

1 Introduction
Today, metalloenzymes are under very intense scrutiny in several laboratories throughout the world. It is quite clear that inorganic chemistry is being enriched by those biochemical studies, since the role of metal ions in living processes often corresponds to new chemistry. More chemistry has been invented by chance over billions of years than by man in 200 years. Thus the study of metalloenzymes gave a formidable impulse to inorganic chemistry throughout the last 30 years. The reverse is also true: inorganic chemists contributed to the understanding of the metalloenzyme function. Illustrations of the back influence of inorganic chemistry on biochemistry can be found in the work of Groves on hemes 〚1〛, Holm on Fe–S clusters 〚2〛, Lippard 〚3〛 and Que 〚4〛 on modelling of di-iron-active site containing enzymes.
A potential application to chemistry resulting from the researches on metalloenzymes is catalysis. It is quite possible to prepare artificial complexes that have a catalytic activity similar to that of metalloenzymes. This has been the case for iron porphyrins able to catalyse the hydroxylation of alkanes as inspired from CytP450 〚5〛, for iron complexes able to catalyse the oxidative cleavage by dioxygen of catechols as do intradiol catechol dioxygenases 〚6〛 and for Cu-phenolato complexes catalysing the oxidation by air of alcohols into aldehydes 〚7〛, so mimicking galactose oxidase.
At this point, we can make a difference between biomimetic and bio-inspired catalysis. Biomimetic chemistry implies a great similarity between the enzyme active site and the artificial catalyst. It is very demanding, since it can be a real challenge to faithfully mimic the active site of an enzyme; such an effort is certainly valuable for the understanding of the function of the enzyme, but it may be not adapted to the research of a low cost and high robustness industrial catalyst. In bio-inspired chemistry, the enzyme is taken as a source of inspiration for the chemist. It can be a good approach for large-scale applications such as depollution, detergents and energy. It also contains a freedom in invention, which is one of the great pleasures of chemistry.
In this paper, we present a review of our own work in collaboration with industry to explore application of bio-inspired catalysis. In the past, with Elf Company, we invented a process to degrade lignin in wood pulp for paper industry 〚8〛. This process was inspired from Mn-dependent ligninases 〚9〛 and used TPA/Mn(II) as catalyst and H2O2 as oxidizing agent. (Abbreviations used: TPA = tris(pyridylmethyl)amine; N4py = N,N-bis(2-pyridylmethyl)-N-(bis-2-pyridylmethyl)amine; hfacac– = hexafluoroacetylacetonate; HBpz3– = tris(pyrazolyl)borate.) We discovered with the Unilever Company new ways for stain bleaching 〚10〛. We will deal here with our work with Elf Company about the degradation of aromatic pollutants 〚11〛, a research inspired from dioxygenases found in bacteria able to grow on aromatic hydrocarbons, and with a new work with Osaka Gas Company about oxidation of alkanes by air in the presence of a reducing agent, a work inspired from the famous Methane MonoOxygenase (MMO) 〚12〛.
2 Degradation of aromatic pollutants
Some bacteria are able to grow on aromatic compounds like naphtalene. They accomplish oxidation of aromatic compounds using a cascade of enzymes schematised in Fig. 1.
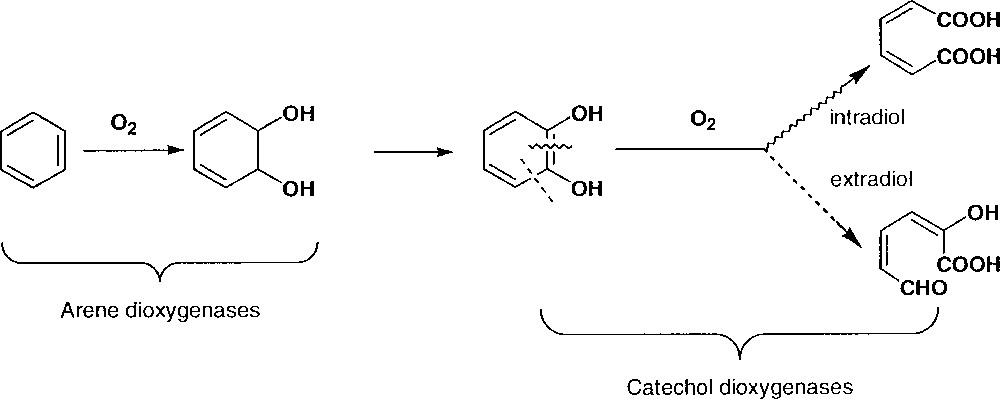
Degradation of aromatics by bacteria.
The first type of enzymes is arene dioxygenases, which use dioxygen and electrons to cis-dihydroxylate aromatic rings. The active site of naphtalene dioxygenase has been studied by X-ray diffraction: it contains one Fe(II) linked to a chelating aspartate, two histidines, a water molecule and possibly an asparagine (Fig. 2) 〚13,14〛.
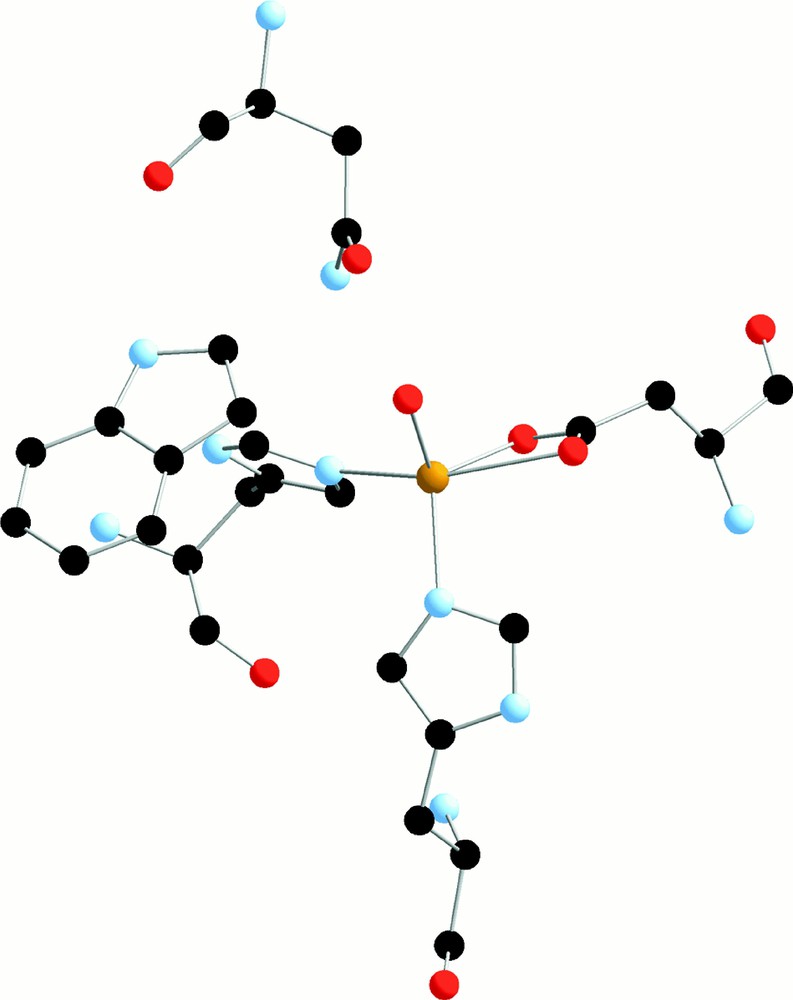
Active site of the naphtalenedioxygenase (from 〚13〛).
The Fe(II) ion can be seen as hexacoordinated or pentacoordinated – if asparagine is not linked – with the water molecule exchangeable with dioxygen. A possible mechanism implies a Fe(III) peroxo or hydroperoxo intermediate 〚15〛. The diol formed is rearomatised to catechol by a dehydrogenase. The catechol is then opened into linear oxidized molecules by catechol dioxygenases, either intradiol (Fe(III) site) or extradiol (Fe(II) site) 〚16〛. For instance, it has been shown 〚17〛 that in intradiol dioxygenase the catecholate anion is chelated to the Fe(III) ion before being attacked by dioxygen (Fig. 3).
Active site of 3,4-protocatechuate dioxygenase (from 〚17〛).
We decided to use a simple pentadentate ligand to prepare a stable Fe complex with a sixth free position available for dioxygen or H2O2 and we synthesized the trispicMeen one described in Fig. 4.
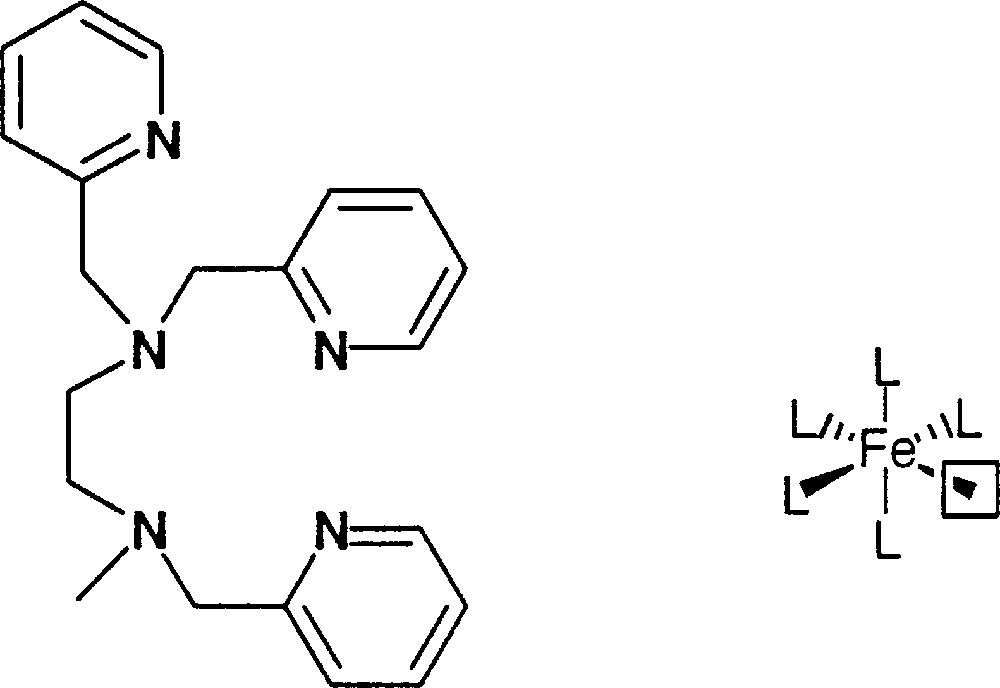
Ligand trispicMeen.
Instead of dioxygen and electrons, we used H2O2. In collaboration with Elf Company, we found that indeed this ligand in presence of Fe(ClO4)3 was very active to catalyse the degradation of aromatics by H2O2 at 40 °C in water 〚11〛. Polyaromatic hydrocarbons such as phenanthrene and fluoanthrene were decomposed into a mixture of polycarboxylic acids.
We tried to isolate the Fe(III) complex but we succeeded only in getting the dinuclear complex 〚(trispicMeen)ClFeIIIOFeIIICl(trispicMeen)〛2+ by reaction of the ligand on the dinuclear 〚Cl3FeIIIOFeIIICl3〛2- complex 〚18〛. On the opposite, we isolated and crystallized with Fe(II) the complex 〚FeII(trispicMeen)Cl〛+. Its structure is displayed in Fig. 5 〚19〛.
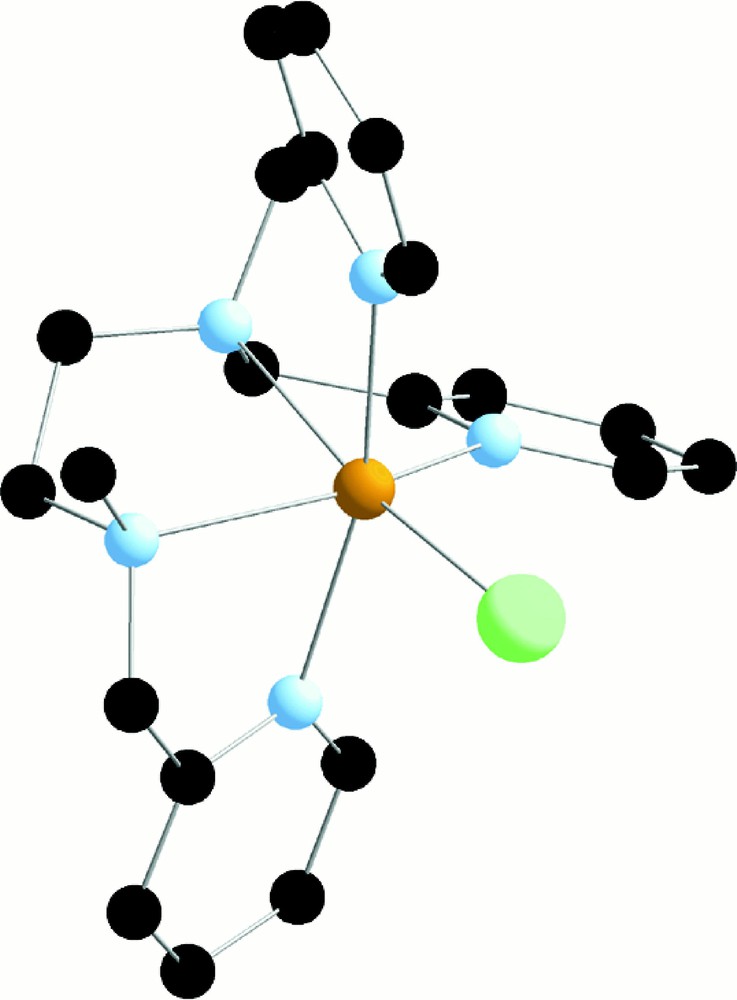
X-ray crystal structure of 〚FeII(trispicMeen)Cl〛+ in 〚FeII(trispicMeen)Cl〛〚BPh4〛 (from 〚19〛).
Fe(II) is surrounded by the pentadentate ligand and the sixth position is occupied by a chloride ion.
Independently, Bernal et al. 〚20〛 studied this complex and similar ones. They discovered that H2O2 gives an intermediate that they identified by UV-vis and EPR spectroscopy as a Fe(III)OOH entity. As this intermediate is a likely candidate to explain the catalysis of oxidation of aromatics we observed, we went further into its characterisation.
As observed by Bernal et al. 〚20〛, the spectrum of a methanol solution of 〚FeII(trispicMeen)Cl〛+ changes spectacularly by addition of an excess of hydrogen peroxide. A strong band appears at 540 nm, which was attributed to a LMCT transition between the HOO– group and Fe(III). This absorption remains for one to two hours at room temperature. EPR confirmed that Fe(II) was oxidised to a low spin Fe(III) very similar to that found in activated bleomycin, which was demonstrated to be (BLM)FeIIIOOH 〚21〛. Other non-heme Fe(III)OOH complexes have been characterized 〚22–27〛.
Resonance Raman spectroscopy using a 568 nm laser excitation allows to observe a vibration at 796 cm–1 attributable to a 16O16O vibration and another at 617 cm–1 attributable to a Fe16O vibration. By using H218O2, those frequencies shifted to 752 cm–1 and 600 cm–1 respectively. Using a very simple diatomic model, one computes 751 cm–1 and 590 cm–1 for the 18O sample, which agrees satisfactorily with the proposed attribution. Introducing some coupling between the vibrators would certainly allow to reach a better agreement. With TPA as ligand, a Fe(III)OOH species was characterised in RR with frequencies at 789 and 626 cm–1 〚28〛 and with the N4py 〚10〛 ligand, whose frequencies were 790 cm–1 and 632 cm–1 〚29〛. All those Fe(III)OOH species seem to have very similar properties. A very likely structure for 〚FeIII(trispicMeen)(OOH)〛2+ is shown in Fig. 6.
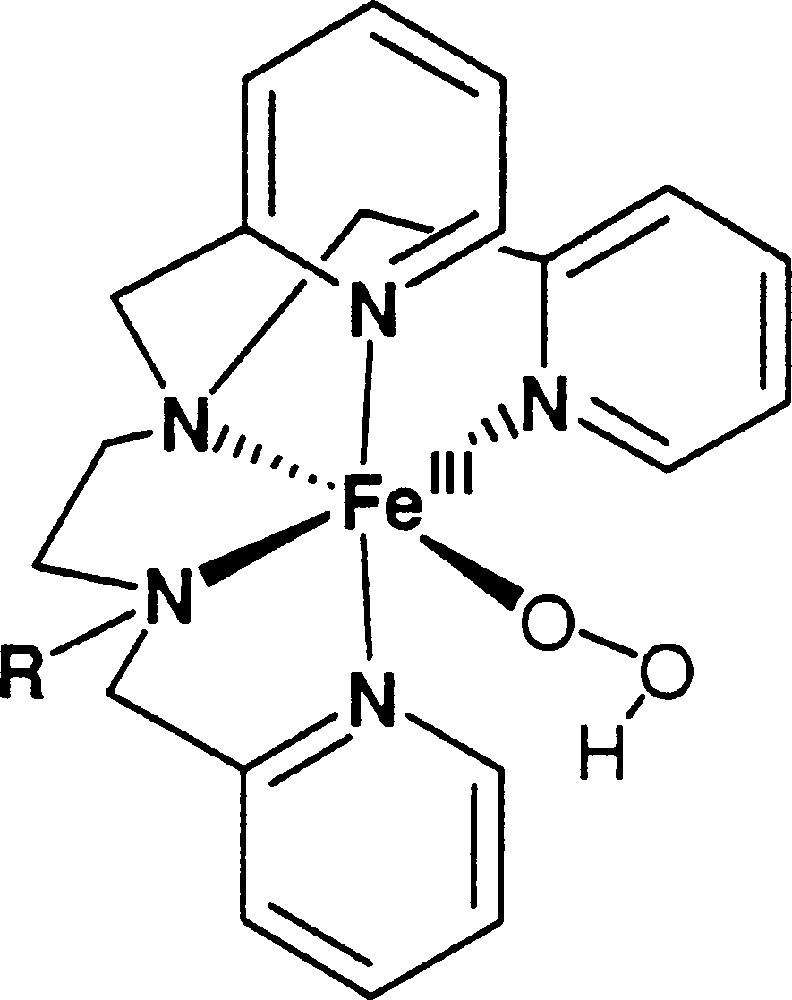
Proposed structure for 〚FeIII(trispicMeen)(OOH)〛2+.
The g values observed by EPR (1.95, 2.12, 2.19) can be rationalised by the model of Taylor 〚30〛 for low spin Fe(III) species. Application of this model gives Δ/λ = –11.31 and V/λ = 4.05, where Δ is the axial distortion energy and V the rhombic one for the 2T2g state 〚31〛. This shows that the ground state can be described (before application of spin orbit coupling) as built on the configuration dyz2dxz2dxy1. The most destabilized orbital is the orbital dxy. The axes x, y and z are such that gx = –2.12, gy = 2.19, gz = –1.95. Evidently one does not know at this stage the orientation of those axes versus the molecule, but the hydroperoxo group is certainly along x or y since this would explain that the dxy orbital is the most destabilised by interaction with the π* orbital of the hydroperoxo group (Fig. 7).
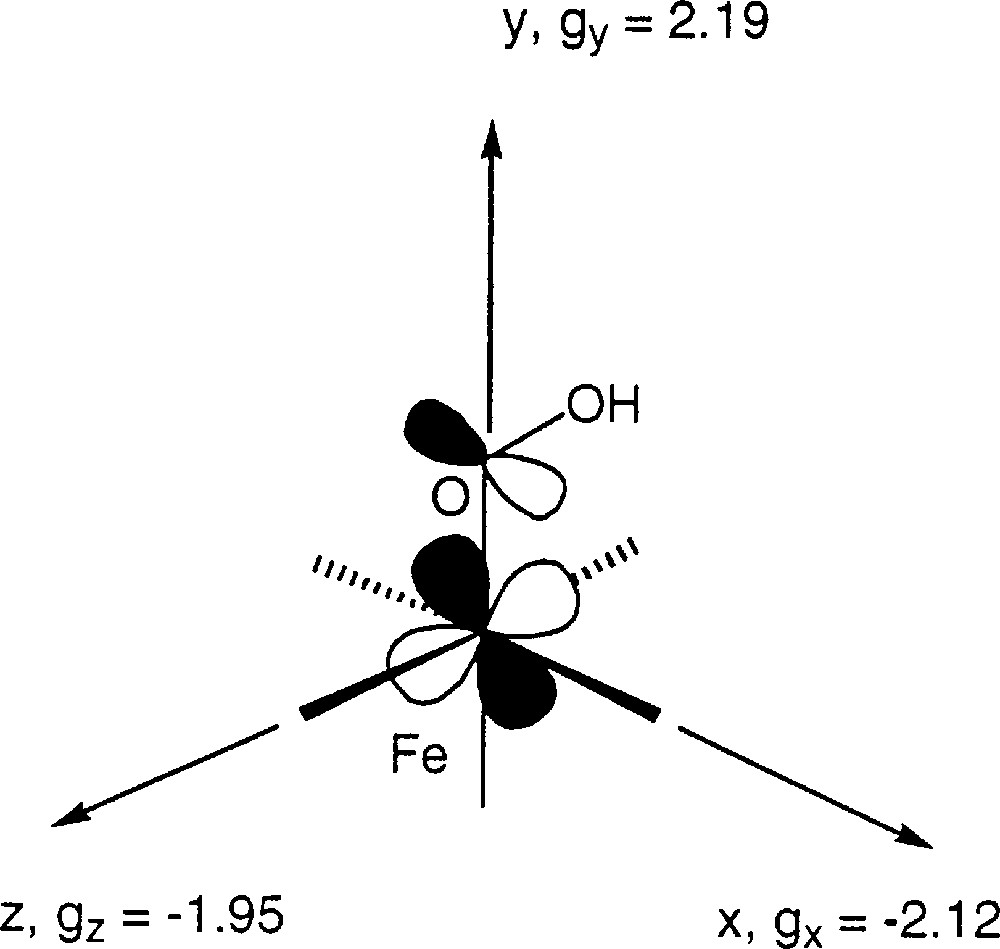
Representation of the dxy orbital and of the hydroperoxo π orbital in the 〚FeIII(trispicMeen)(OOH)〛2+ complex.
When one applies this model to bleomycin, one obtains Δ/λ = –8.74 and V/λ = 3.04. This suggests that the dxy orbital is less perturbed by the hydroperoxo group in BLM than that in the 〚FeIII(trispicMeen)(OOH)〛2+. This could be related to a shorter distance between Fe and the hydroperoxo group in 〚FeIII(trispicMeen)(OOH)〛2+.
The 〚FeIII(trispicMeen)(OOH)〛2+ model complex is so stable than deprotonation can be achieved in MeOH solution by adding NEt3 〚32〛. This can easily be detected by UV–vis: the maximum of absorption in the visible shifts from 540 nm to 740 nm. This is indicative of a lower energy for the charge transfer from the peroxo group to Fe(III). EPR establishes that upon deprotonation Fe(III) turns high spin (g = 7.5, g = 5.9, E/D = 0.08). In RR, vibrations were found at 819 cm–1 and 470 cm–1 shifting to 776 cm–1 and 454 cm–1 by using 18O2H2 〚25〛. Possible structures of the 〚FeIII(trispicMeen)(OO)〛+ derivative are described in Fig. 8.
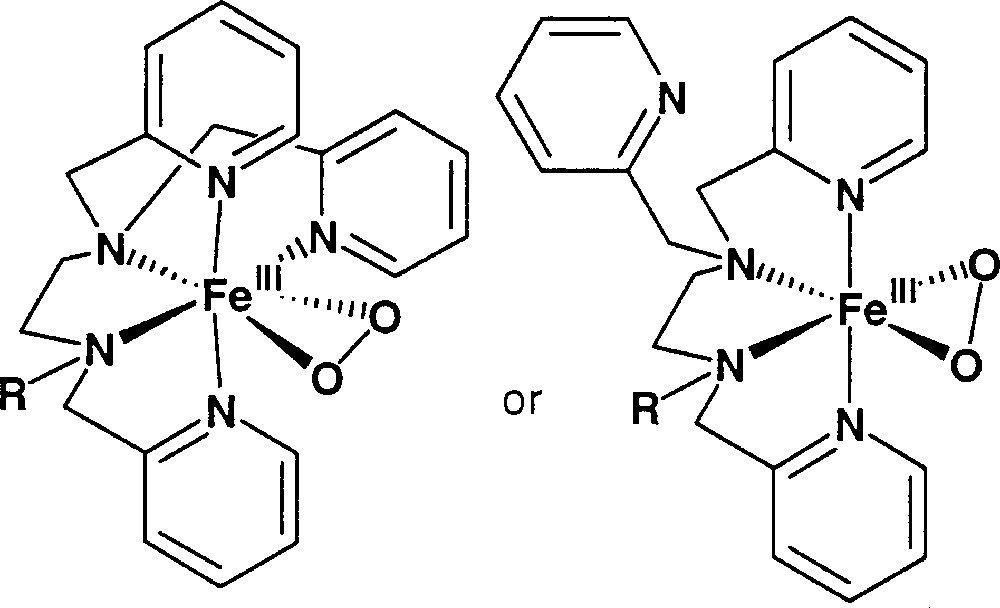
Proposed structures for 〚FeIII(trispicMeen)(OO)〛+.
Similar results were obtained by Jensen et al. 〚33〛. Using the ligand N4py, Ho et al. 〚34〛 identified the following frequencies: 827 and 495 cm–1, shifting to 781 and 478 cm–1. The FeO bond seems much weaker in 〚FeIIIO2〛+ than in 〚FeIIIO2H〛2+, while for the OO bond the reverse is true.
As for oxidation of aromatics by the system trispicMeen/Fe/H2O2 it is possible that the aromatics are first hydroxylated by OH radicals formed from the Fe(III)OOH entity. This would lead to phenols and catechols.
Another possibility is that the Fe(III)OOH intermediate itself cis-dihydroxylates an aromatic group, as represented in Fig. 9.
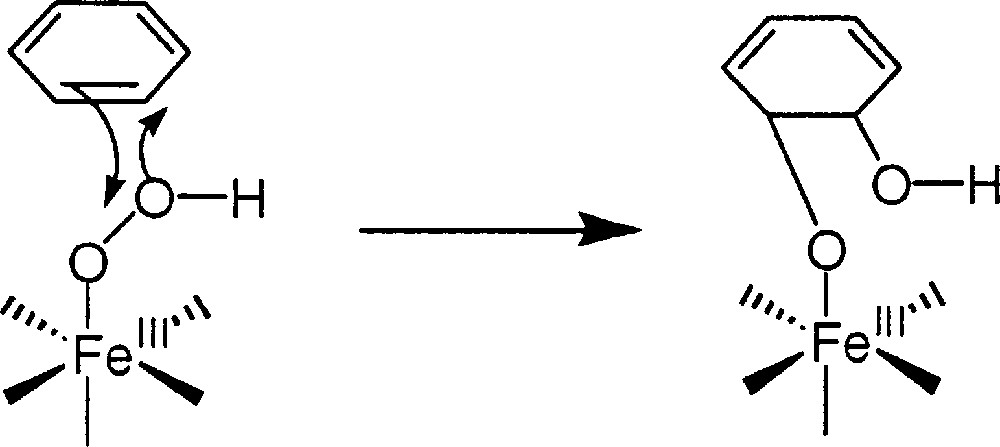
Proposed cis-dihydroxylation by the Fe(III)OOH species.
The diol formed could be reoxidised to catechols in the experimental conditions used.
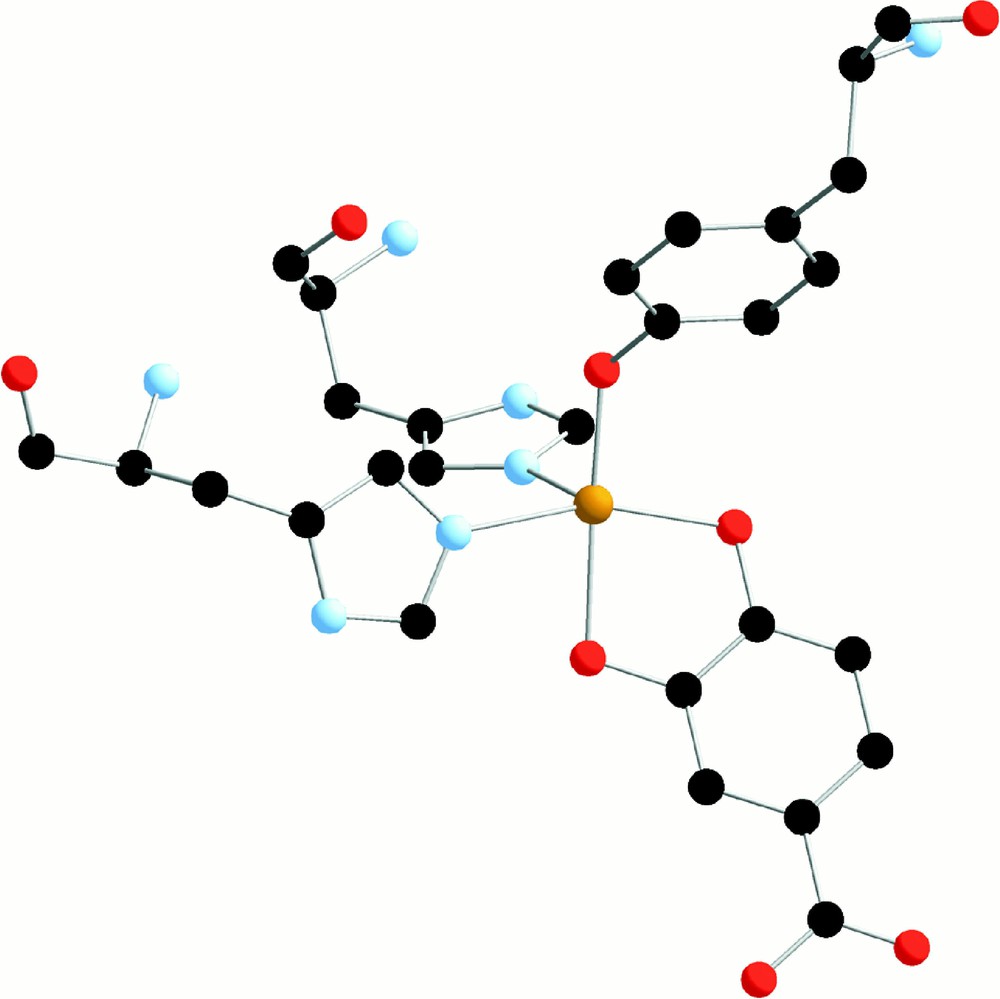
Once the catechols formed by either pathway, their consecutive degradation is easy to understand. We have shown that catecholate can chelate Fe(III) complexes with trispicMeen and similar ligands 〚35〛. For instance, we show in Fig. 10 the structure of the complex 〚(bispicMe2en)FeIII(DBC)〛+, where bispicMe2en is the tetradentate analogue of trispicMeen and DBC stands for the dianion of 3-5-diterbutylcatechol.
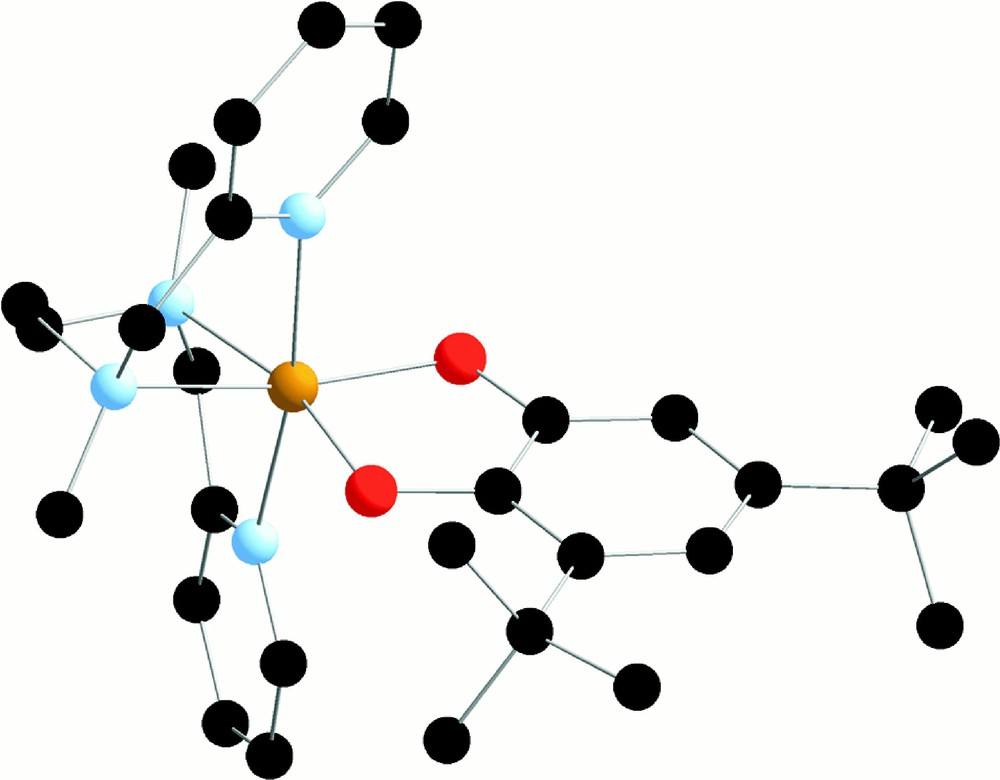
X-ray crystal structure of 〚FeIII(bispicMe2en)(DBC)〛+ in 〚FeIII(bispicMe2en)(DBC)〛〚BPh4〛 (from 〚35〛).
For simplicity, the ligand bispicMe2en will be noted below L42. Dioxygen breaks the intradiol carbon–carbon bond of the catecholate linked to Fe(III) in a manner similar to what is observed with intradiol catechol dioxygenases and schematised in Fig. 11 〚36〛.
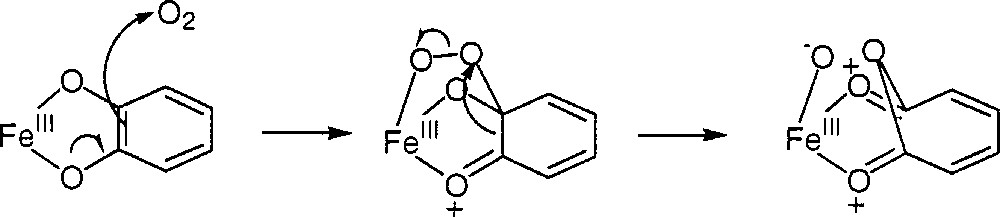
Intradiol carbon–carbon bond breaking similar to the mechanism observed with intradiol catechol dioxygenases (from reference 〚36〛).
In summary, the reactivity of our catalytic system can be understood by the hydroxylation of aromatic hydrocarbons by the Fe(III)OOH intermediate and the subsequent chelation to Fe(III) of the catechols formed, which induces the breakage of the intradiol carbon–carbon bond by dioxygen as in intradiol catechol dioxygenases.
3 Alkane hydroxylation
Methane is predicted to be a very important source of energy in the future. One possible application is its transformation into methanol, which has the advantage to be a liquid and thus to be easier to handle. Methanol could then be used as carburant as such or in fuel cells. Methanol can be prepared from syn gas at high pressure and high temperature but a direct conversion of methane into methanol at ambient pressure and temperature would be extremely valuable.
Such a transformation is accomplished by bacteria that use methane as a source of C and energy. The first transformation operated by these bacteria is the oxidation of methane into methanol according to the equation:
The enzyme responsible for this transformation is Methane Monooxygenase (MMO) and has been wonderfully studied by two American groups, those of Lipscomb 〚12〛 (University of Minnesota) and Lippard 〚37〛 (MIT). The enzyme contains a dinuclear iron site, which in its native form is in the Fe(III)Fe(III) oxidation state. A highly simplified catalytic cycle is shown in Fig. 12
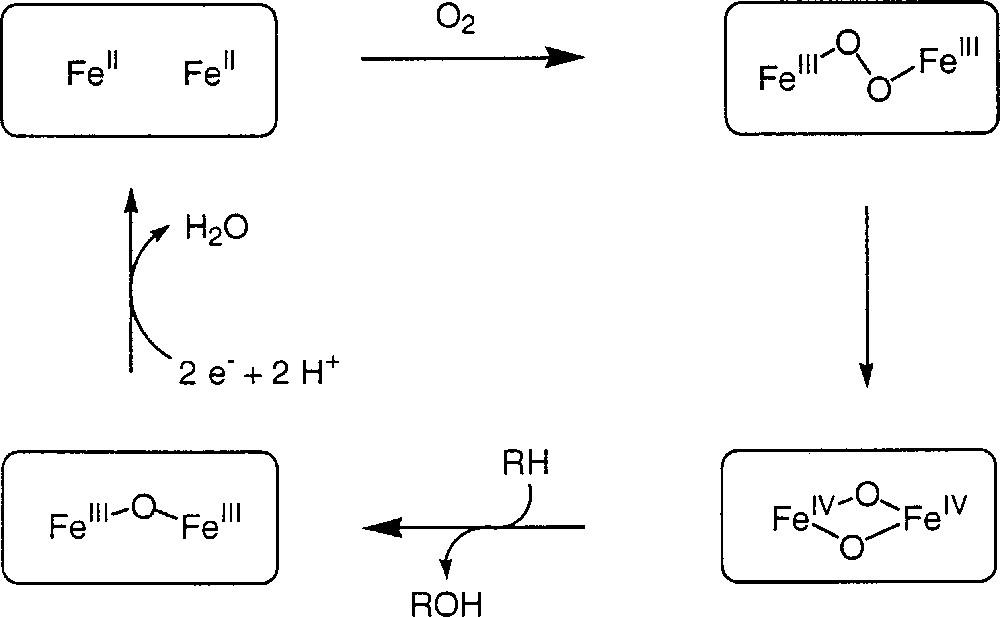
Simplified catalytic cycle of Methane Monooxygenase (MMO).
The active form (called intermediate Q) is a dinuclear Fe(IV)O2Fe(IV) identified by different spectroscopies 〚38〛, which is known as the diamond core. Efforts are being done to chemically model this fantastic enzyme 〚3〛. One of the best achievements is the isolation of Fe(IV)O2Fe(III) entities, which have never been identified before and even thought of 〚39〛.
Consideration of the structure of intermediate Q invites us to use tetradentate ligands. We selected bispicMe2en and bispicMe2pn to study the influence of the length of the linear tetradentate ligand. We note these ligands L42 and L43 respectively for simplicity (Fig. 13).
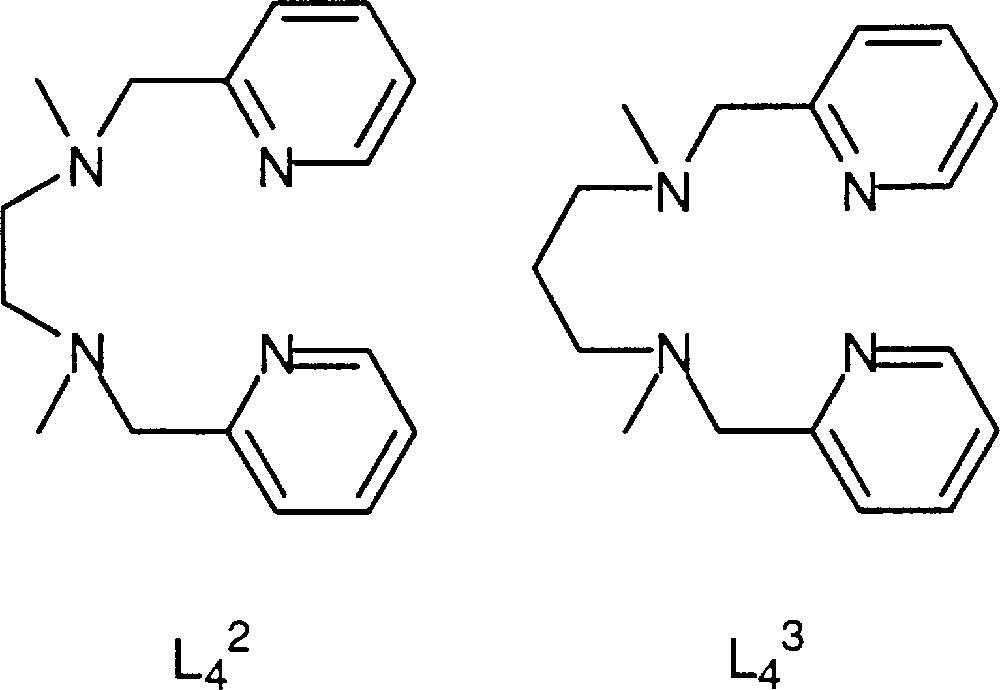
L42 and L43 ligands.
Complexes of Fe with the ligand L42 or derivatives have already been studied by us and others. A dinuclear 〚L42FeII(AcO)2FeIIL42〛2+ complex has been described by Hazell et al. 〚40〛. We have ourselves described L42FeIICl2 and 〚L42FeIIICl2〛+ complexes in which the methyl groups have been replaced by benzyl ones 〚41〛. Arusalmy et al. 〚42〛 have studied the chemistry of L42 with hydrogens in place of methyl groups. Reaction of L42 with Fe(III)(ClO4)3 gives the dinuclear 〚L42(OH)FeIIIOFeIII(OH2)L42〛3+ complex, in which the hydroxyl group and the water molecule are linked by an hydrogen bond 〚43〛. Dissolution of this complex in acetonitrile gives the 〚L42FeIIIO(OH)FeIIIL42〛2+ complex, which presents the diamond core identified in MMO. This reaction can be reversed by addition of water. The 〚L42FeIIIO(AcO)FeIIIL42〛3+ complex has been described by Okuno et al. 〚44〛. The complex 〚L42FeII(MeCN)2〛2+ has been described by Chen and Que 〚45〛.
We found that reaction of one equivalent of L42 with one equivalent of FeIIICl3 gives the symmetric dinuclear complex 〚L42ClFeIIIOFeIIIClL42〛2+, the structure of which is displayed in Fig. 14 〚46〛.
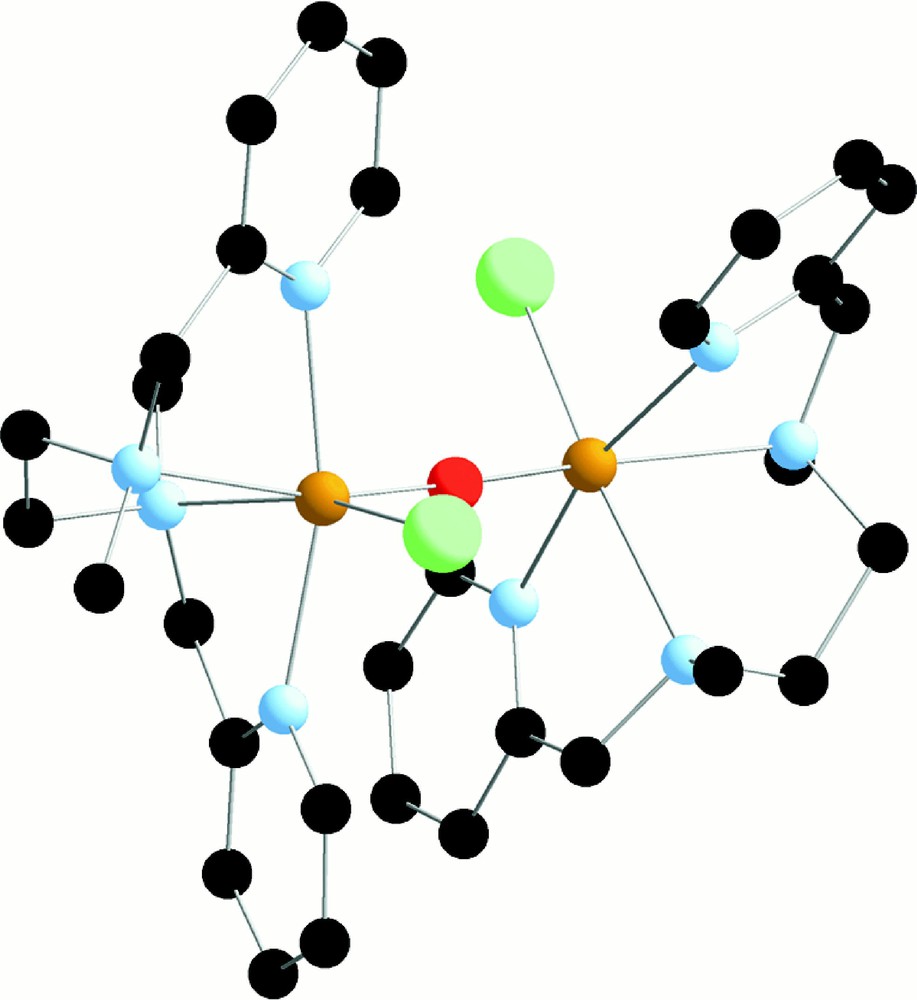
X-ray crystal structure of 〚L42ClFeIIIOFeIIIClL42〛2+ in 〚L42ClFeIIIOFeIIIClL42〛(Cl2).
Several symmetric complexes of this kind have already been obtained 〚18,42,47–49〛.
The ligand L43 has not been very much studied 〚50〛. To our surprise, we found that its reaction with FeIIICl3 gives the asymmetric dinuclear L43ClFeIIIOFeIIICl3 complex (Fig. 15) and not the symmetric dinuclear one. Other asymmetric complexes have been described 〚51,52〛. It has to be noted that in 〚L42ClFeIIIOFeIIIClL42〛2+ the ligand is in a cis α conformation, while in L43ClFeIIIOFeIIICl3 the ligand has a cis β conformation. The aptitude of L43 type ligands to adopt the cis β conformation has already been noted 〚50〛. The formation of the asymmetric dinuclear complex versus the symmetric one may be due to an instability of the complex between L43 and Fe(III) or to a steric hindrance between the L43 ligands in the symmetric complex.
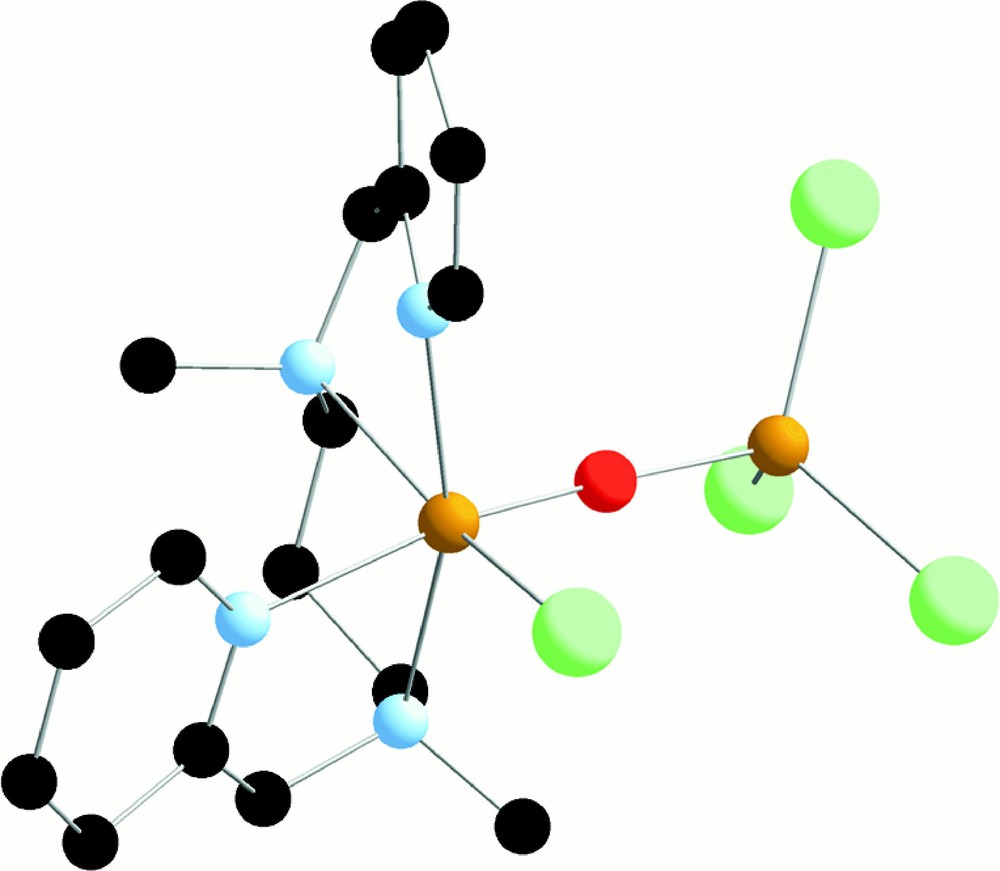
X-ray crystal structure of L43ClFeIIIOFeIIICl3.
The complex 〚L42ClFeIIIOFeIIIClL42〛2+ was found inactive in catalysis of alkane oxidation with dioxygen and reductant. On the opposite, the asymmetric complex L43ClFeIIIOFeIIICl3 gave (in 16 h and using O2 in the presence of trimethylhydroquinone as an electron donor, in MeCN) almost uniquely hexane-derived alcohols with the following turnovers: 1.1 for 1-ol, 3.7 for 2-ol and 5.2 for 3-ol. Those results were improved by fixation of the catalyst on silica and also in presence of acetate (Table 1). They are quite exceptional, although we have no indication on the mechanism of this oxidation.
Oxidation of n-hexane by O2 with L43 catalyst/trimethylhydroquinone/acetic acid. The yields are given as turnover numbers (T.N.) based on the FeIII catalytic system after 16 hours. Reaction conditions: 1 atm O2 at 25 °C in 2.6 ml of dry acetonitrile; final concentration for the catalyst in the reaction mixture 0.33 m mol l–1. catalyst/n-hexane/trimethylhydroquinone/acetic acid 1/3000/350/250. L43 designates the ligand N,N’-dimethyl-N,N’-bis(2-pyridylmethyl)propane-1,3-diamine.
| System | 1-ol | 2-ol | 3-ol | 2-one | 3-one |
| L43ClFeIIIOFeIIICl3 | 1.1 | 3.7 | 5.2 | 0.1 | 0.1 |
| L43 grafted on silica + FeCl3 | 2.0 | 7.1 | 10.0 | 0.4 | 0.6 |
| L43 grafted on silica+〚Fe2OCl6〛2– + AcO– | 4.0 | 13.4 | 18.2 | 1.1 | 1.2 |
The rare examples of non-heme model systems able to catalyse alkane oxidation by O2 and a reducing agent, have been quoted in a recent review by Costas et al. 〚4〛. The only one giving a high alcohol to ketone ratio is the complex (HBpz3)(hfacac)FeIIIOFeIII(hfacac)(HBpz3) studied by Kitajima et al. in 1988 〚53〛.
A highly speculative hypothesis to explain the catalysis we observed would be the reduction of L43ClFeIIIOFeIIICl3 by the trimethylhydroquinone, followed by reoxidation by dioxygen to give a Fe(IV)O intermediate capable to transfer an oxygen atom into a CH bond. If this is the case, the poor yields observed here can be understood by the fact that the Fe(IV)O intermediate is generated in the presence of the reductant. It has to be noted that, in MMO, these steps are carefully separated. In order to explore such an exciting scheme, we have synthesised the Fe(II) complexes with L42 and L43. Indeed it is possible to obtain L43FeIICl2 and L42FeIICl2 as monocrystals (Fig. 16).
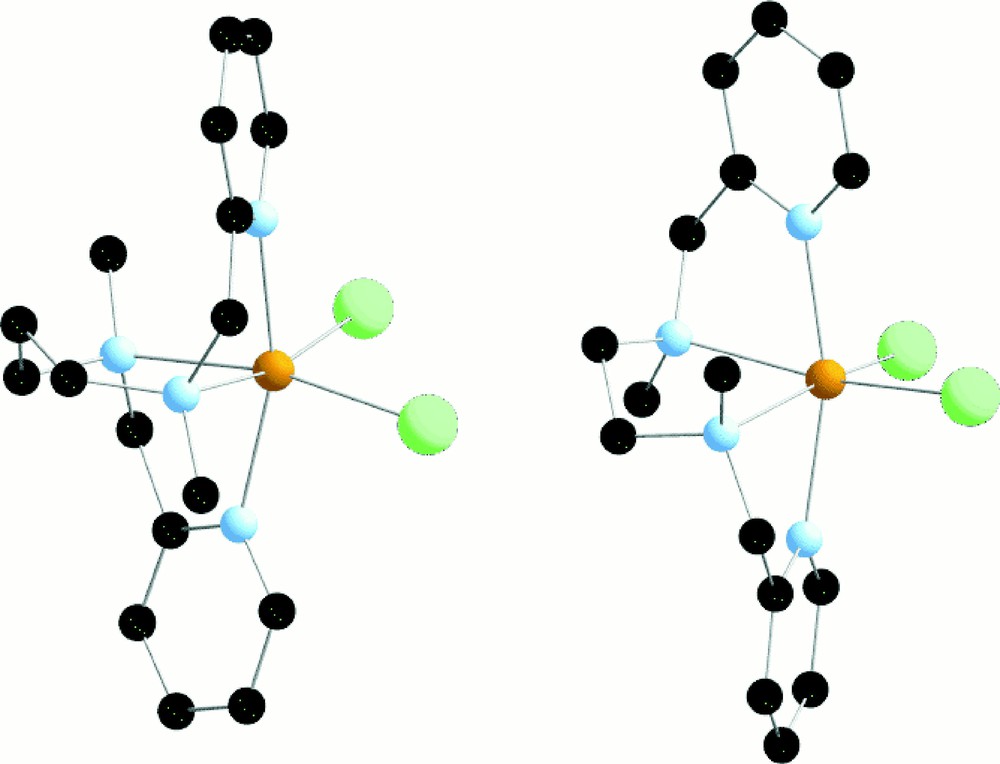
X-ray crystal structure of L42FeIICl2 and L43FeIICl2.
These monomers can be obtained by direct reaction of the ligands with FeCl2 or by electrochemical reduction of the Fe(III) dimers.
We then studied the reactivity of these Fe(II) complexes with dioxygen. Indeed we noted that L42FeIICl2 is stable in MeCN under air, while L43FeIICl2 is oxidised into L43ClFeIIIOFeIIICl3, although the stoichiometry of this oxidation is not yet clear. Why, of the two similar Fe(II) complexes, only L43FeIICl2 reacts with dioxygen? Electrochemistry allowed us to answer that question.
As shown in Fig. 17, the oxidation in MeCN of L43FeIICl2 (0.28 V/SCE) occurs at a higher potential than that of L42FeIICl2 (0.18 V/SCE). Moreover, a striking difference between the two complexes appears on the reverse scan. Upon reduction of the Fe(III) species, two new waves are observed at negative potentials for L43FeIICl2, but not for L42FeIICl2 (Figs. 17 and 18).
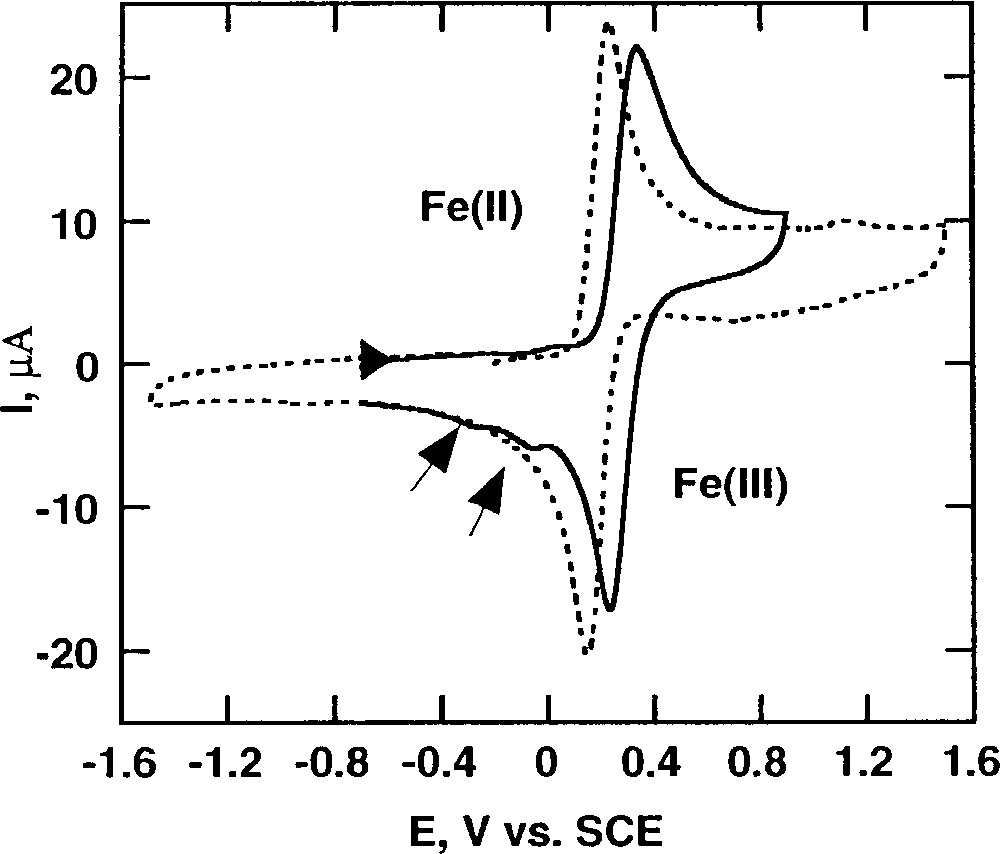
Cyclic voltammetry of 1 mM acetonitrile solutions of L43FeIICl2 (solid line) and L42FeIICl2 (dotted line) in the presence of 0.1 M TEAP at 20 °C. Scan rate: 100 mV s–1.
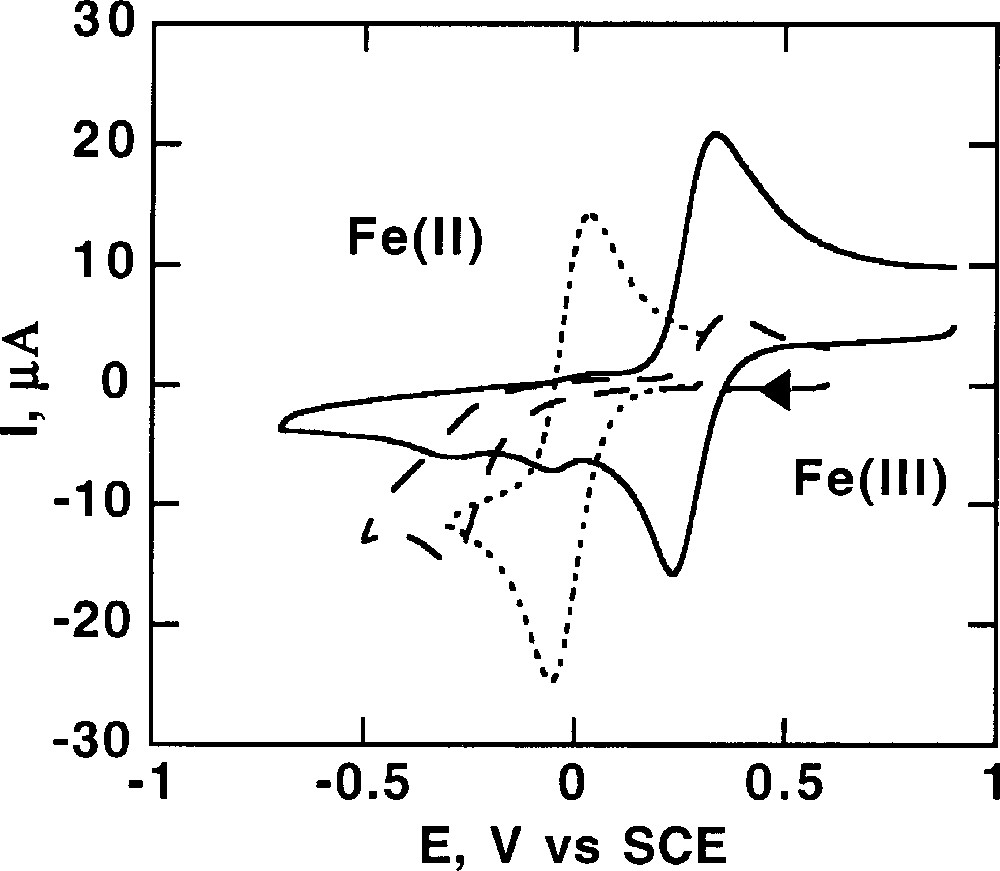
Cyclic voltammetry of 1 mM acetonitrile solutions of L43FeIICl2 (solid line) starting from the Fe(III) form, FeIIICl4– (dotted line) and L43FeIIIOFeIIICl3 (dashed line) in the presence of 0.1 M TEAP at 20 °C. Scan rate: 100 mV s–1.
The first wave is attributed to the reduction of FeCl4– (–0.05 V/SCE) and the more negative one to the reduction of L43ClFeIIIOFeIIICl3 (–0.29 V/SCE) (Fig. 18). The equilibrium displayed in Fig. 19 explains these observations.
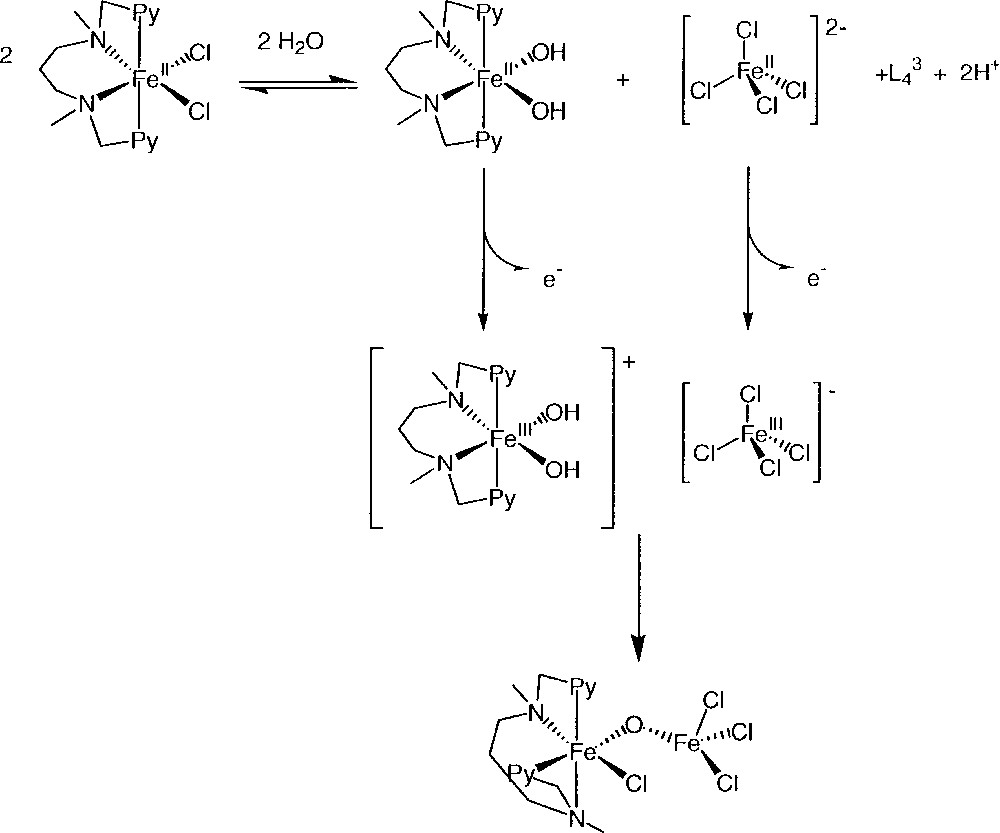
Equilibria involved in acetonitrile for L43FeIICl2.
The complex L43FeIICl2 partly dissociates into L43FeII(OH)2, L43 and FeIICl42–. The electrochemical oxidation of L43FeII(OH)2 and FeIICl42– leads to 〚L43FeIII(OH)2〛+ and FeIIICl4– which form by condensation L43ClFeIIIOFeIIICl3. The reactivity with O2 of L43FeIICl2 is thus related to a slight instability of this complex in MeCN. It is intriguing to note that the ligand L43, which allows oxidation of Fe(II) by O2, is precisely the one that led to the catalytic oxidation of hexane. Further research is being done on these systems, in particular to test the stoichiometric oxidation of alkanes by O2 + L43FeIICl2.
Finally, it may be important to associate both Fe atoms close to each other in an assembling ligand, since in MMO they are maintained in proximity through all the catalytic cycle 〚12〛. Using the hexadentate ligand L634M depicted in Fig. 20, we have obtained the (L634M)FeII2Cl4 dinuclear complex, the structure of which is displayed in Fig. 21.
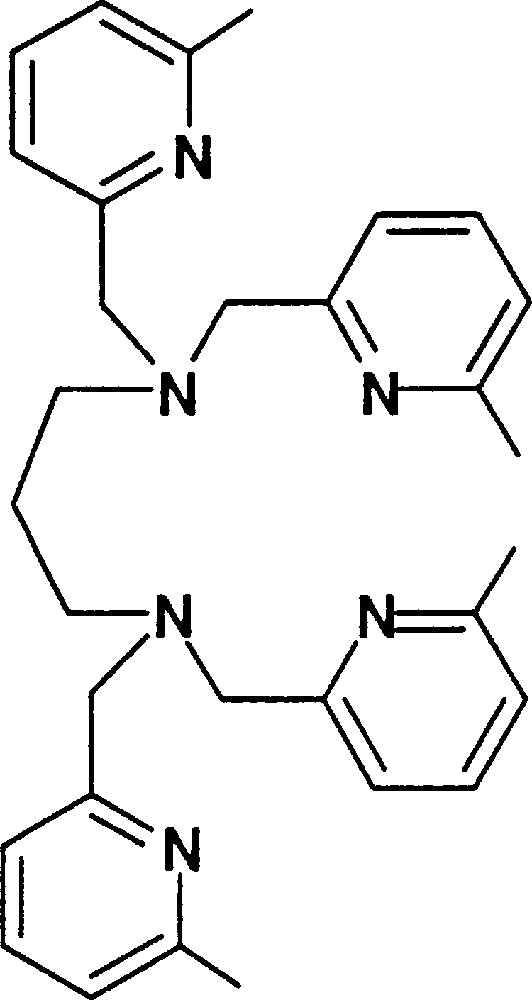
L634M ligand.
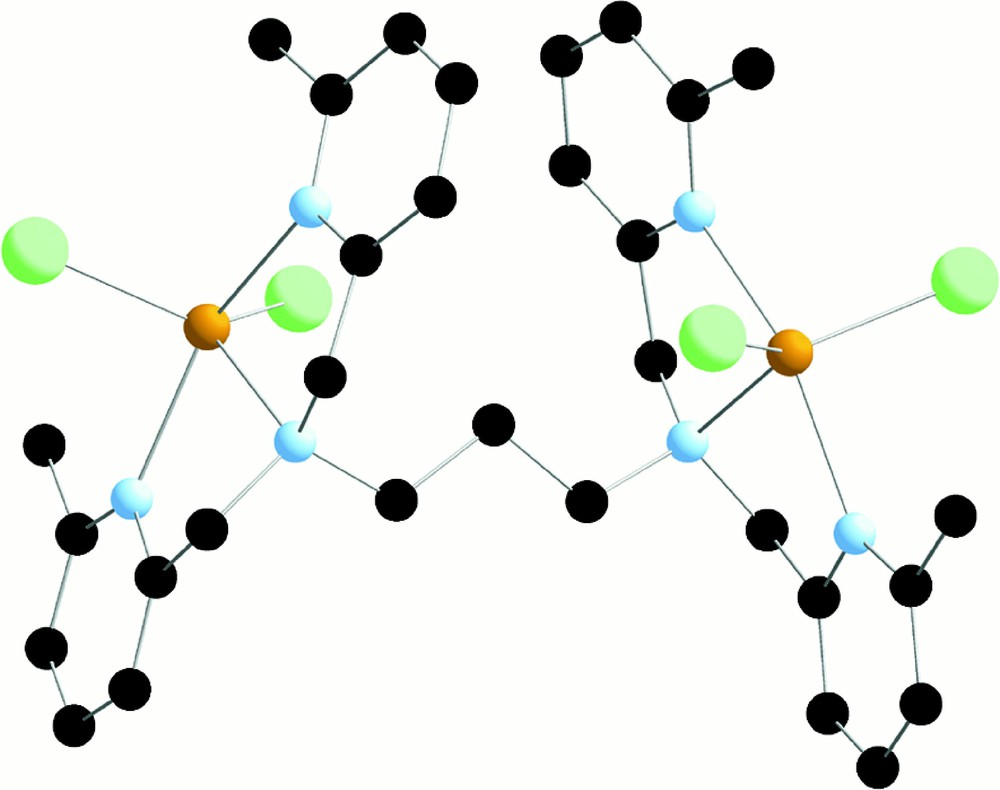
X-ray crystal structure of (L634M)FeII2Cl4.
In this structure, Fe(II) ions are pentacoordinated with two chloride and the tertiary amino group in the equatorial plane and the two lutidine groups in axial positions. The interesting aspect of this complex is that both metal ions are maintained inside a single assembling ligand.
In acetonitrile in the presence of air, this complex is oxidised slowly, to give crystals of a tetranuclear (L634M)FeIII4O3Cl6, which contains four Fe(III) in a row bridged by oxo groups (Fig. 22).
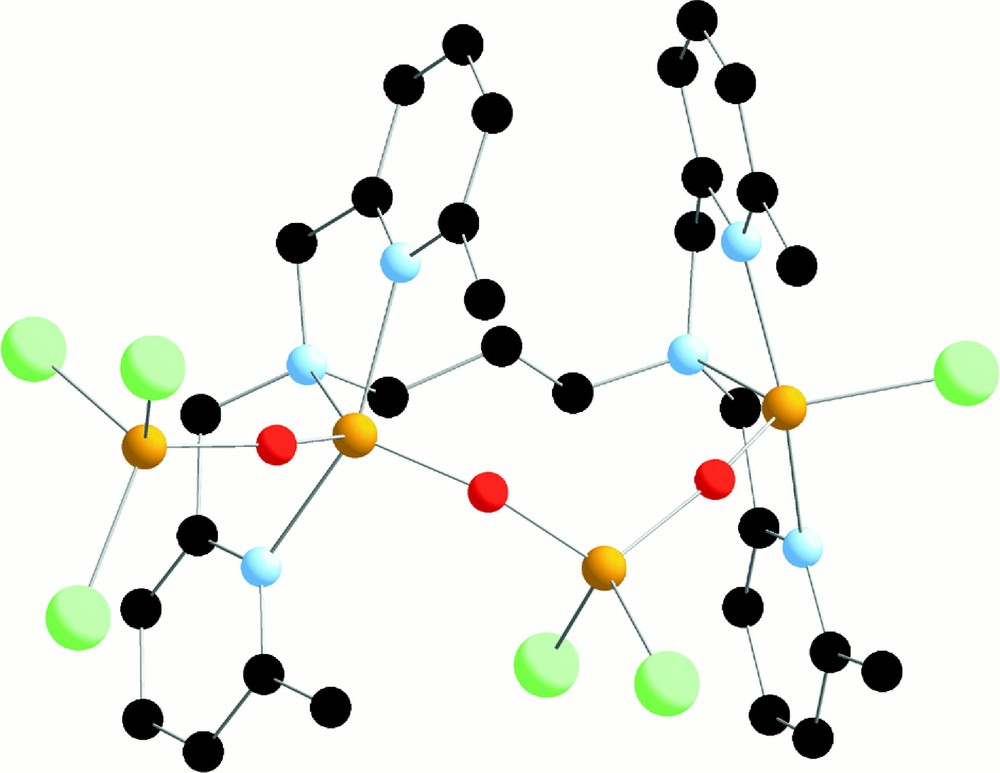
X-ray crystal structure of (L634M)FeIII4O3Cl6.
The reactivity with O2 of the pentacoordinated Fe(II) sites of (L634M)FeII2Cl4 is very interesting and the mechanism of the formation the tetranuclear species is under study. Certainly, assembling ligands could lead to better catalysts by maintaining cooperativity between the Fe ions at every step.
4 Conclusion
We have shown that Fe(III)OOH and Fe(III)OO species are quite accessible by using pentadentate ligands that protect Fe and nevertheless let one position free for peroxo ligation. These species are sufficiently oxidising to degrade aromatic pollutants and could be grafted on silica to get interesting catalysts. In the future, it could be possible to modify those molecules in order to achieve specific cis-dihydroxylation of aromatics. Such results have been already presented by Chen et Que using a bulky tetradentate ligand 〚54〛. Evidently mutants of bacteria (with the dehydrogenase blocked) can be efficiently used to prepare cis-diols 〚55〛. But bio-inspired catalysts could be simpler to use and less expensive.
From an academic point of view, it is also interesting to accumulate knowledge on those iron–peroxo species, which till recently were regarded as rare and to understand their cleavage mode and their reactivity.
Oxidation of methane into methanol is a real challenge for chemists. Biotechnology solutions could be found, but there is certainly place for research of bio-inspired catalysts. Recent advances are encouraging. The mechanism of MMO is not far from being understood thanks to the contributions of the groups of Lispcomb, Que and Lippard. One difficulty with models is to separate the reduction step of the Fe(III)Fe(III) state to the Fe(II)Fe(II) one from the step implying oxidation of Fe(II)Fe(II) by dioxygen. Another difficulty will be the source of the electrons, if one wants to use industrially a mono-oxygenase strategy. We arrive then at the question of the quest of electrons, which inevitably, if we look at Nature, seems to orient research toward photovoltaics and artificial photosynthesis. This opens another exciting chapter of bio-inspired chemistry.
Supplementary Material
Crystallographic data for the structures shown in Figs. 14, 15 and 16 have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication No. CCDC 171970 (L42FeIICl2), No. CCDC 171971 (L43FeIICl2), No. CCDC 171972 (L42ClFeIIIOFeIIIClL42)Cl2, No. CCDC 171973 (L42ClFeIIIOFeIIICl3).
Acknowledgements
We thank Elf and Unilever Companies and New Energy Development Organization and Osaka Gas Company, both in Japan, for financial support. Resonance Raman measurements were made in collaboration with Pr. P. Hildebrandt from Max-Planck Institut in Mülheim, Germany.