

1 Introduction
Coordination of electrophilic Mn(CO)3 group to an arene modifies its reactivity, allowing regioselective transformations impossible to perform in the case of the uncomplexed arenes 〚1, 2〛. The main synthetic methods reported for the preparation of cationic (η6-arene)Mn(CO)3 complexes involve the direct coordination of Mn(CO)3 to the corresponding free arene (Fig. 1, path a). When R is an electron-donating group such as alkyl, alkoxy or hydroxy substituent, complexation may occur in high yield 〚3〛. Likewise arenes bearing weak electron-accepting groups such as chloride or non-conjugated carbonyl groups can be coordinated 〚3, 4〛, but for arenes substituted by resonance electron withdrawing groups, the strong electron deficiency of the ring prevents direct complexation of the metal, whatever the method employed 〚5〛. An alternative route for functionalised (η6-arene)Mn(CO)3+ complex syntheses could be an ipso nucleophilic aromatic substitution (Fig. 1, path b), which is known to occur on manganese complexes substituted by leaving group (LG), but only in the case of oxo, thio and amino nucleophiles 〚6〛. More generally, nucleophiles add anti to the metal leading to neutral and stable (η5-cyclohexadienyl)Mn(CO)3 complexes that might generate cationic complexes by hydride abstraction (Fig. 1, path c). But this endo-hydride abstraction remains unsuccessful because of the difficult approach of a bulky electrophile, e.g triphenylmethyl carbocation, to the metal side.
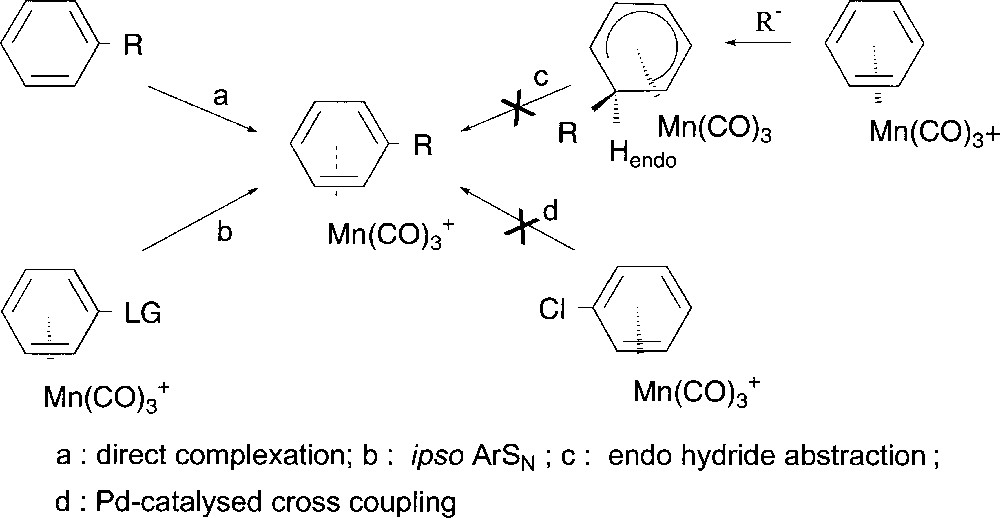
Preparation of substituted (η6-arene)Mn(CO)3+ complexes.
As part of our ongoing interest on cross coupling reactions 〚7, 8〛, we have shown the efficiency of bimetallic activation starting from (η6-chloroarene)Cr(CO)3 under smooth conditions avoiding side product formation and keeping intact the metal entity 〚9〛. All our attempts to extend such catalytic transformations to cationic tricarbonyl(η6-chloroarene)manganese complexes have unfortunately failed 〚10〛 up to now (Fig. 1, path d). Indeed, it has been shown that the catalytic process stops at the early oxidative addition step.
We considered that an alternative method to obtain substituted (η6-arene)Mn(CO)3+ complexes could consist in the selective functionalisation of (η5-chlorocyclohexadienyl)Mn(CO)3, without alteration of the metal fragment. The corresponding (η5-cyclohexadienyl)Mn(CO)3 complexes could then undergo exo-hydride abstraction 〚11–13〛 to afford new substituted (η6-arene)Mn(CO)3+ complexes that cannot be prepared directly by paths a to d (Fig. 1).
We now wish to report the synthesis of substituted (η5-cyclohexadienyl) complexes B by Pd-catalysed cross coupling reaction starting from (η5-chlorocyclohexadienyl)Mn(CO)3 A, and then the transformation of these new complexes B into the corresponding cationic η6 complexes C (Fig. 2).
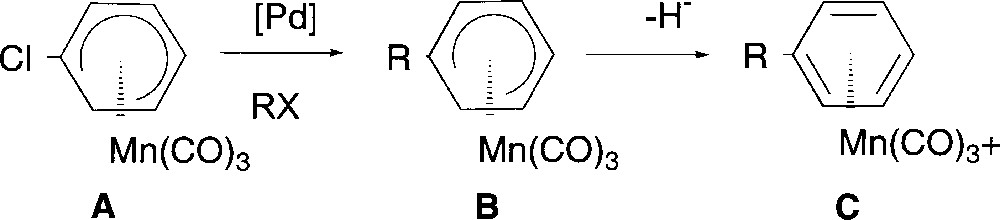
General synthetic strategy.
2 Pd-catalysed functionalisation of (η5-chlorocyclohexadienyl)Mn(CO)3 complexes
(η5-chlorocyclohexadienyl)Mn(CO)3 complexes are prepared by hydride addition in THF to (η6-chlorobenzene)MnCO)3+ complexes 1 (Fig. 3) leading generally to a mixture of separable regioisomers ortho 2 and meta 3 with respect to the Cl 〚14–16〛. In the case of (η6-p-chloroanisole)Mn(CO)3+, this addition afforded regioselectively complex 2 (Table 1, entry 2).
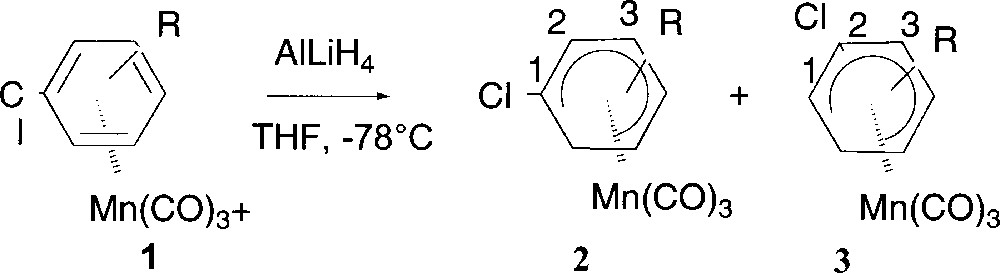
Preparation of (η5-chlorocyclohexadienyl)Mn(CO)3 complexes.
| Entry | R | Complex 1 | Ratio 2:3 | Yield (%) |
| 1 | H | 1a | 100:0 | 80 |
| 2 | 4-OMe | 1b | 100:0 | 95 |
| 3 | 4-Me | 1c | 60:40 | 90 |
| 4 | 2-Me | 1d | 25:75 | 91 |
We first investigated the Stille cross coupling reaction and we showed that complexes 2a and 2c coupled with phenyltin reagents using 35 % Pd2(dba)3 in the presence of 10 % AsPh3 〚17〛 in DMF at room temperature to give the expected complexes 4a and 4c (Fig. 4). Extending the reaction conditions to thienyltin reagents complexes 5a–5d were obtained. The results gathered in Table 2 show that both substitution (entries 1, 7, 8 and 9) and steric hindrance (entries 10 and 11) of starting complex 2 did not affect the course of the reaction. Similarly, the reactivity of the chlorine atom towards the palladium catalysis in the oxidative addition does not seem to depend on its position in the π system 〚18〛. Indeed, identical reaction conditions applied to complex 2a (entry 1) or 3a (entry 12) afforded the expected coupling product 4a in 57% or 5’a in 58% yield respectively. In contrast, it is worthy to note that the reaction course is greatly affected by the nature of the ligand. As shown in entries 3–6, using phosphorous ligands (PPh3, dppf, P(OEt)3, PtBu3) instead of AsPh3 led in all cases to the decomposition of the starting material. The same reaction run without catalyst gave no arylation product (Table 2, entry 2), indicating that complexes 4 arose from a Pd-catalysed process.
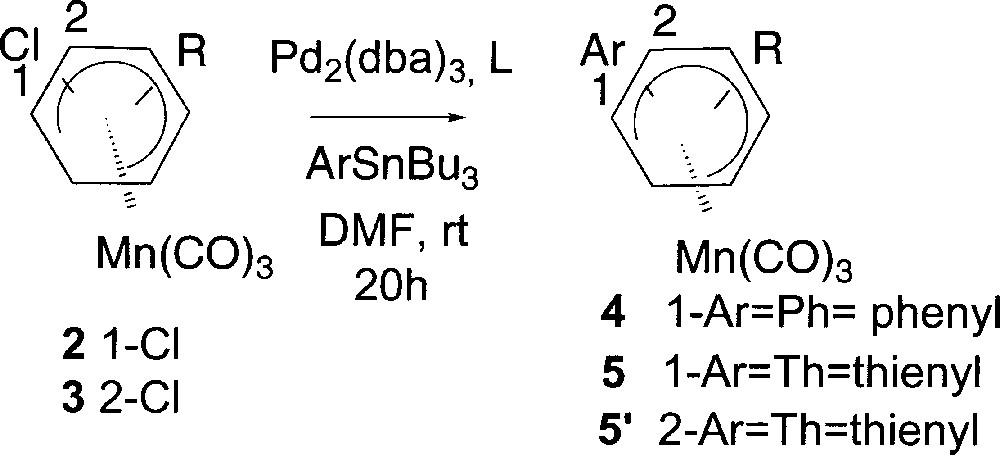
Stille cross coupling reaction of (η5-1-chlorocyclohexadienyl)Mn(CO)3 complexes.
| Entry | Complex | R | Ar | L | Yield (%) |
| 1 | 2a | H | Ph | AsPh3 | 4a (57) |
| 2 | 2a | H | Ph | — | — |
| 3 | 2a | H | Ph | PPh3 | — |
| 4 | 2a | H | Ph | dppf | — |
| 5 | 2a | H | Ph | P(OEt)3 | — |
| 6 | 2a | H | PtBu | — | |
| 7 | 2a | H | Th | AsPh3 | 5a (51) |
| 8 | 2b | 4-OMe | Th | 5b (78) | |
| 9 | 2c | 4-Me | Ph | 4c (45) | |
| 10 | 2c | 4-Me | Th | 5c (48) | |
| 11 | 2d | 2-Me | Th | 5d (53) | |
| 12 | 3a | H | Th | 5’a (58) |
We next turned our attention to the preparation of (η5-cyclohexadienyl)Mn(CO)3 complexes substituted by conjugated electron-withdrawing groups. We first synthesized (η5-cyclohexadienyl)Mn(CO)3 complexes 6 substituted by conjugated keto, ester and amide groups using a Pd-catalysed carbonylation of neutral (η5-chlorocyclohexadienyl)Mn(CO)3 (Fig. 5). Complex 2b reacted under CO atmosphere in the presence of Pd2(dba)3 35% and AsPh3 10% and 2-tributylstannylthiophene to deliver after 2 h complex 6a in 90% yield (Table 3, entry 1). Under the same conditions, complex 2c gave complex 6b in 90% yield (Table 3, entry 2). This reaction has been extended to the preparation of alkyl ketones using Bu4Sn as nucleophile (Table 3, entry 3).
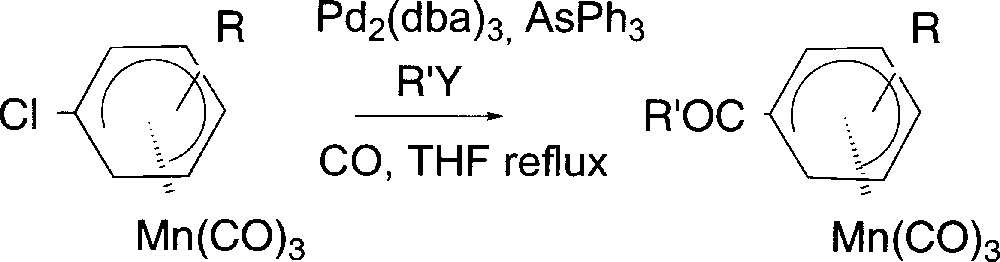
Carbonylation of (η5-1-chlorocyclohexadienyl)Mn(CO)3 complexes.
| Entry | Complex | R | R’Y | COR’ | Yield (%) |
| 1 | 2b | 4-OMe | ThSnBu3 | COTh | 6a (90) |
| 2 | 2c | 4-Me | ThSnBu3 | COTh | 6b (90) |
| 3 | 2c | 4-Me | SnBu4 | COBu | 6c(56) |
| 4 | 2b | 4-OMe | HN(C2H4)2O | CON(C2H4)2O | 6d (48) |
Since the reaction pathway may involve an acyl palladate as an intermediate, we thought it could be trapped by classical nucleophiles 〚19, 20〛 in order to prepare carboxylic acid derivatives. Indeed reacting morpholine with complex 2b under the same catalytic conditions afforded amide derivative 6d (entry 4) in 48% yield 〚21〛.
3 Synthesis of unprecedented cationic (η6-arene)Mn(CO)3 complexes
In order to use these Pd-catalysed reactions as alternative methods for the preparation of functionalised cationic η6 manganese complexes, it was crucial, at this point of our study, to ascertain if the new neutral η5 complexes could be easily transformed into the corresponding cationic complex. The hydride abstraction proceeded very nicely in CH2Cl2 at room temperature (Fig. 6). The rearomatisation of complex 5c gave quantitatively the unprecedented complex 7c (Table 4, entry 1), which cannot be obtained by direct complexation of the corresponding free arene, since the manganese entity coordinates either the phenyl or the thienyl ring or both, giving a mixture of three inseparable products.
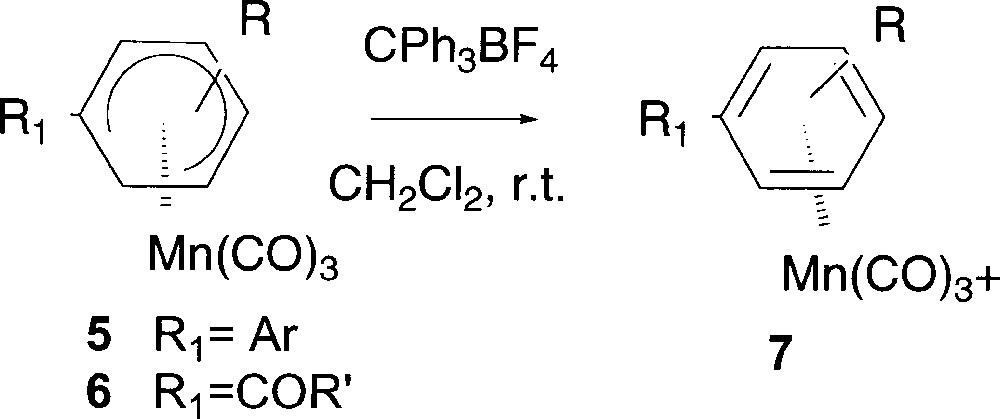
Preparation of cationic complexes 7 by rearomatisation.
| Entry | Complex | R | R1 | Yield (%) |
| 1 | 5c | 4-OMe | Th | 7c (>99) |
| 2 | 6a | 4-OMe | COTh | 7a (95) |
| 3 | 6b | 4-Me | COTh | 7b (70) |
| 4 | 6d | 4-OMe | CON(C2H4)2O | 7d (85) |
Performing the hydride abstraction from complexes 6 arose the matter of the coexistence of two strong acceptor groups (the Mn(CO)3+ entity and the carbonyl function) on the same ring. But the rearomatisation could be achieved in good yields from 70 to 95% yield by using CPh3BF4 as hydride abstracting reagent (Table 4, entries 2–4) to deliver stable cationic Mn complexes 7 bearing ketone or amide substituent.
As a conclusion we developed a high yield two step methodology to prepare unprecedented (η6-arene)Mn(CO)3+ complexes starting from (η5-chlorocyclohexadienyl)Mn(CO)3 complexes consisting in Pd coupling reactions followed by exo-hydride abstraction.
4 General experimental procedure
4.1 Stille cross-coupling reaction: preparation of tricarbonyl(4-methyl(1-thienyl)cyclohexa dienyl)manganese complex 5c
Pd2dba3 (0.019 g, 0.02 mmol) and AsPh3 (0.21 g, 0.07 mmol) were added to a solution of complex 2c (0.063 g, 0.2 mmol) in degased DMF (7 ml). After 10 min, 2-tributylstannyl thiophene (0.071 g, 0.2 mmol) was introduced. The reaction mixture was stirred at room temperature for 20 h, poured into ice cold water (100 ml) and extracted with 50 ml pentane, the organic phase was washed with water (2 × 50 ml), dried over magnesium sulphate, filtered. Solvents were evaporated under reduced pressure. The residue obtained was purified by silica gel chromatography (pentane) to deliver complex 5c as a yellow solid in 48% yield.
RMN 1H (CDCl3): 1.90 (3H, s, CH3); 2.51 (1H, d, J = 12 Hz, H6exo); 3.15(1H, d, J = 6 Hz, H5); 3.24 (1H, dd, J = 6 and 12 Hz, H6endo); 5.22 (1H, d, J = 6 Hz, H2); 5.74 (1H, d, J = 6 Hz, H3); 6,83 (1H, t, J = 5 Hz, H10); 6,90 (1H, t, J = 5 Hz, H9); 7,17 (1H, d, J = 5 Hz, H8).
Elemental analysis: calculated, C 53.51, H 3.53; found, C 53.59, H 3.75.
4.2 Carbonylation reaction: preparation of (4-methoxy-cyclohexadienyl)Mn(CO)3thiophen-2-yl methanone complex 6a
Pd2dba3 (44 mg, 0,048 mmol) and AsPh3 (51 mg, 0,17 mmol) were added to a solution of complex 2c (135 mg, 0,48 mmol) in THF (10 ml). CO was bubbled into the reaction mixture, which was stirred at room temperature for 15 min, before introducing 2-tributylstannyl thiophene (343 mg, 1 mmol). The mixture was stirred at 60 °C overnight, then poured into ice-cold water (100 ml) and extracted with 50 ml ethyl ether. The organic phase was washed with water (2 × 50 ml), dried over magnesium sulphate and filtered. The solvents were evaporated under reduced pressure. The residue obtained was purified by silica gel chromatography (pentane 90%, ether 10%) to deliver complex 6a as a yellow solid in 90% yield.
RMN 1H (CDCl3): 2.36(1H, d, J = 13 Hz, H6exo); 3.20 (1H, dd, J = 6 et 2 Hz, H5); 3.50 (1H, dd, J = 6 et 13 Hz, H6endo); 3.48 (3H, s, OCH3); 6.03 (1 H, d, J = 6 Hz, H2); 6.14 (1H, d, J = 6 Hz, H3); 7.07 (1H, dd, J = 4 and 5 Hz, Ar–H); 7.48 (1H, dd, J = 4 and 5 Hz, Ar–H); 7,55 (1H, m, Ar–H).
Elemental analysis: calculated, C 50.29, H 3.12; found, C 50.84, H 3.71.
4.3 Hydride abstraction: preparation of tricarbonyl(4-methoxy(1-thienyl)arene)manganese complex 7a
A solution of complex 6a (108 mg; 0.15 mmol) in CH2Cl2 (5 ml) is introduced into a solution of CPh3BF4 (96 mg, 0.3 mmol) in CH2Cl2 (5 ml). The reaction mixture was stirred overnight, half the solvent was then evaporated. After cooling at 0°C, anhydrous Et2O was added, a yellow solid precipitated. The solution was filtered and washed with anhydrous Et2O and dried, complex 7a was obtained as a yellow solid in 95% yield.
RMN 1H (acetone d6): 4.27 (3H, s, OMe); 6.57 (2H, d, J = 7.5 Hz, H2 and H6); 7.37 (1H, m, Ar–H); 7.66 (2H, d, J = 7.5 Hz, H3 and H5); 8.09 (1H, m, Ar–H); 8.23 (1H, m, Ar–H).
Elemental analysis: calculated, C 40.42, H 2.42; found, C 40.52, H 2.59.