

1 Introduction
La synthèse des composés α,β-insaturés de type accepteurs de Michaël est essentiellement effectuée selon la condensation de Knoevenagel, mais des produits secondaires sont souvent observés, sans que leur origine ne soit toujours clairement établie. À titre d’exemple, la condensation entre le cyanoacétate d’éthyle et l’acétophénone conduit majoritairement à l’ester activé 1, mais la dihydropyridone 2 se forme aussi (Fig. 1) 〚1, 2〛.
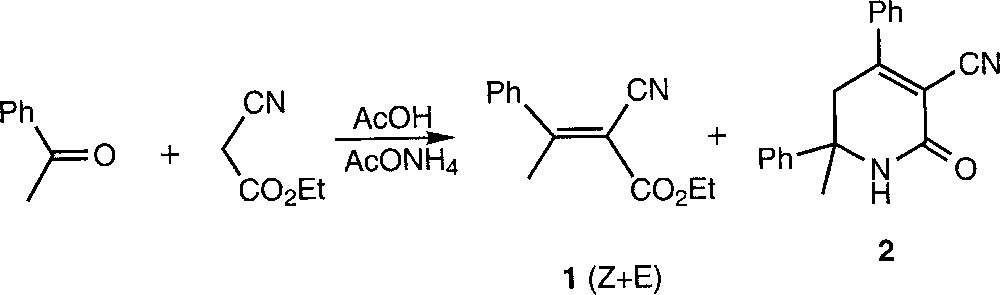
Condensation entre le cyanoacétate d’éthyle et l’acétophénone, conduisant majoritairement à l’ester activé 1, mais avec également formation de dihydropyridone 2.
Dans le cadre d’une étude réalisée en partenariat avec l’industrie, notre premier objectif était de préparer l’ester activé 1 selon le schéma de la Fig. 1. Les conditions retenues dans la variante de la réaction de Knoevenagel 〚3〛, connue sous le nom de Knoevenagel–Cope, se prêtent bien à une application industrielle, compte tenu du coût et du caractère peu polluant des réactifs utilisés (acide acétique et acétate d’ammonium). Nous avons préféré cette méthode à celle, très utile à l’échelle du laboratoire, qui utilise l’alumine basique en quantité relativement abondante et nécessite une quantité importante de solvant dans la phase finale d’extraction 〚4〛. Afin d’optimiser la réaction décrite dans la Fig. 1, il nous fallait remplacer le solvant benzène par un solvant moins toxique et limiter la formation du produit secondaire 2. C’est pour atteindre cet objectif que nous avons cherché à préciser l’origine de la dihydropyridone 2. Nous montrerons dans cette note que ce travail nous conduit aussi à proposer les conditions de Knoevenagel–Cope pour préparer certaines cétones α,β-éthyléniques.
2 Résultats et discussion
Un examen de la littérature fait apparaître que la dihydropyridone 2 est encore le produit secondaire lorsque l’acétophénone est condensée avec le cyanacétamide 〚5〛. Cette dihydropyridone 2 est également formée au cours de la réaction entre la dypnone et le cyanacétamide 〚6〛. On retrouve, par ailleurs, cette même dihydropyridone 2 lorsque l’ester activé 1 est placé dans l’éthanol saturé en ammoniac 〚7〛. Nous avons repris la réaction de la Fig. 1 dans un milieu acide acétique–acétate d’ammonium (conditions de Knoevenagel–Cope), mais en remplaçant l’acétophénone par l’éthylénique 1. On vérifie bien que la réaction est réversible, puisque l’on observe la formation d’acétophénone, mais on n’observe que très peu de dihydropyridone 2, qui se forme en moindre quantité que dans les conditions de la réaction de la Fig. 1. Ce résultat exclut que le composé 2 ait pour origine directe l’ester 1. Les traces de composé 2 observées proviendraient plutôt de la présence d’acétophénone, associée à la réversibilité de la réaction de Knoevenagel. En revanche, la formation de 2 pourrait s’expliquer par une réaction entre le cyanoacétate d’éthyle et la dypnone. Il a en effet été montré qu’une cétone éthylénique analogue (oxyde de mésityle) réagit avec l’ammoniac pour donner un bon rendement en pyridinone 3 〚8〛 (Fig. 2).

Réaction d’une cétone éthylénique analogue (oxyde de mésityle) avec l’ammoniac conduisant à la pyridinone 3 〚8〛.
Nous avons été confortés dans cette hypothèse en examinant le spectre de RMN 1H du produit brut issu de la réaction de Knoevenagel–Cope de la Fig. 1. On retrouve bien tous les signaux décrits pour la dypnone 〚9, 10〛. Si la dypnone est bien un intermédiaire clé conduisant au produit secondaire 2, les conditions de Knoevenagel–Cope devraient permettre d’accéder à la dypnone 4a, à condition d’opérer en l’absence de cyanacétate d’éthyle. C’est bien ce que nous avons observé en utilisant les conditions décrites dans la partie expérimentale. Le rendement en produit pur isolé 4a (29%) peut paraître faible, mais nous avons volontairement limité le temps de réaction pour éviter la formation de dihydropyridine 7 (Fig. 3). Dans ces conditions, l’acétophénone de départ n’ayant pas réagi peut être recyclée et la méthode devient intéressante sur le plan industriel, compte tenu du caractère non polluant et peu coûteux des réactifs utilisés. Les cétones α,β-insaturées sont des accepteurs de Michaël très utiles en synthèse, mais leur préparation est délicate et nécessite parfois l’utilisation de réactifs toxiques 〚11〛 ou des temps de réaction importants en présence d’alumine basique 〚9 et réf. citées〛, dont la nature est essentielle 〚12〛. À titre d’exemple, nous avons vérifié que l’autocondensation de cétones dans les conditions de Knoevenagel–Cope peut être une alternative utile pour préparer certaines cétones α,β-éthyléniques avec un bon degré de pureté (Tableau 1).
Formation de la dihydropyridine 7 avec formation de 2 comme produit secondaire.
Rendement de l’auto-condensation de quelques cétones dans les conditions de Knoevenhagel–Cope.
| R | R’ | Temps (h) | Produit | Rdt (%) | Cétone de départa |
| C6H5– | CH3 | 8 | 4a | 29 (90% E–10% Z) | 43% |
| p-MeO–C6H4– | CH3 | 24 | 4b | 35 (E) | 33% |
| p-Cl–C6H4– | CH3 | 5 | 4c | 45 (E) | 33% |
| 2 | 4d | 60 (E) | b |
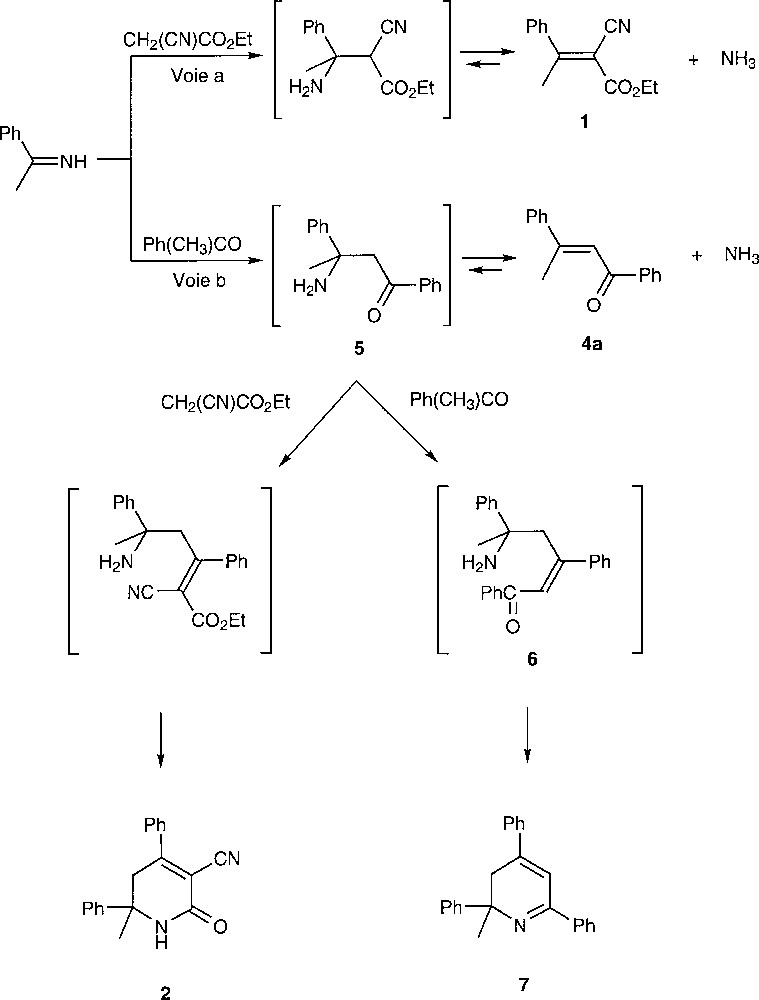
La formation de la dihydropyridine 7, qui accompagne la dypnone 4a lorsque les temps de réaction sont prolongés au-delà de 8 h, peut s’interpréter par une addition d’ammoniac sur 4a pour donner l’intermédiaire 5, qui réagit ensuite avec une nouvelle molécule de benzophénone, conduisant ainsi à 6 puis à 7. La formation de 7, que nous avons isolé et caractérisé sous forme de picrate, est un argument supplémentaire pour proposer l’aminocétone 5 comme intermédiaire conduisant au produit secondaire 2 dans les conditions de Knoevenagel–Cope (Fig. 3).
Il est clair que la formation des composés 2 ou 7 nécessite la présence d’ammoniac dans le milieu réactionnel. Pour éviter leur formation et obtenir un meilleur rendement en ester activé 1, nous avons voulu remplacer l’acétate d’ammonium par du carbonate de sodium, également intéressant sur le plan industriel. Aucune réaction n’est observée avec ce catalyseur, même après des temps de réaction importants. L’utilisation d’acétate de triéthylammonium à la place de l’acétate d’ammonium ne nous permet pas non plus d’obtenir l’ester activé 1. Il est vraisemblable que l’imine Ph(CH3)C=NH est, comme l’avait déjà proposé Knoevenagel 〚3〛, le premier intermédiaire conduisant à 1 (Fig. 3, voie a). Dans ces conditions, si on souhaite optimiser la synthèse de 1 dans les conditions de Knoevenagel–Cope, les seuls paramètres ajustables sont le solvant, la température et le temps de réaction. Nous avons retenu le cyclohexane comme solvant et maintenu une ébullition durant 30 h avec distillation azéotropique de l’eau formée (méthode B, partie expérimentale). On obtient alors un rendement de 80% en ester activé 1 purifié.
3 Conclusions
Ce travail nous a permis d’optimiser le rendement en ester activé 1. Nous avons également caractérisé la dihydropyridone 2 qui accompagne 1 en faible quantité et nous avons émis l’hypothèse que sa formation a pour origine la dypnone que nous avons décelée dans le produit réactionnel brut. Cette hypothèse est confortée par le fait que nous avons pu préparer cette dypnone en utilisant les conditions de Knoevenagel–Cope en l’absence de cyanoacétate d’éthyle. En prolongeant les temps de réaction, cette dypnone évolue vers la dihydropyridine 7. À notre connaissance, nous montrons ici pour la première fois que les conditions de Knoevenagel–Cope sont utilisables pour préparer certaines cétones α,β-éthyléniques.
4 Partie expérimentale
Les spectres de RMN 1H et 13C ont été enregistrés respectivement à 200 (ou 300 MHz) et à 75 MHz dans des solutions de CDCl3. Le TMS est la référence interne. Les spectres de masses HR ont été enregistrés au Centre de mesures physiques de l’Ouest, en utilisant la technique d’ionisation électronique. Les points de fusion ont été mesurés sur un banc Kopfler et sont non corrigés. Les résultats des analyses élémentaires ont été obtenus par le laboratoire central de microanalyse du CNRS (Lyon). Les spectres IR dans KBr sont déterminés par IR–TF.
4.1 Réaction entre le cyanoacétate d’éthyle et l’acétophénone
4.1.1 Méthode A
La synthèse est effectuée d’après la méthode connue 〚1〛 en remplaçant le benzène par le toluène. L’huile obtenue est analysée par RMN 1H et apparaît être un mélange d’ester 1 (Z + E ; 65%), d’acétophénone (8%), de cyanoacétate d’éthyle (18%), de dihydropyridone 2 (9%) et de dypnone 4a (traces). 2 précipite lentement et peut être isolée, puis recristallisée, dans l’éthanol (Pf = 215–216 °C). La structure de 2 a été clairement établie par RMN 1H et 13C (l’isomère postulé dans la référence 3 peut être exclu). RMN 1H (DMSO-d6) : δ (ppm) = 1,37 (s, 3H), 3,23 et 3,46 (AB, 2H, J = 18,3 Hz), 7,16–7,39 (m, 10H) et 8,77 (s, 1H) ; RMN 13C (DMSO-d6) : δ (ppm) = 30,1 (s), 44,6 (t), 57,1 (s), 107,1 (s), 116,2 (s), 125,8 (d), 127,9 (d), 128,2 (d), 129,3 (d), 129,7, 132 (d), 136,8 (s), 146,1 (s) 162,3 (s) et 165,5 (s) ; IR (KBr) 2900–3200 cm–1 (large), 2222 cm–1.
4.1.2 Méthode B
L’acétophénone (36 g, 0,3 mol), le cyanoacétate d’éthyle (34 g, 0,3 mol), l’acide acétique (54 g, 0,9 mol) et l’acétate d’ammonium sec (24 g, 0,3 mol) sont maintenus à reflux dans le cyclohexane (240 ml) pendant 30 h en éliminant l’eau qui se forme à l’aide d’un Dean et Stark. Après refroidissement, on lave successivement par de l’eau (100 ml), par une solution de Na2CO3 (1 N, 100 ml), puis le solvant est séché sur Na2SO4 et évaporé. L’huile obtenue (55,4 g) contient simplement des traces du composé cyclique 2, de cyanoacétate d’éthyle et d’acétophénone. La purification de cette huile sous pression réduite permet d’isoler le composé 1 (Eb = 140 °C à 0,1 torr ; Rdt : 80% Z + E).
4.2 Cétones α,β-éthyléniques : dypnone 4a
À une solution d’acétophénone (144 g, 1,2 mol) dans le toluène (500 ml) sont ajoutés l’acide acétique (108 g, 1,8 mol), et l’acétate d’ammonium sec (46,2 g, 0,6 mol). Le milieu est porté à reflux (8 h) et l’eau est éliminée par un Dean et Stark. Après refroidissement, on lave par de l’eau (200 ml), et la phase organique est séchée sur Na2SO4 et évaporée. L’huile obtenue (140 g) est purifiée sous pression réduite (0,04 torr). L’acétophénone n’ayant pas réagi est obtenue (63 g à 60 °C), avec 4a (Eb = 144 °C ; 38,8 g ; Rdt : 29%, dont 90% E et 10% Z). Les cétones éthyléniques 4b, 4c et 4d ont été obtenues selon ce protocole en utilisant les temps de réaction précisés dans le Tableau 1. Les analyses centésimales effectuées pour les composés 4a–4d sont en adéquation avec leurs formules brutes. Par ailleurs, les points de fusion et les analyses spectrales RMN 1H de ces composés sont conformes aux données de la littérature 〚9, 10〛. L’acétophénone n’ayant pas réagit peut être recyclée.
4.3 2,3-dihydro-2-méthyl-2,4,6-triphényl-pyridine 7
L’acétophénone (14,4 g, 0,12 mol), l’acide acétique (10,8 g, 0,18 mol), et l’acétate d’ammonium sec (4,8 g, 0,06 mol) sont maintenus à reflux dans le toluène (50 ml) pendant 2 h, en éliminant l’eau qui se forme à l’aide d’un Dean et Stark. Puis l’acide acétique (10,8 g, 0,18 mol) et l’acétate d’ammonium sec (4,8 g, 0,06 mol) sont à nouveau ajoutés et le reflux est prolongé durant 17 h. Après refroidissement, on lave successivement par de l’eau (20 ml), par une solution de Na2CO3 (1 N, 30 ml), puis la phase organique est séchée sur Na2SO4 et évaporée. L’huile obtenue (11 g, 85%) est caractérisée par RMN. RMN 1H : δ (ppm) = 1,66 (s, 3H), 2,97 (AB, 2H, J = 12,1 Hz), 6,86 (s, 1H) et 7,36–7,98 (br, 15H) ; RMN 13C : δ (ppm) = 28,4 (q, J = 128 Hz), 37,31 (t, J = 129 Hz), 60,0 (s), 115,4 (d, J = 141,2 Hz), 125,8, 126, 126,5, 127, 128,3, 128,6, 129, 129,4 et 130. SM : C24H21N ; PM = 323,1674 ; obs. : 323,1682. L’analyse centésimale est obtenue à partir du picrate de 7. A une solution de 7 (1,9 g, 0,003 mol) dans l’acétone (20 ml) est ajoutée une solution de l’acide picrique (4,8 g, 0,021 mol) dans l’acétone (5 ml). Après quelques minutes, le précipité est filtré et recristallisé dans l’acétone (0,7 g, Rdt : 42%, Pf = 234 °C). Anal. calc. pour C30H24N4O7 : C, 65,21 ; H, 4,38 ; N, 10,14. Trouvée : C, 65,33 ; H, 4,30 ; N, 10,18.