. En revanche, les protons NH centraux sont influencés par la longueur des liens ; leurs déplacements chimiques en RMN donnent une mesure du degré de distorsion du macrocycle porphyrinique. Alors que les analyses spectroscopiques en solution ne donnent qu’une moyenne de toutes les conformations que la porphyrine peut adopter, la diffraction des rayons X permet de caractériser des conformères très différents, piégés au sein du réseau cristallin.
6 Partie expérimentale
Les différentes chiroporphyrines bridées ont été préparées selon la méthode décrite dans la référence 〚7〛. Tous les composés ont été caractérisés par spectrométrie de masse en mode électro-nébulisation (Finnigan MAT LCQ), par spectroscopie UV-visible (Perkin Elmer Lambda 9) et par RMN (Bruker AC 200 et Varian Unity 400). Les structures cristallines ont été résolues à l’aide d’un diffractomètre Smart-CCD de Bruker à basse température (T = –80 °C), en utilisant les méthodes directes (programme SHELXTL5.05).
Remerciements
Nous remercions le Centre national de la recherche scientifique et le Commissariat à l’énergie atomique pour le financement de ces travaux.