

1 Introduction
Dimetallated polyynes, or polyynediyl complexes, constitute a fascinating class of molecules for which only limited examples with bridges containing more than four carbon atoms have been reported so far [1–18]. In particular, representatives that contain two redox-active metal centers provide, when sufficiently stable in several redox states [5,7,8,11,13,14,16,19,20], valuable test compounds for understanding how carbon-rich bridges can mediate the electronic interactions between the two inorganic endgroups as a function of redox state [18,21]. Such insights are crucial in the field of molecular electronics in order to be able to develop efficient nanoscopic molecular-based devices made from such organometallic building blocks for processing or storing information at the molecular level [22].
In this connection, symmetrically-substituted homodinuclear rhenium and iron complexes like (η5-C5Me5)Re(NO)(PPh3)(CC)n(PPh3)(NO)Re(η5-C5Me5) (1n) or (η5-C5Me5)Fe(η2-dppe)(CC)n(η2-dppe)Fe(η5-C5Me5) (2n) were independently demonstrated to present remarkable electronic delocalization properties in their mono-oxidized, namely the mixed-valent (MV) state (n ≥ 2) [16,23–25]. Cyclic voltammetry data along with spectroscopic studies in the various redox states showed that the electronic communication in 1n or 2n decreased with increasing chain length until it was no longer operative. Note that “electronic communication” is taken here in the broad sense, i.e., including not only the influence electronic delocalization, but also all other contributions like for instance those originating from magnetic exchange interactions and more generally the so-called synergistic effects [26,27]. We could also show in subsequent studies that the heterodinuclear butadiynediyl analogue [(η5-C5Me5)Re(NO)(PPh3)-(CC)2(η2-dppe)Fe(η5-C5Me5)][PF6] (32[PF6]) featuring each of these endgroups presents a dominant localization of the electronic vacancy on the iron terminus, but that the electronic delocalization is not fully interrupted in these non-symmetric compounds [28,29]. Given that studies of electronic communication in unsymmetrical polyyne-bridged complexes have been quite rare to date [18,21,30], we were curious to study higher homologues of 32 in order to better understand the influence of longer bridges on the electronic structure of this peculiar class of dimetalla-polyynes.
Moreover, in view of the decreasing kinetic stability for such complexes upon chain extension, especially in oxidized states, we wondered whether complexation to bulky dicobalt hexacarbonyl units might afford less labile systems. The dicobalt hexacarbonyl fragment is indeed well known to bind to alkyne ligands, introducing a sterically crowded “Co2(CO)6” fragment coordinated in a π-fashion [9,31–37]. Accordingly, we therefore report in this communication (i) the synthesis and characterization of two higher homologues of 32 with hexatriynediyl (33) and octatetraynediyl bridges (34), (ii) cyclic voltammetry and preparative experiments directed at defining the redox chemistry of 33 and 34, including the isolation of the cation radical of the former, and (iii) the synthesis and characterization of a [Co2(CO)6] adduct for the former (4).
2 Synthesis and characterization of (η5-C5Me5)Re(NO)(PPh3)(CC)3(η2-dppe)Fe(η5-C5Me5) (33) and (η5-C5Me5)Re(NO)(PPh3)(CC)4(η2-dppe)Fe(η5-C5Me5) (34)
In previous work, the butadiynediyl complex 32 was prepared by the condensation of the butadiynyl complex (η5-C5Me5)Re(NO)(PPh3)(CC)2H (62) and (η5-C5Me5)Fe(η2-dppe)(Cl) (5) in the presence of alcoholic KPF6 at room temperature, and related routes. This transformation required extended reaction times (>48 h), and is believed to involve a vinylidene-like intermediate analogous to 32-v[PF6] as shown in Fig. 1. Although this putative species has not yet been isolated nor characterized, its transient formation is, however, highly probable in light of the known reactivity of related terminal polyynyl complexes with 5 [9,18]. Tautomers derived from 1,3 shifts would also logically be possible.
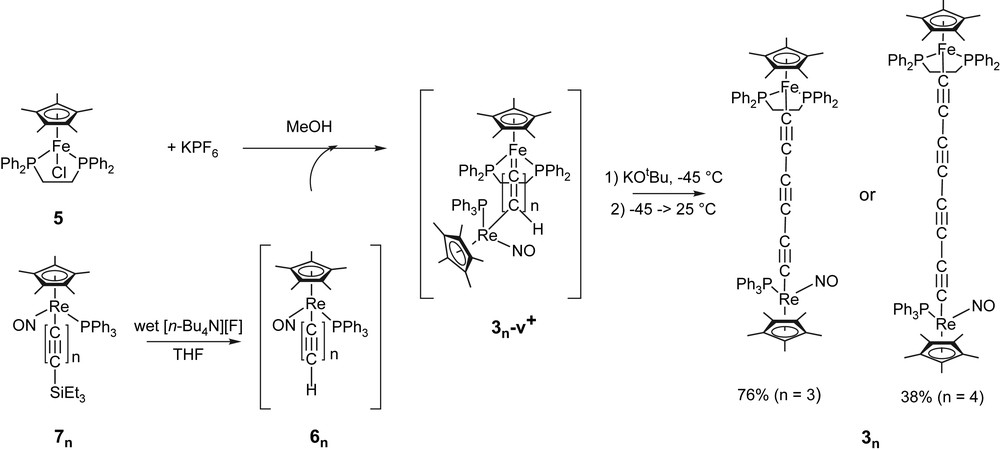
Syntheses of 3n (n = 3, 4). Species that were generated in situ are represented in brackets.
The syntheses of the title compounds 33 and 34 were approached similarly, but with some strategic modifications. First, since longer chain complexes are often more labile, the reactions were conducted at −40 °C, and in the presence of the base KOtBu to accelerate the deprotonation of 3n-v[PF6]. Second, the polyynyl complexes 6n were generated in situ from the silylated precursor (η5-C5Me5)Re(NO)(PPh3)(CC)nSiEt3 (7n) and wet [Bu4N][F]/THF [16]. While butadiynyl complexes are generally easy to isolate, it is noteworthy that higher homologues become increasingly more labile.
As shown in Fig. 1, the hexatriynediyl complex 33 could be isolated in good yield via this protocol (76%). As a check on the presumed pathway, we confirmed that 33 could also be obtained from 5 and isolated 63 in Et3N/THF (10:1 v/v) at room temperature, following a related workup. This afforded 33 in 37% yield, but due to the additional step required, the workup of this reaction was never optimized. The higher homologue 34 was then isolated using the first synthetic procedure from (η5-C5Me5)Fe(η2-dppe)(Cl) (5) and (η5-C5Me5)Re(NO)(PPh3)(CC)SiEt3 (74) in 38% yield. Both compounds are moderately air-sensitive orange (33) and dark-orange (34) solids.
Both 33 and 34 were characterized by FAB-MS and exhibit strong molecular ions. Conjugated triynes and tetraynes of Cs symmetry are expected to have three and four infrared-active νCC stretches, respectively. Accordingly, 33 and 34 present several absorptions of unequal intensities in the 1940–2100 cm−1 region. Additionally, the strong νNO stretch expected near 1650 cm−1 is apparent for each complex (Table 1). Upon lengthening of the carbon chain, this stretch shifts to higher wavenumbers, in line with decreased back donation into the nitrosyl π∗ orbitals, or a less electron-rich rhenium fragment. This might have been expected for two reasons. First, upon extension of the sp carbon chain, the increasingly remote iron center becomes a less effective donor. Second, longer polyynes are also more electron withdrawing, as evidenced by the progressively decreasing pKa values of H(CC)nR species [38]. NMR spectroscopy (1H, 31P and 13C) also supports the assigned structures. Interestingly, the 31P NMR spectrum of 33 exhibits an AB pattern (3JPP = ca. 10 Hz), consistent with diastereotopic dppe phosphorus atoms. Separate signals were not detected under normal conditions for the shorter 32, presumably due to much broader peaks attributable to the presence of paramagnetic impurities. However, distinct peaks could be observed when a slight excess of reducing agent was introduced in the NMR tube [29]. This splitting is no longer detected for 34, presumably because of the longer carbon bridge which renders the Fe(II) endgroup less sensitive to the asymmetry on the Re(I) terminus.
Selected infrared data for 3n (n = 2, 3, 4), 33[PF6] and 4 (cm−1, CH2Cl2)
| Complexa | νCC | νNO |
| [Re]C4[Fe] (32) | 2095 sh, 2058 m, 1956 m | 1620 vs |
| [Re]C6[Fe] (33) | 2092 m, 2050 s, 1942 m | 1635 vs |
| [Re]C8[Fe] (34) | 2100 vs, 2045 w, 1971 w(sh), 1944 s | 1655 vs |
| [Re]C6[Fe]/[Co2] (4) | 2069 s, 2031 vs, 2002 vsb | 1636 s |
| ([Re]C6[Fe])[PF6] (33[PF6]) | 2003 s, 1892 vs, 1801 s | 1654 s |
a [Re] = (η5-C5Me5)Re(NO)(PPh3) and [Fe] = (η2-dppe)Fe(η5-C5Me5).
b Some of these represent overlapping νCO bands.
UV–vis spectra of 33 were recorded in different solvents (CH2Cl2, THF, C6H6) and presented no significant solvatochromy. As expected, the absorptions of 33 are slightly shifted to longer wavelengths in comparison to that of 32 and become more intense [16].
3 Complexation of 33 by Co2(CO)8: synthesis of (η5-C5Me5)Re(NO)(PPh3)(CC){(CC)[Co2(CO)6]}(CC)(η2-dppe)Fe(η5-C5Me5) (4)
Coordination of a dicobalt hexacarbonyl unit to one of the bridging alkyne groups appeared as a simple way to us to bring some steric protection to the carbon-rich bridge. While 33 might constitute a sterically more congested substrate than 34, it features an odd number of triple bonds. If one assumes that coordination of the MCC linkage is disfavored, then there is only one possible adduct involving the central CC moiety. In contrast, 34 could afford two adducts. Thus, the reaction of 33 and the cobalt hexacarbonyl precursor Co2(CO)8 was investigated. Both species were combined in CH2Cl2 at room temperature in 1:1 molar ratio as shown in Fig. 2. Infrared monitoring showed completion of the reaction after 1 h. Workup gave the target complex (η5-C5Me5)Re(NO)(PPh3)(CC){(CC)[Co2(CO)6]}(CC)(η2-dppe)Fe(η5-C5Me5) (4) as an air-sensitive green solid in 82% yield.
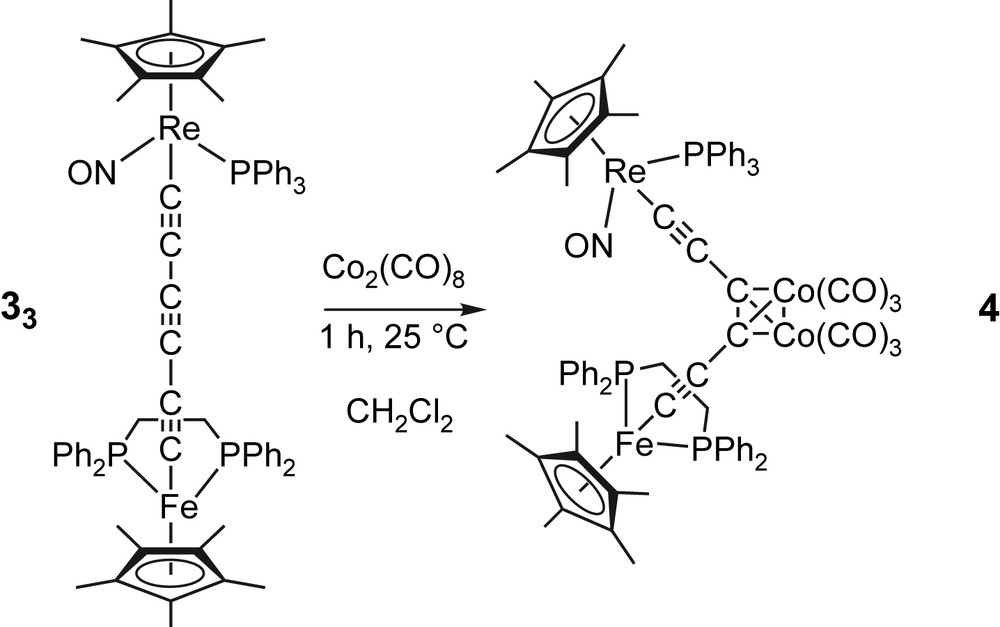
Synthesis of 4.
The 31P NMR spectrum of 4 showed formation of exclusively one compound, while the IR spectra showed several new peaks in the νCO region, unambiguously revealing the presence of the cobalt carbonyl fragment. Unfortunately, the precise assignment of the νCO bands is difficult due to νCC bands in the same spectral region. The UV–vis spectrum of 4 exhibits a new broad and low-energy absorption near 688 nm responsible for the green color of the compound. The structural assignment is also strongly supported by FAB-MS, which reveals a molecular ion 4+ at 1561 amu as well as the presence of ions resulting from the successive loss of six carbonyl groups and two cobalt atoms. Note also that a related adduct obtained from Co2(CO)8 and (η5-C5Me5)Ru(PPh3)2(CC)3(PPh3)2Ru(η5-C5Me5) was reported by Bruce and co-workers in 2000. In this complex, the coordination of dicobalt hexacarbonyl to the central CC moiety was unequivocally demonstrated by X-ray crystallography [35].
4 Cyclic voltammetry of 33, 34 and 4
Cyclic voltammograms of the preceeding complexes were recorded from −1.1 to +1.4 V vs SCE in CH2Cl2 to see how the structural modifications have affected the redox properties and the stability of the oxidized states at the electrode. All exhibited two quasi-reversible oxidations corresponding to the formation of the monocation and the dication. Concerning the all-carbon bridged species, the redox potentials of these oxidations are separated by 0.56 V (Fig. 3 and Table 2) and 0.50 V in 33 and 34, respectively. Both ΔE0 values are smaller than that previously found for 32 (0.73 V). In line with the previous study of the latter complex [29], the values of the redox potentials suggest that the first oxidation occurs on the Fe(II) center. Upon lengthening the all-carbon spacer, both redox potentials are rendered more positive, the shift being roughly twice as large for the redox potentials corresponding to the first oxidation. This anodic shift can be related to the increase of the bridging carbon character of the HOMO in 34, resulting from the well known increasing electronegativity of the sp carbon chains upon lengthening [10]. Alternatively, the anodic shift can also be explained by the fact that the iron-centered oxidation becomes less and less influenced by the more and more “remote” electron-rich rhenium center due to a less effective transmission of inductive/mesomeric effects by the carbon-rich bridge upon chain extension. Similar trends were already observed in the corresponding homodinuclear series [16,18]. Notably, in the 3n series, the first or iron-centered oxidation is each time more positive than the first oxidation in the homodinuclear iron analogue 2n (i.e., −0.63, −0.42, −0.23 V for n = 1, 2, 3, respectively) [10,18], while the second or rhenium-centered oxidation is each time more negative than the second oxidation in the homodinuclear rhenium analogue 1n (i.e., 0.54, 0.48, 0.52 V for n = 1, 2, 3, respectively) [16]. This indicates that oxidation of the iron endgroup is more difficult than in the corresponding homodinuclear compounds, while oxidation of the rhenium terminus is always easier than in the corresponding homodinuclear compounds. Proper understanding of the electronic phenomena affecting the energy gap between the two oxidations would require the examination of the corresponding mononuclear model complexes (η5-C5Me5)Fe(η2-dppe)(CC)nR and (η5-C5Me5)Re(NO)(PPh3)(CC)nR for n = 2, 3 and 4, R being an alkyl group or a proton [29]. However, the higher homologues (n = 2, 3) are currently not available in the case of iron.
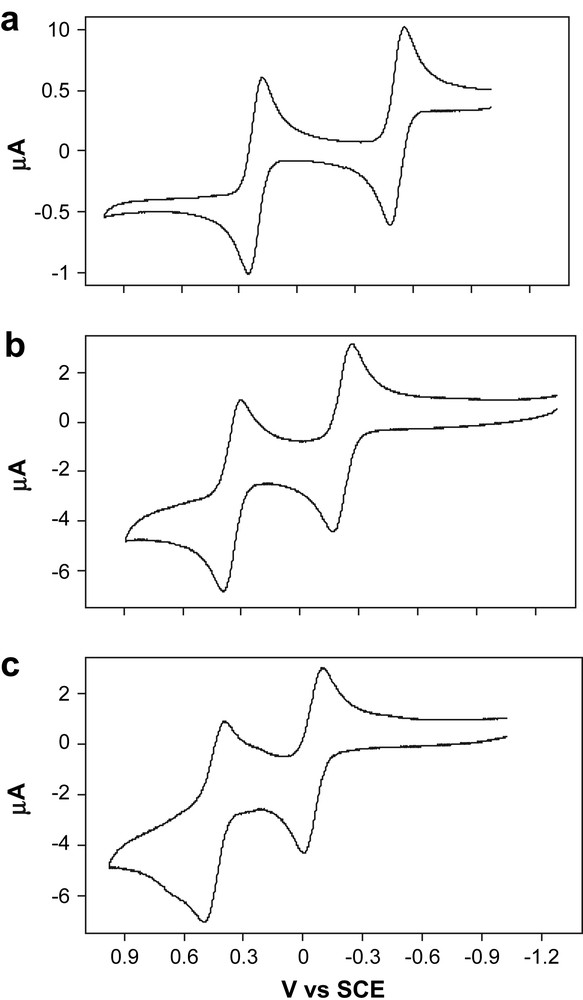
Cyclic voltammograms for 3n (a, n = 2; b, n = 3; c, n = 4).
Cyclic voltammetry data for 3n (n = 2, 3, 4) and 4a,b
| Compoundc | ΔEp (mV) | E0 (V) | ΔE0 (V) | Kcond |
| [Re]C4[Fe] (32) | 70 | −0.50 | 0.73 | 2.36 × 1012 |
| 70 | +0.23 | |||
| [Re]C6[Fe] (33) | 90 | −0.24 | 0.56 | 3.10 × 109 |
| 90 | +0.32 | |||
| [Re]C8[Fe] (34) | 90 | −0.05e | 0.50 | 2.98 × 108 |
| 100 | +0.45e | |||
| [Re]C6[Fe]/[Co2] (4) | 70 | −0.18 | 0.61 | 2.18 × 1010 |
| 70 | +0.43 |
a Conditions: 0.1 M [Bu4N][PF6]; CH2Cl2; 100 mV/s, E0 vs SCE and ferrocene used as internal standard (E0 for Fc/Fc+ taken at 0.46 V [43]).
b ia/ic ≈ 1 for all peaks, except for the second oxidation of 34 at +0.45 V, for which ia/ic ≈ 0.8.
c [Re] = (η5-C5Me5)Re(NO)(PPh3) and [Fe] = (η2-dppe)Fe(η5-C5Me5).
d Comproportionation constants corresponding to Eq. 1 at 298 K.
e [Bu4N][BF4] used instead of [Bu4N][PF6].
Cyclic voltammetry of the cobalt hexacarbonyl adduct 4 also showed two reversible oxidations at −0.18 and 0.43 V (ΔE0 = 0.61 V), which can be assigned to the iron- and rhenium-centered oxidation by analogy with the previous data. As might have been anticipated, both oxidations are more difficult than in the parent complex 33 by reason of the coordination of the electron-withdrawing [Co2(CO)6] to the bridge. The [Co2(CO)6(CC)] fragment is not electroactive in the potential range investigated. This was expected, since the reduction of this unit was very unlikely below 0.9 V vs SCE by reason of the strongly electron-releasing nature of the organometallic endgroups of the heterodimetalla-triyne 33 [39,40]. Interestingly, the anodic shift of the second oxidation is slightly larger than that of the first one, resulting in an increased peak separation (0.61 V) for 4 in comparison to 33 (0.56 V). Here also, an in-depth understanding of the way the electronic communication is modified awaits further studies.
| [3n]++ + [3n] ⇄ [3n]+ + [3n]+ | (1) |
| (RT/F)log(Kcon) = ΔE0 | (2) |
The comproportionation constants corresponding to the equilibrium given in Eq. 1 are also computed in Table 2 according to Eq. 2. These data clearly show that the thermodynamic stabilities of the MV complexes diminish upon chain extension in the series 3n, and comparison between 33 and 4 suggests that complexation of the [Co2(CO)6] fragment somewhat enhances the thermodynamic stability of the MV complex. In any case, the Kcon values remain large enough to allow for selective isolation of the MV parents by chemical oxidation of the neutral species 3n in solution, should these MV complexes prove to be sufficiently stable in the kinetic sense. Note that the values obtained for a given heterodinuclear MV complex are usually larger than the values of the corresponding homodinuclear MV complexes [16,18]. In this context, progressing from the homodinuclear series to the conjugal [29] series provides a simple means to increase the stability of the MV complexes.
5 Radical cations of 33 and 4: isolation of [(η5-C5Me5)Re(NO)(PPh3)(CCCCCC)(η2-dppe)Fe(η5-C5Me5)][PF6] (33[PF6])
The above cyclic voltammetry data indicate that the monoxidized species 33+ should be accessible upon chemical oxidation of 33 with the ferricinium ion. Hence, 33 was combined with ∼0.9 equiv of ferricinium hexafluorophosphate in THF at low temperature (Fig. 4). Workup at low temperature (−80 °C) gave the expected radical cation 33[PF6] as an analytically pure green solid in 91% yield. When this reaction was performed in dichloromethane rather than in THF, a complex mixture of unidentified products was obtained.
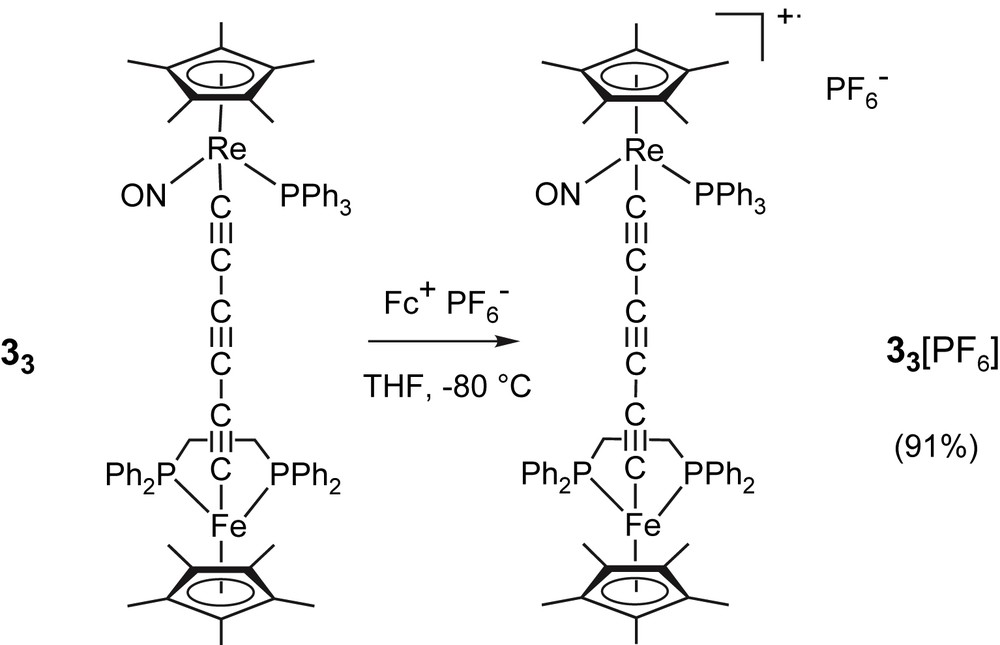
Synthesis of 33[PF6].
The infrared spectrum of 33[PF6] showed three νCC stretches at much lower frequencies than for the parent neutral complex 33 (Table 1), in line with a reduced bond order in the carbon bridge, a feature usually observed upon oxidation of dimetalla-polyyne complexes. The νNO stretch is also shifted from the neutral compound to higher frequency by 17–22 cm−1, indicating a reduced back-bonding into the nitrosyl ligand-centered π∗ orbital. This shift is, however, smaller than that observed upon oxidation of 32 (Δν = 40 cm−1 in CH2Cl2) [29]. NMR confirmed the isolation of a paramagnetic species, but proved of little help in terms of characterization, except for a characteristic 31P NMR signal for the PF6− anion. However, ESR spectra in CH2Cl2/ClCH2CH2Cl mixtures (1:1 v/v) clearly confirmed the metal-centered nature of the cation radical product (Fig. 5) [41]. Thus, the low temperature ESR spectrum of 33[PF6] is quite similar to that previously observed for 32[PF6], except that the hyperfine coupling to rhenium is no longer so well resolved, rendering proper simulation problematic [29]. The presence of hyperfine coupling to rhenium is indicated by the asymmetry of the signal, and a rough estimate of the (Aiso)Re value can be obtained from the spectrum recorded at room temperature. An upper bound of 60 G can be set, slightly less than in 32[PF6] (65 G). Extrapolated values of g-tensors (g1 = 1.919, g2 = 2.010, g3 = 2.225, and Δg = g3 − g1 = 0.306) were very close to those previously found for 32[PF6] (g1 = 1.920, g2 = 2.008, g3 = 2.236, and Δg = g3 − g1 = 0.316), which are typical for an iron-centered radical. In spite of their closeness, these values nevertheless suggest a slight diminution of spin density on rhenium upon bridge extension, in line with the mean g value, which is closer to the free electron value for 33[PF6] (2.051) than for 32[PF6] (2.054).
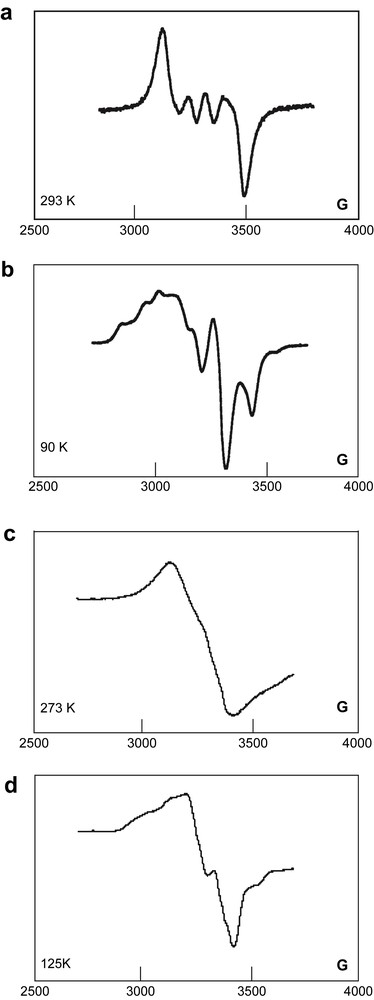
Room temperature (solution) and low temperature (frozen glass) ESR spectra of 32 (a, b) and 33 (c, d) in CH2Cl2/ClCH2CH2Cl mixtures (1:1 v/v).
The chemical oxidation of 4 was then attempted following a strictly similar experimental protocol at low temperature. In that case, a mixture of deeply colored (blue-green) unidentified species was obtained, as revealed by infrared spectroscopy and cyclic voltammetry.
6 Conclusion
We have briefly reported in this communication on the synthesis and characterization of two higher homologues (33 and 34) of the known heterodinuclear complex (η5-C5Me5)Re(NO)(PPh3)(CC)2(η2-dppe)Fe(η5-C5Me5) (32), as well as a hexacarbonyl dicobalt adduct (4) of the hexatriynediyl complex. These dimetalla-polyyne complexes can be isolated in a rather straightforward way in their neutral Fe(II)/Re(I) state using a synthetic route inspired by that previously used for 32, while 4 is isolated by reaction of 33 with Co2(CO)8. All of these compounds exhibit the expected spectroscopic features in relation with their proposed structure. Cyclic voltammetry shows that the electronic communication between the organometallic endgroups as well as the thermodynamic stability of the mixed-valent complex along the series 32/33/34 is strongly influenced by bridge extension or by complexation of the [Co2(CO)6] fragment. A better understanding of the precise way these properties are altered by these structural modifications awaits gathering of additional spectroscopic data from the oxidized species. In that connection, preliminary experiments have revealed that the MV complex 33[PF6] was isolable after performing a chemical oxidation using ferricinium at low temperature. However, contrary to our initial expectations, chemical oxidation of 4 using the same experimental protocol does not lead to a stable species, in spite of the steric protection provided by the bulky [Co2(CO)6] fragment. Work is underway to answer these fundamental questions.
7 Experimental
7.1 General experimental
All manipulations were carried out under argon atmospheres using standard Schlenk techniques. Transmittance FT-IR spectra were recorded using a Bruker IFS28 spectrometer. UV–vis–near-IR spectra were recorded on an UVIKON XL spectrometer. All NMR experiments were conducted on Bruker or Varian spectrometers operating at 200 or 300 MHz for 1H. Cyclic voltammograms were recorded using an EG&G potentiostat (M.263) on platinum electrodes as detailed in Table 2. MS analyses were performed at the “Centre Regional de mesures physiques de l'Ouest” (CRMPO, University of Rennes, France) or at the University of Utah, USA, on a high-resolution MS/MS ZABSpec TOF Micromass Spectrometer. Elemental analyses were conducted at the Centre for Microanalyses of the CNRS at Lyon-Solaise, France. The complexes (η5-C5Me5)Fe(η2-dppe)(Cl) (5) [42] and (η5-C5Me5)Re(NO)(PPh3)(CC)nSiEt3 (6n) [16] were obtained as previously reported.
7.2 Synthesis of (η5-C5Me5)Re(NO)(PPh3)(CCCCCC)(η2-dppe)Fe(η5-C5Me5) (33)
Method A: A Schlenk flask was charged with MeOH (30 mL), (η5-C5Me5)Fe(η2-dppe)(Cl) (5) (0.130 g, 0.208 mmol), and KPF6 (0.044 g, 0.239 mmol). The complex (η5-C5Me5)Re(NO)(PPh3)(CCCCCCH) (63) was generated separately in situ from (η5-C5Me5)Re(NO)(PPh3)(CCCCCCSiEt3) (73; 0.152 g, 0.190 mmol) and [Bu4N][F] (1.0 M in THF/5 wt% H2O; 0.038 mL, 0.038 mmol, 20 mol%) in THF (5 mL) [16]. The solution of 63 was transferred via cannula to the former solution with stirring and the resulting homogeneous dark solution was cooled to −45 °C. Then KOtBu (0.023 g, 0.205 mmol) was added. The flask was removed from the cold bath, wrapped with aluminum foil, and stirred for 12 h. An orange suspension separated from the dark green medium. The flask was cooled to −45 °C and the solvent was removed from the resulting powder using a cannula that contained a filter. The residue was washed with MeOH (3 × 5 mL) and pentane (2 × 5 mL), and dried under oil pump vacuum to give 33 as a moderately air-sensitive orange powder (0.185 g, 0.145 mmol, 76%). FAB+-MS (m/z in amu, 3-NBA/CH2Cl2): 1275 (33+, 100%), 877 (33+−C5Me5−PPh3, 10%), 614 ([(C5Me5)Re(NO)(PPh3)]+, 25%), 589 ([(C5Me5)Fe(dppe)]+, 65%). FT-IR (ν in cm−1, nujol/KBr) νCC = 2098 m, 2052 s, 1942 m, νNO = 1626 vs. 31P{1H} NMR (δP in ppm, C6D6, 121 MHz): 100.2, 100.1 (2d, JPP = 9.2, 10.6 Hz, 2P, dppe), 21.2 (s, 1P, PPh3). 1H NMR (δH in ppm, C6D6, 300 MHz): 8.1–7.7 and 7.4–7.0 (2m, 35H, C6H5), 2.53 (m, 2H, CH2), 1.76 (m, 2H, CH2), 1.63 (s, 15H, C5(CH3)5), 1.47 (s, 15H, C5(CH3)5). 13C{1H} NMR (δC in ppm, C6D6, 75.5 MHz): 136.5 (t, JCP = 40 Hz, FeCC), 140.0–127.3 (overlapping signals, ), 115.9 (s, ReCC), 102.8 (s, FeCC), 100.3 (s, (C5(CH3)5)Re), 93.3 (d, JCP = 17 Hz, ReCC), 88.8 (s, (C5(CH3)5)Fe), 69.7 and 50.5 (2s, CCCCCC), 31.1 (m, CH2), 10.3 and 10.0 (2s, (C5(CH3)5)Fe and (C5(CH3)5)Re). UV–vis (λ in nm [ɛ in cm−1 M−1], CH2Cl2): 298 [57 500], 342 (sh) [55 100], 396 (sh) [25 300].
Method B: A Schlenk flask charged with Et3N (30 mL), THF (3 mL), 5 (0.073 g, 0.117 mmol), and NaBPh4 (0.045 g, 0.131 mmol). Then 1.0 equiv of 63 [16] (0.080 g, 0.117 mmol) was added and the mixture was stirred at room temperature for 16 h. This gave a dark deposit and a yellow-orange solution. The volatiles were evaporated using an oil pump and the residue was extracted with Et2O (3 × 10 mL). After filtration of the extract, the solvent was removed by oil pump vacuum. The red-orange residue was washed with MeOH (3 × 5 mL) and pentane (3 × 5 mL), and dried by oil pump vacuum to give 33 as an orange powder (0.055 g, 0.043 mmol, 37%).
7.3 Synthesis of (η5-C5Me5)Re(NO)(PPh3)(CCCCCCCC)(η2-dppe)Fe(η5-C5Me5) (34)
A Schlenk flask charged with MeOH (30 mL), 5 (0.180 g, 0.28 mmol), and KPF6 (0.058 g, 0.32 mmol). The complex (η5-C5Me5)Re(NO)(PPh3)(CCCCCCCCH) (64) was generated separately in situ from (η5-C5Me5)Re(NO)(PPh3)(CCCCCCCCSiEt3) (74; 0.220 g, 0.260 mmol) and [Bu4N][F] (1.0 M in THF/5 wt% H2O; 0.104 mL, 0.104 mmol, 40 mol%) in THF (5 mL) [16]. The solution of 64 was transferred via cannula to the former solution with stirring and the resulting homogeneous dark solution was cooled to −45 °C. Then KOtBu (0.033 g, 0.32 mmol) was added. The flask was removed from the cold bath, wrapped with aluminum foil and stirred for 12 h. An orange suspension separated from the dark green medium. A workup identical to that used for 33 (method A) gave 34 as a moderately air-sensitive orange powder (0.130 g, 0.100 mmol, 38%).
FAB+-MS (m/z in amu, 3-NBA/CH2Cl2): 1299 (34+, 100%), 901 (34+−C5Me5−PPh3, 10%), 614 ([(C5Me5)Re(NO)(PPh3)]+, 25%), 589 ([(C5Me5)Fe(dppe)]+, 65%). FT-IR (ν in cm−1, nujol/KBr): νCC = 2098 m, 2046 s, 1971 sh, 1948 s, νNO = 1655 vs. 31P{1H} NMR (δP in ppm, C6D6, 121 MHz): 99.1 (br s, 2P, dppe), 21.2 (s, 1P, PPh3). 1H NMR (δH in ppm, C6D6, 300 MHz): 7.87, 7.39, and 7.25–6.90 (3m, 35H, C6H5), 2.45 (m, 2H, CH2), 1.66 (m, 2H, CH2), 1.53 (s, 15H, C5(CH3)5), 1.39 (s, 15H, C5(CH3)5). 13C{1H} NMR (δC in ppm, C6D6, 75.5 MHz): 145.8 (t, JCP = 39 Hz, FeCC), 139.5–127.0 (overlapping signals, ), 115.2 (s, FeCC), 102.3 and 71.0 (s, FeCCCC), 102.6 (d, 2JPC = 17 Hz, ReCC), 100.8 (s, (C5(CH3)5)Re), 89.1 (s, (C5(CH3)5)Fe), 68.2, 59.1, and 50.0 (3s, ReCCCC), 10.3 and 10.1 (2s, (C5(CH3)5)Fe and (C5(CH3)5)Re).
7.4 Synthesis of (η5-C5Me5)Re(NO)(PPh3)(CC){(CC)[Co2(CO)6]}(CC)(η2-dppe)Fe(η5-C5Me5) (4)
A Schlenk flask was charged with CH2Cl2 (10 mL) and 33 (0.050 g, 0.039 mmol). Then Co2(CO)8 (0.013 g, 0.038 mmol) was added with stirring. After several minutes the solution started to turn green, and was monitored by IR. After 1 h (complete conversion), the solvent was removed by oil pump vacuum. The residue was extracted with pentane (5 × 10 mL). After filtration, the solvent was removed from the filtrate by oil pump vacuum (drying the residue for several hours) to give 4 as green powder (0.050 g, 0.031 mmol, 82%). FAB+-MS (m/z in amu, o-NPOE/CH2Cl2): 1561 (4+, 18%), 1477 (4+−3CO, 14%), 1421 (4+−5CO, 10%), 1393 (4+−6CO, 67%), 1334 (4+−Co−6CO, 11%), 1275 (4+−2Co−6CO, 100%). FT-IR (ν in cm−1, nujol): νCC and νCO = 2068 s, 2030 vs, 1999 vs, νNO = 1642 s. 31P{1H} NMR (δP in ppm, C6D6, 81 MHz): 99.9 (s, 2P, dppe), 21.6 (s, 1P, PPh3). 1H NMR (δH in ppm, CDCl3, 200 MHz): 8.0–7.8 and 7.7–7.0 (2m, 35H, C6H5), 2.67 (m, 2H, CH2), 2.12 (m, 2H, CH2), 1.84 (s, 15H, C5(CH3)5), 1.48 (s, 15H, C5(CH3)5). UV–vis (λ in nm [ɛ in cm−1 M−1], CH2Cl2): 270 [72 000], 332 [61 400], 688 [5 400].
7.5 Synthesis of [(η5-C5Me5)Re(NO)(PPh3)(CCCCCC)(η2-dppe)Fe(η5-C5Me5)][PF6] (33[PF6])
A Schlenk flask charged with 33 (0.074 g, 0.058 mmol) and [(η5-C5H5)2Fe][PF6] (0.017 g, 0.051 mmol, 88 mol%). Then THF (20 mL) was added, and the flask was cooled to −80 °C. After 8 h, the sample was concentrated to 5 mL, and pentane (50 mL) was added at low temperature. The resulting precipitate was vigorously stirred. After 0.2 h, the solvent was removed using a cannula that contained a filter. The residue was washed at low temperature with toluene (2 × 5 mL) and Et2O (2 × 5 mL), and dried by oil pump vacuum to give 33[PF6] as a green powder (0.066 g, 0.046 mmol, 91%). Calcd for C70H69F6FeNOP4Re: C, 59.20; H, 4.90; N, 0.99. Found: C, 59.30; H, 5.14; N, 0.95. FT-IR (ν in cm−1, nujol): νCC = 1999 s, 1882 vs, 1793 s, νNO = 1648 s. UV–vis (λ in nm [ɛ in cm−1 M−1], CH2Cl2): 236 [66 400], 266 [45 500], 312 [23 300], 414 [14 400], 688 [22 500], 798 [30 200], 858 [33 500].
Acknowledgements
We thank the CNRS for a fellowship grant to S.S., OTAN (F.P., C.L., J.A.G.), the Deutsche Forschungsgemeinschaft (SFB 583; J.A.G.), the US National Science Foundation (CHE-9732605 and CHE-0719267; J.A.G., W.E.M.), and the Polish State Committee for Scientific Research (4 T09A 148 24; S.S.) for support.