

1 Introduction
This thematic issue of Comptes Rendus celebrates a GECOM–CONCOORD Symposium held in Albé (Alsace) 13–18 May 2001. Our oral presentation described a variety of developments involving complexes in which sp carbon chains span two transition metals, I, a good background for which is available in a series of full papers published by our laboratory 〚1–6〛. This subject has also been of much interest in other research groups 〚7–10〛. Rather then republish results that are thoroughly described elsewhere, two unique themes are presented in this written contribution. The first involves an unanticipated by-product from an sp carbon-chain extension reaction. The second involves the structural characterization of monoplatinum complexes with C6–C8 sp carbon chains.
Complexes of the type I can exist in several valence forms (e.g., M–C, M=C, or M≡C systems) and electronic ground states (e.g., singlet/triplet). They exhibit numerous fascinating physical and chemical properties 〚1–10〛. In many cases, interactions between the metal endgroups play key roles. In order to accurately analyse endgroup–endgroup interactions, properties of monometallic model complexes LnMCxRm must be precisely defined. Two recent papers provide good illustrations. The first, a collaboration with Lapinte 〚11〛, details a variety of manifestations of iron–rhenium interactions as a function of oxidation state in heterobimetallic C4 complexes 〚(η5-C5Me5)Re(NO)(PPh3)(C4)(η2-dppe)Fe(η5-C5Me5)〛n+·n PF6– (n = 0–2). Analyses required many measurements on mono-iron and monorhenium model compounds, and the analogous di-iron and dirhenium C4 complexes. The second is a theoretical analysis of odd carbon-chain complexes 〚(η5-C5R5)(NO)(PRˈ3)ReCxMLn〛n+ (x = 3, 5, 7, 9), and analogous monometallic species 〚11〛. Structural, electronic, and thermodynamic data revealed strong metal–metal interactions in some cases (MLn = Mn/Re(CO)2(η5-C5H5)), but not others (MLn = W(OMe)3) 〚12〛.
We recently communicated a new series of PtCxPt complexes (x = 8, 12, 16) with polyynediyl ((C≡C)n) chains and square-planar (p-tol)(Ph3P)2Pt, (p-tol)(p-tol3P)2Pt, or (C6F5)(p-tol3P)2Pt endgroups, illustrated by the general structure II 〚13–16〛. These are useful building blocks for more elaborate species, and are rapidly being extended, as detailed in our oral contribution. Thus, the corresponding monoplatinum complexes must also be thoroughly characterized. Accordingly, this paper reports the crystal structures of four such compounds, including one derived from an unexpected addition of the non-platinum sp terminus to acetone.
2 Results
The Hay oxidation represents one of the most useful methods for the symmetrical or cross coupling of terminal alkynes 〚17, 18〛. An excess of O2 and 0.20–0.25 equivalents of CuCl/TMEDA in acetone are frequently employed. As previously described 〚14〛, reaction of the readily available 1,3-butadiynyl complex trans-(C6F5)(p-tol3P)2PtC≡CC≡CH (1) and an eight-fold excess of HC≡CSiEt3 under such Hay conditions gave the cross-coupled target complex trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CSiEt3 (2) in 53% yield (Fig. 1). Under standard conditions for protodesilylation 〚5, 6, 13〛, 2 was treated with n-Bu4N+ F– in wet THF (0.20 equiv). The 1,3,5-hexatriynyl complex trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CH (3) was isolated as a white powder in 85% yield after a low-temperature workup 〚14〛. It darkened within a few minutes at room temperature, but was stable at –18 °C for several days.
Syntheses of platinum complexes.
The enhanced lability of longer hydrogen-capped polyynes is well documented 〚5, 13〛. Thus, coupling conditions were sought that would allow 3 and related compounds to be used in situ. Accordingly, an acetone solution of 2 was treated with n-Bu4N+ F– in wet THF to generate 3, followed by ClSiMe3 and then excess HC≡CSiEt3 under the Hay coupling conditions used above 〚14〛. Workup gave the 1,3,5,7-octatetraynyl complex trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CC≡CSiEt3 (4) in 30% yield 〚14〛. When ClSiMe3 was omitted, only traces of 4 formed. We hypothesize that fluoride ion must be scavenged prior to coupling. The yield of 4 is lowered due to the formation of some homocoupling product trans,trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CC≡CC≡CC≡CPt(Pp-tol3)2(C6F5) (5). This C12 or dodecahexayndiyl complex is of considerable interest and use in its own right, but can be prepared in higher yield under homocoupling conditions in the absence of HC≡CSiEt3 〚14〛.
In one such sequence, SiMe4 was accidentally substituted for ClSiMe3. Silica gel chromatography gave a 35:65 mixture of 5 and a new species 6 (58% total). Further purification attempts were not successful. The 1H NMR spectrum of the mixture showed two closely spaced C6H4CH3 signals, as well as a singlet (δ 1.53) associated with the new compound 6. The 13C NMR spectrum was not particularly informative, so the mixture was crystallized from CHCl3/ethanol. The crystal structure of 5 had been previously determined 〚14〛, so a crystal of a different colour and morphology was selected in the hopes that it would correspond to the new compound 6.
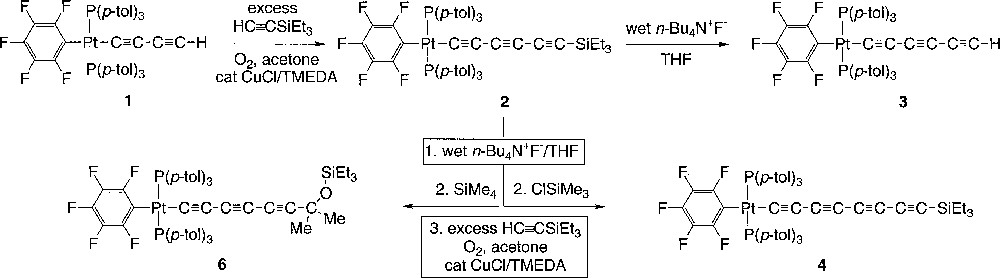
The X-ray structure was determined, and general data are summarized in Table 1. The ORTEP diagram, which shows 6 to have the structure trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CC(Me)2OSiEt3, is given in Fig. 2. In a formal sense, 6 is derived from the 1,2-addition of the sp-carbon–silicon bond of 2 to acetone. However, when 2 was refluxed in acetone (45 min), no reaction occurred. Key bond lengths and angles are listed in Table 2. An ion corresponding to this mass could be found in the mass spectrum of the mixture. Furthermore, a 13C NMR signal with an appropriate chemical shift and intensity for the acetone-derived methyl groups was evident (δ 32.7).
Summary of crystallographic data.
| Complex | 6 | 4 | 2 | 7 |
| Formula | C63H63F5OP2PtSi | C62H57F5P2PtSi | C60H57F5P2PtSi | C55H52P2PtSi |
| Diffractometer | Nonius KappaCCD | Nonius KappaCCD | Nonius KappaCCD | Nonius KappaCCD |
| Temperature 〚K〛 | 173(2) | 173(2) | 173(2) | 200(0.1) |
| Wavelength 〚Å〛 | 0.710 73 | 0.710 73 | 0.710 73 | 0.710 73 |
| Crystal system | monoclinic | triclinic | triclinic | triclinic |
| Space group | C2/c | P1 | P1 | P1 |
| a 〚Å〛 | 18.9881(2) | 12.415 30(10) | 12.9303(2) | 12.6693(2) |
| b 〚Å〛 | 17.9089(2) | 14.2459(2) | 13.2663(2) | 12.8115(2) |
| c 〚Å〛 | 34.1429(4) | 17.0738(3) | 15.8682(2) | 17.2017(3) |
| α 〚°〛 | 90 | 91.9530(4) | 87.9570(10) | 80.9290(9) |
| β 〚°〛 | 95.8280(10) | 95.3030(4) | 88.2170(10) | 80.5370(9) |
| γ 〚°〛 | 90 | 109.4490(6) | 89.3000(10) | 60.7590(8) |
| V 〚Å3〛 | 11550.5(2) | 2828.52(7) | 2718.77(7) | 2389.25(7) |
| Z | 8 | 2 | 2 | 2 |
| dcalc 〚g cm–3〛 | 1.399 | 1.388 | 1.415 | 1.387 |
| Absorption coefficient 〚mm–1〛 | 2.562 | 2.612 | 2.715 | 3.063 |
| Crystal size 〚mm3〛 | 0.20 × 0.20 × 0.02 | 0.30 × 0.20 × 0.15 | 0.30 × 0.30 × 0.10 | 0.31 × 0.28 × 0.10 |
| θ limit 〚°〛 | 1.20 to 26.04 | 1.75 to 25.01 | 1.28 to 27.46 | 1.20 to 24.39 |
| Index ranges (h, k, l) | –23 to 23, –22 to 22, –42 to 42 | –14 to 14, –16 to 16, –20 to 20 | –16 to 16, –17 to 17, –18 to 20 | 0 to 14; –12 to 14; –19 to 19 |
| Reflections collected | 22 108 | 18 804 | 22 881 | 7816 |
| Independent reflections | 11 361 〚R(int) = 0.0524〛 | 9916 〚R(int) = 0.0187〛 | 12 290 〚R(int) = 0.0245〛 | 7816 |
| Reflections 〚I > 2 σ(I)〛 | 7845 | 9018 | 10724 | 7491 |
| Data/restraints/parameters | 11361/0/658 | 9916/0/640 | 12290/0/622 | 7816/0/569 |
| Goodness-of-fit on F2 | 0.996 | 1.048 | 1.121 | 1.310 |
| Final R indices 〚I > 2 σ(I)〛 | R1 = 0.0432, wR2 = 0.0993 | R1 = 0.0268, wR2 = 0.0637 | R1 = 0.0260, wR2 = 0.0619 | R1 = 0.0295, wR2 = 0.0959 |
| R indices (all data) | R1 = 0.0835, wR2 = 0.1306 | R1 = 0.0317, wR2 = 0.0660 | R1 = 0.0355, wR2 = 0.0756 | R1 = 0.063, wR2 = 0.1264 |
| Δp (max), e Å–3 | 0.919 | 1.175 | 0.614 | 2.515 |
Molecular Structure of trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CC(Me)2OSiEt3 (6).
Comparison of key bond lengths and angles.
| 6 | 4 | 2 | 7 | |
| Pt–C1 | 1.992(6) | 1.986(3) | 1.985(3) | 2.000(5) |
| C1≡C2 | 1.206(8) | 1.224(5) | 1.226(4) | 1.227(8) |
| C2–C3 | 1.379(9) | 1.356(5) | 1.362(4) | 1.360(8) |
| C3≡C4 | 1.204(8) | 1.219(5) | 1.210(5) | 1.207(9) |
| C4–C5 | 1.363(9) | 1.355(5) | 1.367(5) | 1.369(8) |
| C5≡C6 | 1.194(8) | 1.211(5) | 1.206(5) | 1.203(8) |
| C6–C7 | 1.483(9) | 1.367(5) | — | — |
| C7≡C8 | — | 1.202(5) | — | — |
| C7–O | 1.397(8)a | |||
| C6–Si or C8–Si | — | 1.848(4) | 1.843(3) | 1.835(6) |
| O–Si | 1.636(6) | — | — | — |
| Pt–P1 | 2.3085(15) | 2.3044(8) | 2.3111(7) | 2.2963(13) |
| Pt–P2 | 2.3012(15) | 2.3049(8) | 2.2994(7) | 2.2996(14) |
| Pt–Cipso | 2.069(6) | 2.061(3) | 2.068(3) | 2.079(6) |
| Pt–C1–C2 | 175.9(5) | 179.3(3) | 178.8(3) | 176.8(5) |
| C1–C2–C3 | 175.4(7) | 177.3(4) | 175.7(3) | 177.0(6) |
| C2–C3–C4 | 178.3(7) | 178.4(4) | 177.3(4) | 178.2(6) |
| C3–C4–C5 | 176.7(8) | 175.1(4) | 174.9(4) | 177.2(6) |
| C4–C5–C6 | 177.3(8) | 175.7(4) | 178.7(4) | 178.5(7) |
| C5–C6–C7 | 174.0(8) | 174.5(4) | — | — |
| C6–C7–C8 | — | 177.7(4) | — | — |
| C6–C7–O | 112.6(6) | |||
| C5–C6–Si or C7–C8–Si | — | 178.6(4) | 178.8(4) | 176.3(6) |
| C7–O–Si | 137.3(4) | — | — | — |
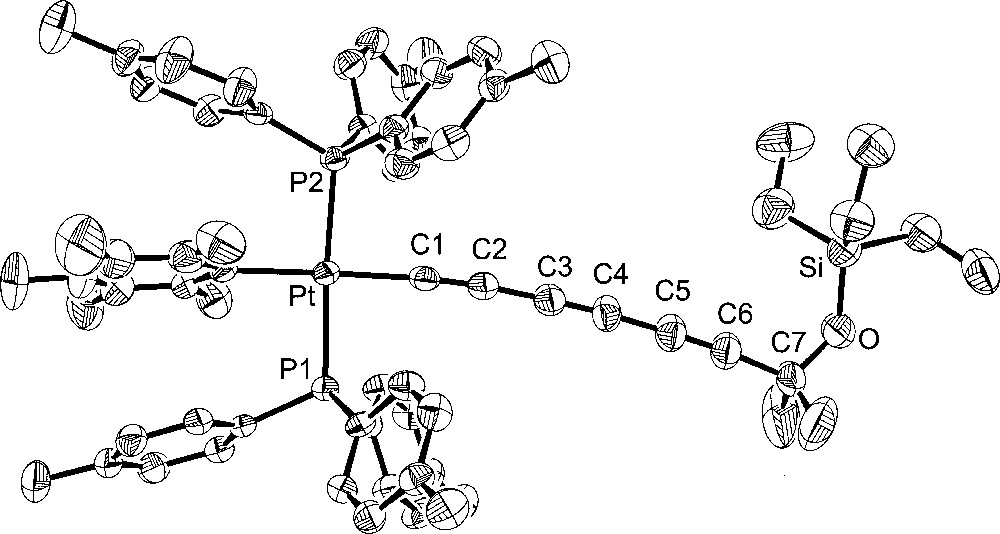
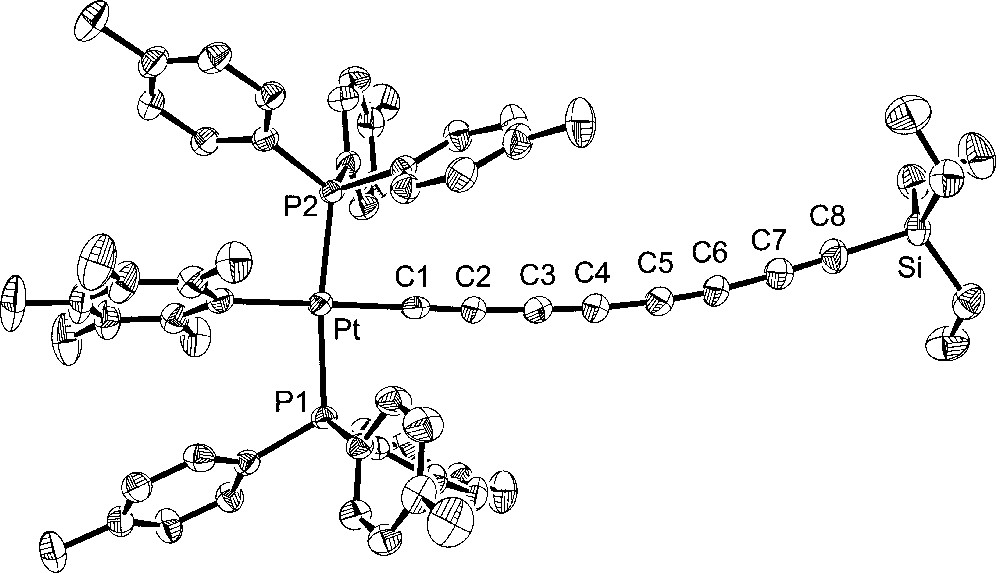
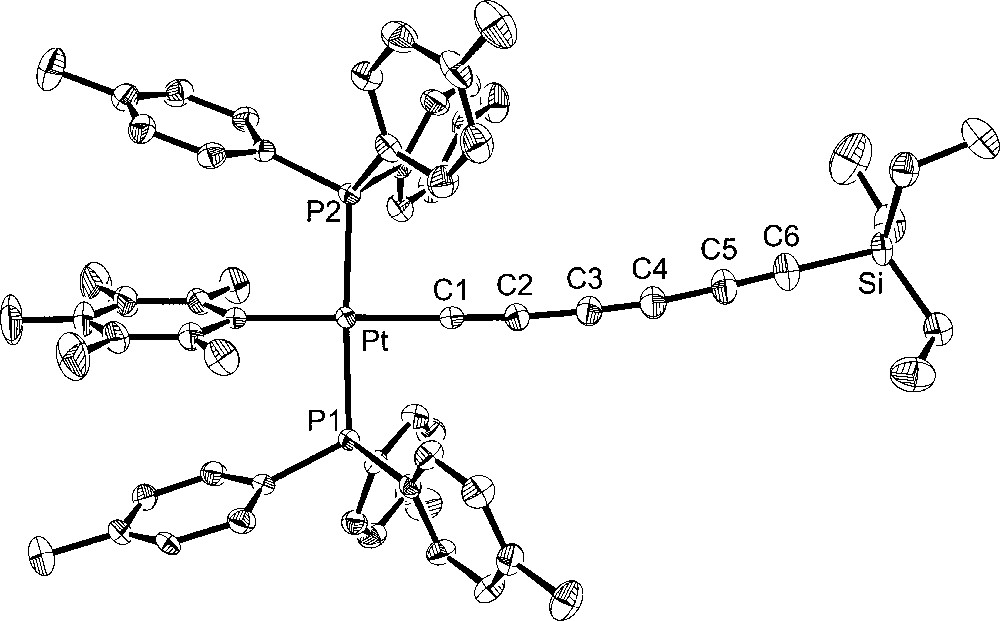
Additional crystal structures were executed for comparison purposes. The first was that of the 1,3,5,7-octatetraynyl complex 4, which features the same number of atoms between platinum and silicon as 6. The second was the 1,3,5-hexatriynyl complex 2, which contains the same number of sp-hybridised carbons as 6. The third was the previously reported 1,3,5-hexatriynyl complex, trans-(p-tol)(Ph3P)2PtC≡CC≡CC≡CSiEt3 (7), which was obtained by a sequence analogous to that used for 2 in Fig. 1 〚13〛. Complexes 2 and 7 differ in the phosphine (p-tol3P vs Ph3P) and aryl (C6F5 vs p-tol) ligands on platinum. The molecular structures are shown in Figs. 3–5, and other data are incorporated into Tables 1 and 2.
Molecular structure of trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CC≡CSiEt3 (4).
Molecular structure of trans-(C6F5)(p-tol3P)2PtC≡CC≡CC≡CSiEt3 (2).
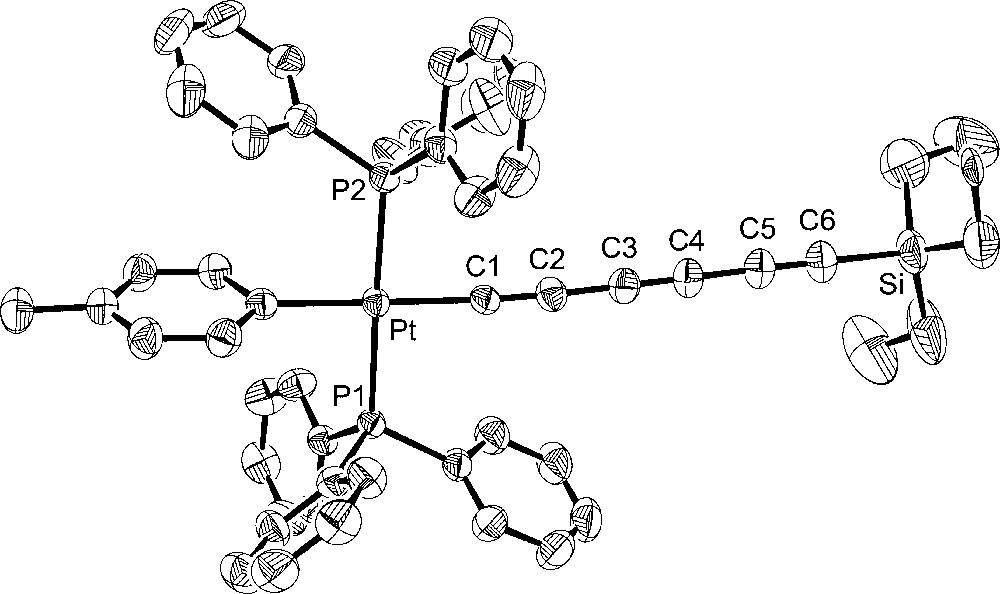
Molecular structure of trans-(p-tol)(Ph3P)2PtC≡CC≡CC≡CSiEt3 (7).
3 Discussion
All of the structurally characterized complexes feature PtC≡CC≡CC≡C linkages. Complexes with 1,3-butadiynyl ligands have recently been comprehensively reviewed by Low and Bruce 〚19〛, and crystal structures of several platinum adducts have been reported. To our knowledge, no higher monoplatinum polyynyl species have been structurally characterized, as verified by an independent search of the Cambridge database. However, the crystal structures of a number of diplatinum complexes of the type II have been determined 〚13, 14, 20〛. Crystallographic data and packing motifs for conjugated octatetraynes with both organic and transition metal endgroups are summarized in detail elsewhere 〚21〛.
Turning to the data in Table 2 and Figs. 2–5, the C8 chain in octatetraynyl complex 4 shows a slight but distinct bending. The average bond angle, 177.1°, is not very different from 180°, but the deviations are complementary and reinforce the curvature. The platinum–silicon distance, 12.6168(10) Å, is 1% shorter than the sum of the nine intervening bond lengths, 12.768 Å. By these criteria, 4 is similar to several other tetraynes, including (η5-C5Me5)Re(NO)(PPh3)(C≡CC≡CC≡CC≡CSiMe3) 〚21〛 and one with cyclobutadienyl cobalt endgroups (average angles 177.5°, 176.7°) 〚22〛. One tetrayne, (η5-C5Me5)Re(NO)(PPh3)(C≡CC≡CC≡CC≡C-p-tol) 〚23〛, exhibits distinctly greater bending (average angle 175.7°). The hexatriynyl complexes 6, 2, and 7 have average bond angles of 176.3°, 177.4°, and 177.3°, but since there are fewer sp carbons the bending (Figs. 2, 4 and 5) is not as visually dramatic.
The Pt–C≡C–C≡C–C≡C bond lengths in homologous complexes 4 and 2 are identical within experimental error. The Pt–Cipso bond lengths and the average of the two Pt–P bond lengths are also identical. Only one or two bond angles show meaningful differences. Furthermore, the space groups are identical, and the unit cells show analogous packing motifs. Somewhat greater bond length and angle differences are evident in 6. Of these, the Pt–C≡C angles are the most noteworthy. The M–C≡C linkages are often among the most bent in the chain, presumably reflecting their lower bending force constants (vs C–C≡C). The angle in 6 (175.9(5)°) is typical. However, 4 and 2 exhibit nearly linear linkages (179.3(3)°, 178.8(3)°). Some of the bond lengths and angles of 7 lie at the extremes of the other compounds, but the differences are not great enough for a distinct endgroup effect to be claimed. In all four compounds, the aryl ligands adopt similar Pt–Cipso conformations, with angles of 73.8°–76.7° between the least squares planes of the aryl ring and platinum (P1–Pt–Cipso–Cortho; 6/4/2/7 76.7°/73.8°/74.8°/74.3°).
The following points are relevant to the formation of 6. First, additions of silicon compounds of the type XSiR3 to carbonyl groups are well known, and can in many cases be catalysed by nucleophiles. The fluoride ion-mediated addition of HC≡CSiMe3 to carbonyl compounds has been explicitly reported 〚24〛. An anionic PtC≡CC≡CC≡C– species would likely be generated when 2 is treated with n-Bu4N+ F– in wet THF (Fig. 1), both in the initial step and then in small equilibrium amounts from the protonation product 3. Curiously, the IR monitoring of several reactions like these has established that only substoichiometric quantities of n-Bu4N+ F– are needed. Following acetone addition, a triethylsilyl group could be transferred from the eight-fold excess of HC≡CSiEt3 or from the other XSiEt3 species present. However, what would exactly inhibit acetone addition/silylation under the conditions involving ClSiMe3, or promote addition/silylation under the conditions utilizing SiMe4, is a matter of speculation. There are simply too many variables, and narrowing the possibilities would require an extensive series of control experiments. For obvious reasons, we wish to avoid a lengthy formal mechanistic study of an unwanted side reaction.
With regard to future research with compounds of the types I and II, chain lengthening will be an important theme. There is no doubt that compounds with very long sp carbon chains have been generated in solution. These include a C32 hexadecayne with SiR3 endgroups 〚25〛, and a C24 dodecayne with t-butyl endgroups 〚26〛. The question that should be answered shortly is whether transition metal endgroups, which are electropositive and commonly quite bulky, will enhance stabilities and allow such compounds to be isolated and thoroughly characterised. The current records stand at C20 for the (η5-C5Me5)Re(NO)(PPh3) endgroup 〚4〛, and C16 for the (C6F5)(p-tol3P)2Pt endgroup 〚14〛. In all of these efforts, the physical and chemical study of monometallic model compounds, as exemplified by the structural and reactivity data in this paper, will play an essential role 〚27〛.
4 Experimental
For the general procedures employed, see references 〚13–16〛.
4.1 Synthesis of 6
A three-necked flask was charged with 2 〚14〛 (0.201 g, 0.174 mmol) and acetone (15 ml), and fitted with a gas dispersion tube and a condenser. A Schlenk flask was charged with CuCl (0.100 g, 1.02 mmol) and acetone (30 ml), and TMEDA (0.060 ml, 0.40 mmol) was added with stirring. After 30 min, stirring was halted and a green solid separated from the blue supernatant. Then n-Bu4N+ F– (1.0 M in THF/5 wt% H2O, 0.040 ml, 0.040 mmol) was added to the solution of 2 with stirring. After 10 min, SiMe4 (0.014 ml, 0.103 mmol) was added. After 20 min, the condenser was cooled to –20 °C. Then O2 was bubbled through the three-necked flask, and the solution was heated to 65 °C. After ca 10 min, HC≡CSiEt3 (0.197 g, 1.404 mmol) was added, followed by portions of the blue supernatant. After 3 h, solvent was removed by rotary evaporation. The residue was extracted with hexane (3 × 5 ml) and then benzene (3 × 5 ml). The extracts were passed in sequence through an alumina column (15 cm). Solvent was removed from the benzene extracts by rotary evaporation and oil pump vacuum. The resulting yellow powder was chromatographed on a silica gel column (15 cm) with 10:90 v/v CH2Cl2/hexane to elute traces of 4 〚6〛 and then 40:60 v/v CH2Cl2/hexane to elute a second band. The latter was taken to dryness by oil pump vacuum to give a yellow powder that was a 65:35 mixture of 6 and 5 (0.114 g, 0.049/0.026 mmol, 28%/30%), as assayed by 1H NMR.
IR (cm–1, powder film) νC≡C 2150 (s), 2038 (m), and bands of 5 〚14〛. NMR (δ, CDCl3, signals of 5 〚14〛 omitted unless noted): 1H 7.45 (m, 12H, o to P), 7.10 (d, 3JHH = 7.4 Hz, 12H, m to P), 2.35 (s, 18H, C6H4CH3), 1.53 (s, 6H, C(CH3)2O), 0.87 (t, 3JHH = 7.8 Hz, 9H, CH2CH3), 0.59 (q, 3JHH = 7.8 Hz, 6H, CH2CH3); 13C{1H} (partial; the signals o/m/p to Pt are very close to those of 5 and 1JCF and 2JCF values could not be determined) 145.2 (dd, o to Pt), 140.3 (s, p to P), 136.9 (dm, p to Pt), 136.2 (dm, m to Pt), 140.3 (s, p to P), 133.8 (virtual t, 2JCP = 6.5 Hz, o to P), 128.3 (virtual t, 3JCP = 5.5 Hz, m to P), 127.1 (virtual t, 1JCP = 30.2 Hz, i to P), 95.5, 79.7, 69.7, 68.2, 66.3, 65.8, 63.1, 61.0, 57.2 (9 s, C≡ of 6 and 5), 54.9 (s, C(CH3)2O, tentative), 32.7 (s, C(CH3)2O), 21.1 (s, C6H4CH3), 6.7 (s, CH2CH3), 5.7 (s, CH2CH3); 31P{1H}, 17.9 (s). MS (m/z; FAB, 3-NBA) 1215 (6-H+, 10%), 970 (〚(C6F5)Pt(Ptol3)2〛+, 50%), 802 (〚Pt(Ptol3)2–H〛+, 50%).
4.2 Crystallography
A CHCl3 solution of 6 and 5 (65:35) was layered with ethanol. After 14 h at room temperature, a mixture of thin colourless plates (6) and yellow powder (5) had formed. One of the former was removed and data were collected as outlined in Table 1. Cell parameters were obtained from 10 frames using a 10° scan and refined with 11 450 reflections. Lorentz, polarisation, and absorption corrections 〚28, 29〛 were applied. The space group was determined from systematic absences and subsequent least-squares refinement. The structure was solved by direct methods. The parameters were refined with all data by full-matrix-least-squares on F2 using SHELXL-97 〚30〛. Non-hydrogen atoms were refined with anisotropically. The hydrogen atoms were fixed in idealized positions using a riding model. Scattering factors were taken from the literature 〚31〛.
A CHCl3 solution of 4 was slowly concentrated by evaporation. After several days at room temperature, one of the yellow needles was removed. A CH2Cl2 solution of 2 was layered with ethanol. After several days at room temperature, a yellow needle was removed. Data were collected and the structures refined and solved exactly as for 6.
A concentrated CH2Cl2 solution of 7 〚13〛 was layered with hexanes and slowly concentrated by evaporation. After two weeks, a colourless needle was removed, and data were collected as outlined in Table 1. Cell parameters were obtained from 10 frames using a 10° scan. The space group was determined from least-squares refinement. Lorentz, polarisation and absorption corrections were applied using DENZO-SMN and SCALEPACK 〚29〛. The structure was solved by standard heavy atom techniques with the SIR97 package and refined with SHELXL-97 〚30〛. Non-hydrogen atoms were refined with anisotropic thermal parameters. Two ethyl groups showed displacement disorder (C81/C81ˈ, C82/C82ˈ, C83/C83ˈ, C84/C84ˈ), which could be solved and refined to a 77:23 ratio. Hydrogen atom positions were calculated and added to the structure factor calculations, using the riding model. Scattering factors, as well as Δfˈ and Δf′′ values, were taken from literature 〚32, 33〛.
Supplementary material
All crystallographic data (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre, CCDC nos. 166712 (6), 166711 (4), 166710 (2), and 134398 (7). Copies of this information can be obtained free on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK. 〚Fax: (internat.) + 44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk〛.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, GL 300/1-1) and Johnson Matthey PMC (platinum loan) for support of this research.