1 Introduction
Les esters entre un sucre et un acide gras sont des tensioactifs1 non-ioniques. Ils sont complètement biodégradables, non toxiques, non irritants pour la peau, sans odeur et sans goût, ce qui permet leur utilisation dans l’industrie alimentaire ou cosmétique, par exemple [2–5]. Depuis les années 1950, les sucroesters sont réalisés principalement par voie chimique [6,7]. Mais cette méthode de synthèse présente plusieurs inconvénients. En premier lieu, la réaction a lieu à haute température dans des solvants toxiques difficiles à enlever [8], ce qui entraîne une non sélectivité dans l’estérification des groupements hydroxyles du sucre [2,9] et en plus par un phénomène de caramélisation, les produits sont ainsi colorés [10] et contiennent des sous-produits nocifs [11]. Depuis 1986, la synthèse des esters de sucre peut être réalisée par voie enzymatique, notamment par des lipases [7,12]. L’avantage est que ce type de réaction peut être conduit dans des conditions douces et des solvants moins nocifs que par la voie chimique [13] et conduisent généralement à des monoesters [6]. Dans le cas de réaction enzymatique, le surfactant peut bénéficier du label « naturel ». Quant à la synthèse des esters de fructose, elle a débuté dans l’année 1995 [14].
Dans cet article, nous présentons nos résultats sur la réaction enzymatique entre le fructose et l’acide laurique principalement mais aussi l’influence de divers paramètres comme le temps de réaction, le solvant, la longueur de chaîne de l’acide gras, le ratio molaire entre le sucre et l’acide gras, et la quantité d’eau initiale dans le milieu. Dans la mesure du possible, nous modéliserons les courbes obtenues afin d’apporter des données cinétiques importantes industriellement. Nous donnons des conditions analytiques permettant un suivi rapide de la réaction.
Le comportement des agents tensioactifs dépend de la longueur de la chaîne grasse : une courte chaîne (en dessous C12) donne un tensioactif hydrophile tandis que les tensioactifs à longue chaîne (au-dessus de C12) sont lipophiles. Le choix de l’acide laurique comme composé test a été dicté par le fait qu’il a une longueur de chaîne intermédiaire mais surtout parce que le laurate de fructose est reconnu pour certaines propriétés d’intérêt, dont notamment un fort effet d’inhibition de croissance sur Streptococcus mutans responsable de la carie dentaire [2]. Les esters de fructose possèdent aussi une activité émulsifiante plus élevée sur les hydrocarbures aromatiques et les huiles que sur les hydrocarbures aliphatiques [15,16]. Grâce à son fort pouvoir de réduction des tensions de surface, le laurate de fructose peut être adopté en substitution des émulsifiants synthétiques [15]. Quant au laurate de fructose, son HLB indique qu’il est neutre et stabilise aussi bien les émulsions eau dans huile que huile dans eau [4]. Tout cela permet l’utilisation de ces esters de fructoses dans l’industrie alimentaire dans la production d’arôme ou bien la maturation de fromage, qui ne sont que quelques exemples parmi tant d’autres [17].
Les lecteurs trouveront les résultats sur l’estérification de l’inuline, qui est une polymère de résidus fructose liés en β-(1- > 2), en annexe dans le matériel complémentaire.
2 Matériel et méthodes
Le biocatalyseur utilisé est la lipase B immobilisée de Candida antarctica Novozym 435 de Novo Industri. Les plaques de chromatographie sur couche mince (CCM) sont des plaques de gel de silice 60 F254 de Merck. Le gel de silice est du silica gel 60 Å SI0 1720 de chez Rocc (70-210 mesh). Les tamis moléculaires (4 Å, 8-12 mesh, Aldrich) sont activés pendant 2 h à 350 °C et laissés une semaine dans un dessiccateur.
L’α-naphtol est préparé de la façon suivante : 21 mL de solution mère (15 % – p/v dans l’éthanol) sont prélevés et dilué dans 13 mL d’acide sulfurique concentré, 81 mL d’éthanol 96 % et 8 mL d’eau.
Le révélateur au permanganate de potassium basique est constitué de 10 g de carbonate de potassium, 1,5 g de permanganate de potassium dans 150 mL d’eau et 1,25 mL d’une solution aqueuse de soude à 10 %.
Le calcul des moyennes et des erreurs (avec niveau de confiance de 95 % de Student-Fisher) a été déterminé par le module One variable statistic Version 1.05 [18] qui fait partie du programme Gabriel Data Analysis [19]. L’analyse des courbes de points a été réalisée par le module Two variables statistic Version 1.31 [20] qui fait partie du même logiciel.
2.1 Solubilité
Les essais ont été effectués dans l’hexane (log P = 3,5), le 2-méthyl-2-butanol (log P = 0,89), la pyridine (log P = 0,71), l’acétate d’éthyle (log P = 0,68), le tertio-butanol (log P = 0,35), l’acétone (log P = –0,23), l’acetonitrile (log P = –0,33) et l’eau. À 20 mL de solvant sont ajoutés 0,5 g de sucre. Le mélange est maintenu à 60 °C sous agitation (100 rpm) pendant cinq heures, centrifugée à 14 000 rpm pendant dix minutes. Un prélèvement de 2 μl du surnageant (environ 10 mL) est développé par CCM (butanol/éthanol/eau 5/3/2 v/v/v - révélation à l’α-naphtol).
La détermination quantitative de la solubilité a été réalisée de la façon suivante : dans 20 mL de solvant, 0,6 g de fructose sont ajoutés, puis le mélange est chauffé à 60 °C sous agitation pendant deux heures. Après une heure à 60 °C sans agitation, 10 mL de surnageant sont prélevés avec précision et transférés dans un ballon de 100 mL préalablement taré, puis le solvant est éliminé à l’évaporateur rotatif.
2.2 Méthodes analytiques
2.2.1 Méthodes analytiques rapides de suivi de la réaction enzymatique – CCM
Une première élution de 2,7 cm est effectuée par le mélange de solvant chloroforme/méthanol/eau (85/13,5/1,5, v/v/v). La plaque est séchée, puis un second développement par du chloroforme/méthanol/acide acétique (98,5/1,5/1, v/v/v) est conduit jusqu’à 5,2 cm. La plaque est de nouveau séchée, puis remise à éluer par le mélange de n-hexane/éther diéthylique/acide acétique (70/30/1, v/v/v) jusqu’à 9 cm. La révélation s’effectue sur la plaque séchée par une pulvérisation d’une solution de fluorescéine (0,2 % dans le méthanol) et une observation sous lampe UV suivi d’une pulvérisation d’une solution d’α-naphtol puis d’une carbonisation à 105 °C pendant cinq minutes. Dans ces conditions, les Rf sont de 0,00, 0,12, 0,18 et 0,41, respectivement, pour le fructose, le monolaurate de fructofuranosyle, les polylaurates de fructofuranosyle et l’acide laurique et de 0,10 pour le monolaurate de glucopyranosyle.
L’autre méthode consiste en une élution par un mélange de solvant dichlorométhane/éthanol (19/1, v/v) suivie d’une révélation à froid par une solution de permanganate de potassium basique. Les Rf sont de 0,00, 0,10, 0,37 et 0,58 respectivement pour le fructose, le monolaurate de fructofuranosyle, les polylaurates de fructofuranosyle et l’acide laurique.
2.2.2 Dosage des acides gras
À 1 mL (qui sera pesé) de prélèvement de la réaction, on ajoute 10 mL d’éthanol absolu et deux gouttes de phénolphtaléine (0,1 % éthanol 96 %). Les acides gras libres sont alors titrés par de la potasse 0,02 M. Chaque échantillon est dosé deux fois.
2.2.3 HPLC
Les analyses HPLC ont été effectuées sur un appareil Agilent Technologies 1200 séries équipé avec un détecteur ELSD Alltech 330 (température : 84,5 °C, débit du gaz : 2,22 slpm). Une colonne Agilent Zorbax SB-C18 (150 × 4,6 mm, 3,5 μm) a été utilisée lors de l’analyse. Le programme du gradient de solvant a été le suivant (débit 0,5 mL min−1) : à 0 min 45/55 (% v acetonitrile/% v eau), à 15 min 100/0, à 25 min 100/0, à 30 min 45/55, puis maintenu pendant dix minutes.
2.2.4 Teneur en eau
La teneur en eau des milieux réactionnels a été caractérisée par titrage de 1 g de ce milieu par un titrateur Mettler Toledo DL31 Karl Fisher [21].
2.2.5 Tension de surface
Les tensions de surface ont été déterminées à l’aide d’un appareil Lauda TVT 1 équipé du logiciel Lauda Drop-Volume-Tensiometer TVT-1 Version 2.0 avec les réglages suivant : température : 20 °C, mode : DYNamic, volume de la seringue : 2,5 mL, rayon : 1,055 mm, densité : 0,998 g.mL−1, nombre de répétitions : deux, nombre de gouttes : huit, réduction : ON, T1 : 0,07 s.μl−1, T2 : 0,8 s.μl−1. La température était contrôlée par un appareil RM 6 de Lauda à plus ou moins 0,5 °C. Les déterminations des tensions de surface ont été effectuées par le module TVT Analyze Version 1.03 [22] qui fait partie du programme Gabriel Data Analysis [19].
2.2.6 ACD/DSC
Les thermogrammes ont été obtenus sur un 2920 MDSC V 2.6A de TA Instruments dont les réglages étaient un balayage de la température de –40 °C à +200 °C avec une vitesse de 5 °C par minute.
2.3 Réaction d’estérification enzymatique
À 20 mL de solvant (2-méthyl-2-butanol), le fructose (0,5044 g ; 2,8 mmoles) et l’acide gras (2,8 mmoles [0,5609 g dans le cas de l’acide laurique]) sont chauffés à 60 °C dans un erlenmeyer fermé par un bouchon à vis recouvert d’une feuille d’aluminium changée toutes les 24 heures. Au bout d’une heure, le tamis moléculaire (2 g) et la lipase Novozym 435 (100 mg) sont ajoutés en une fois. Le mélange réactionnel est agité grâce à un agitateur orbitalaire (100 rpm) thermostaté par un bain-marie à 60 °C pendant 48 heures. Au bout de ce temps, le mélange réactionnel est filtré pour éliminer le tamis moléculaire et l’enzyme. 40 mL d’eau distillée sont versés dans le filtrat. Ce dernier est extrait par 2 × 10 mL de dichlorométhane et la phase organique est éliminée dans un évaporateur rotatif. Le produit restant est alors redissous dans 10 mL de chloroforme ou dichlorométhane en vue de sa purification qui est décrite au paragraphe suivant.
Un blanc est réalisé dans les mêmes conditions sans enzyme.
2.4 Purification des esters
Trente grammes de gel de silice G60, préalablement conditionnée dans 70 mL de chloroforme, sont déposés, pour atteindre une hauteur de 20 cm, dans une colonne en verre de 2,0 cm de diamètre. Soixante millilitres de chloroforme sont élués avant le dépôt de l’échantillon. Le produit (dissous dans 2 mL de chloroforme) est déposé en tête de colonne. La colonne est lavée par 130 mL de chloroforme (récupération de l’acide gras), puis par 120 mL du mélange chloroforme/méthanol (90/10, v/v) (obtention des polyesters). Le monoester est enfin élué par 120 mL du mélange chloroforme/méthanol/eau (64/10/1, v/v/v). La pureté des esters, contrôlée par HPLC, est supérieure à 99 %.
Une autre méthode consiste à introduire dans une colonne de 6 cm de diamètre, 150 g de silice dans 250 mL d’un mélange dichlorométhane/éthanol 19/1 v/v. 450 mL du même mélange sont élués afin de récupérer l’acide laurique. Ensuite, 100 mL de solvant 18/2 v/v sont utilisés comme tampon (sinon la colonne sèche prématurément) et 700 mL de mélange 17/3 v/v servent à éluer les polylaurates et ensuite le monolaurate de fructose. La pureté est alors déterminée par CCM et aucun spot autre que celui du produit n’est visible.
3 Résultats et discussion
3.1 Recherche du solvant adéquat
Avant de se lancer dans la synthèse, il nous a semblé important de déterminer le meilleur solvant, notamment au niveau de la solubilisation des substrats. Le problème associé à cette étude est que le solvant doit à la fois solubiliser le sucre (polaire), l’acide gras (apolaire) ainsi que l’enzyme sans la dénaturer. Des essais de solubilité ont été effectués dans l’hexane, le 2-méthyl-2-butanol ou alcool tertio-amylique, la pyridine, l’acétate d’éthyle, le tertio-butanol, l’acétone, l’acetonitrile et l’eau. Les solvants fortement polaires (ayant un log P < –0,5), tel que le DMSO par exemple, n’ont pas été testés car ceux-ci sont connus pour conduire au phénomène de water-stripping, c’est-à-dire un déplacement de l’eau liée à l’enzyme vers le solvant [23,24], sachant que les lipases y sont fortement sensibles contrairement aux protéases [25]. Il a été trouvé clairement que le fructose est soluble dans l’eau et la pyridine, qu’il est insoluble dans l’hexane (ce qui est en accord avec les résultats de Spiliotis [9]) et qu’il est partiellement soluble selon l’ordre suivant : 2-méthyl-2-butanol > t-butanol > acétonitrile > acétone > acétate d’éthyle (figure S 1 dans le matériel complémentaire).
Le tableau S 1 donne un relevé de la littérature de la solubilité du fructose en fonction du solvant et de la température ainsi que nos valeurs expérimentales, vu la disparité des valeurs bibliographiques. Il a été trouvé une solubilité de 10,0 g.L−1 à 60 °C dans le 2-méthyl-2-butanol, ce qui est proche de la valeur donnée par Chamouleau (11,0 g.L−1) [6] alors que dans le tertio-butanol, il a été trouvée une valeur de 15,9 g.L−1 à 60 °C, ce qui est en accord avec la valeur de Yoo (15,6 g.L−1) [26].
Pour la suite le 2-méthyl-2-butanol a été utilisé de préférence, qui présente de plus l’avantage d’être autorisé pour le secteur alimentaire [8] ; des traces ne seraient par conséquence pas un obstacle.
3.2 Mise au point d’analyse pour le suivi de la réaction d’estérification enzymatique
Il est important de développer des méthodes analytiques rapides pour suivre la réaction enzymatique.
3.2.1 Méthode rapide basée sur la chromatographie sur couche mince
Les analyses par CCM ont été réalisées afin de permettre un suivi qualitatif rapide de la synthèse des esters de sucre. La mise au point de cette analyse a permis de trouver une méthode qui révèle sur la même plaque chromatographique les substrats (sucre et acide gras) et les produits (mono et diesters) de la réaction. Tous les essais ont été réalisés sur plaques de gel de silice 60 F254.
Le premier système chromatographique utilisé est une adaptation de la littérature [2,3]. L’éluent était un mélange de solvants (chloroforme/méthanol/acide acétique/eau – 80/15/8/2, v/v/v/v) et la révélation ayant été réalisée par pulvérisation d’acide sulfurique à 50 % puis chauffage à 105 °C. Par ce système, on aperçoit seulement les spots du fructose et des esters de fructose (données non montrées).
Un second système a donc été testé, basé sur le mélange de solvants éther pétrole/éther éthylique/acide formique – 6/4/0,15, v/v/v suivi d’une révélation par UV après pulvérisation de fluorescéine. Dans ce cas, seul les spots d’acide gras sont révélés (données non montrées).
Le troisième système a été mis au point par Soultani et al. [3] (chloroforme/méthanol/acide acétique/eau – 80/15/8/2, v/v/v/v ; révélation par pulvérisation d’α-naphtol 50 %, puis carbonisation à 105 °C) et ne permet de détecter que les spots du fructose et de ses esters (données non montrées).
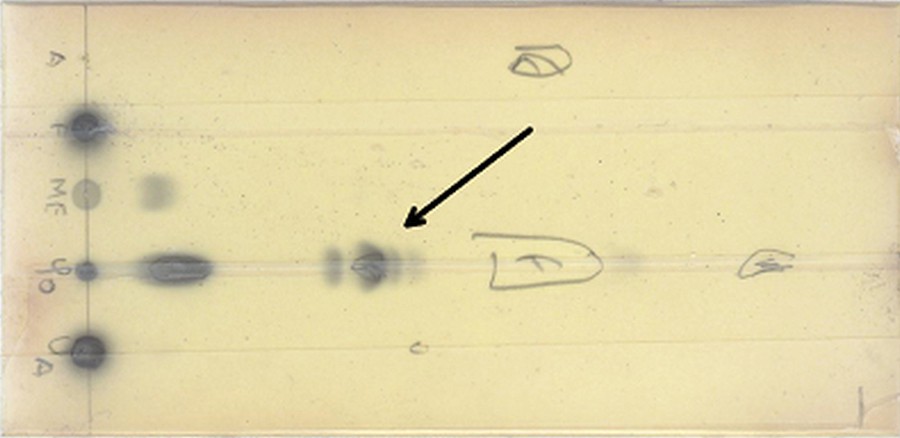
L’objectif fixé a été finalement atteint en modifiant le principe d’une triple élution et une double révélation mis au point par Castillo et al. [27]. Cela consiste en une première élution de 2,7 cm par du chloroforme/méthanol/eau (85/13,5/1,5, v/v/v) suivie d’un séchage de la plaque. Un second développement de 5,2 cm est réalisé par un mélange de chloroforme/méthanol/acide acétique (98,5/1,5/1, v/v/v). La plaque est à nouveau séchée, puis éluée une dernière fois par un mélange de n-hexane/éther diéthylique/acide acétique (70/30/1, v/v/v) sur une hauteur de 9 cm. Une fois la plaque séchée, la double révélation consiste en une pulvérisation d’une solution de fluorescéine et une observation sous lampe UV suivie d’une pulvérisation d’une solution d’α-naphtol suivie d’une carbonisation à 105 °C. Grâce à ce système, on peut observer les spots des réactifs de départ (sucre et acide gras) ainsi que les produits de la réaction (mono et polyesters). Les Rf ont été calculés à partir de la Fig. 1 et sont de 0,00, 0,12, 0,18 et 0,41 respectivement pour le fructose, le monolaurate de fructofuranosyle, les polylaurates2 de fructofuranosyle et l’acide laurique. Dans les mêmes conditions, le monolaurate de glucopyranosyle a un Rf de 0,10 (données non montrées).
Ce système de triple élution et d’une double révélation est pratique pour avoir rapidement les informations sur tous les composés présents en même temps, mais se révèle trop lent dans le cas d’un suivi d’une réaction. Pour cela, une autre méthode plus rapide a été développée. L’objectif a été atteint par un mélange dichlorométhane/éthanol (19/1 v/v) et une révélation par du permanganate de potassium basique (Fig. 2). L’avantage de ce solvant est qu’il sera utilisé directement plus tard lors de la purification par colonne.
3.2.2 Méthode pour la détermination de la conversion d’acide gras
Les acides gras libres ont été dosés par de la soude ou par de la potasse. Cette méthode permet d’évaluer le rendement de la réaction. L’Éq. (1) donne le pourcentage de conversion.
| (1) |
Cette méthode a été utilisée par la suite pour calculer le taux de conversion lors de l’optimisation de la réaction.
3.2.3 Méthode pour la quantification et l’identification des produits synthétisés – HPLC
Le système chromatographique était constitué d’une colonne C18 couplée à un détecteur ELSD (détecteur évaporatif à diffraction de lumière) qui est un détecteur permettant de faire du quantitatif. Différents essais de phases mobiles ont été testés afin d’obtenir une séparation des différents réactifs et produits de la réaction d’estérification enzymatique. Le premier essai effectué a consisté à essayer ce qui a été trouvé dans la littérature et en particulier la publication de Scheckermann [14] qui utilise comme éluant l’acetonitrile/méthanol/eau (35/60/5, v/v/v) à 40 °C (figure S 2 (a)). N’obtenant pas de bonne résolution, le mélange acetonitrile/eau (65/35, v/v) à 35 °C a été testé et donne là aussi une résolution trop faible (figure S 2 (b)). Enfin une excellente séparation (figure S 2 (c)) a été obtenue par un gradient acetonitrile/eau décomposé comme suit : à 0 min 45/55 (% v acetonitrile/% v eau), à 15 min 100/0, à 25 min 100/0, à 30 min 45/55, maintenu pendant dix minutes. Dans ces conditions, le fructose a un temps de rétention de 2,4 minutes, le monolaurate de fructofuranosyle de 10,1 et 10,8 minutes, l’acide laurique 16,8 minutes et les polylaurates de fructofuranosyle de 24,5 minutes. Dans les mêmes conditions, le monopalmitate de fructosyle a un temps de rétention de 14,0 minutes et le dipalmitate de fructose un temps de 19,3 minutes (données non montrées).
3.3 Réaction d’estérification enzymatique
La première étape a été une adaptation des conditions opératoires de Chamouleau et al. [6], ces derniers réalisant la synthèse du palmitate de fructofuranosyle dans 200 mL de 2-méthyl-2-butanol catalysée par la lipase de C. antarctica.
Schématiquement les conditions expérimentales que nous avons utilisées sont les suivantes : à un mélange équimolaire (2,8 mmol) de fructose et d’acide laurique dans 20 mL de solvant sont ajoutés 100 mg d’enzyme Novozym 435 et 2 g de tamis moléculaire activé 4 Å. Le mélange est agité (100 rpm) pendant 48 h à 60 °C. Par la suite, nous nous sommes rendus compte de l’importance de l’eau dans le processus enzymatique et nous proposons les modifications suivantes : l’erlenmeyer doit être à vis et le col est en plus protégée par une feuille d’aluminium changée toute les 24 heures. Grâce à ces modifications, dans le cas de l’alcool tertio-amylique (ou 2-méthyl-2-butanol), la conversion d’acide gras est d’environ 78 %. Un blanc était réalisé dans les mêmes conditions sans enzyme.
La purification des produits obtenus sera décrite plus loin (Section 3.4).
Nous présentons ici l’optimisation de certains paramètres, tels que le temps de réaction, la longueur de chaîne, etc. afin d’avoir les meilleures conditions pour cette réaction.
3.3.1 Influence du temps de réaction
Le premier paramètre que nous avons étudié est le temps de réaction. Pour cela, nous avons mesuré le taux de conversion de l’acide laurique à différents intervalles de temps (Fig. 3). Le graphe obtenu montre que l’on tend vers un plateau maximum vers 80 %. L’augmentation de la conversion n’est que de 8 % (passant de 70 à 78 %) entre la 24e et la 72e heure ; il nous semble donc qu’un temps de réaction de 48 heures est un bon compromis (taux de conversion 76 %).

Cinétique de la réaction enzymatique entre le fructose et l’acide laurique (X : temps (h), Y : conversion molaire de l’acide laurique (%)).
La courbe peut être ajustée par une corrélation hyperbolique (Éq. (2) avec un écart-type S de 1,51 [19,20]).
| (2) |
La comparaison avec les valeurs trouvées dans la littérature se trouvent au point suivant. Dans tous les cas, nos valeurs sont supérieures à celles de Soultani et Sadeber mais légèrement inférieure à celle de Sin, qui est une transestérification sans production d’eau comme sous-produit.
3.3.2 Influence du solvant
Bien que dans la littérature de nombreux solvants de réaction sont cités (l’acétone [2,8], le 2-méthyl-2-butanol [2,3,7,10,13,14], le tertio-butanol [2,9], l’éthyl-méthyl-cétone [2], le n-hexane [9] et la pyridine [16]), peu de données concernent la conversion de l’acide laurique. À notre connaissance, seuls Soultani et al. annoncent une conversion de 57,5 % dans le 2-méthyl-2-butanol sans donner de temps de réaction [3], Sabeder et al. donnent dans le même solvant un taux de conversion de 55 % après 72 heures [2], Sin et al. donnent un taux de consommation du fructose par transestérification dans la pyridine de 86 % après trois jours [16] (ou 90 % en sept jours [15]) et Arcos et al. indiquent un taux de conversion de 55 ou 100 % en 72 heures dans l’acétone [8].
Dans nos conditions expérimentales, à 48 heures, nous obtenons un taux de conversion de l’acide laurique de 76 % dans le 2-méthyl-2-butanol, de 74 % dans le tertio-butanol et seulement de 12 % dans le n-hexane. Sabeder obtient des taux de conversion proche dans le 2-méthyl-2-butanol et le tertio-butanol pour la synthèse du palmitate de fructose [2]. Comme le 2-méthyl-2-butanol est non toxique et utilisable comme solvant alimentaire capable de solubiliser partiellement les sucres et dont les produits de la réaction ne cristallisent pas immédiatement [2], c’est ce solvant qui fut choisi pour la suite de ce travail.
Dans cette partie, l’analyse de la composition des produits obtenus a montré que dans le 2-méthyl-2-butanol 24,7 % de monolaurate de fructose et 11,0 % de polylaurates ont été trouvés. Une composition similaire a été trouvée dans le tertio-butanol (respectivement 24,3 % et 9,8 % pour le mono et les polylaurates), alors que dans le n-hexane seuls 2,6 % de polylaurates ont été détectés.
3.3.3 Influence de la longueur de chaîne de l’acide
L’influence de la longueur de chaîne de l’acide gras n’a qu’un impact limité sur le taux de conversion. Celui-ci étant respectivement de 76, 73,6 et 73,3 % pour l’acide laurique (C12), l’acide palmitique (C16) et l’acide caprique (C10). Nos résultats sont en accord avec ceux de Scheckermann lequel observe une faible variation du taux de conversion, même s’il note que le taux de conversion est inversement proportionnel avec la longueur de chaîne [14]. En revanche, Soultani obtient de meilleurs taux de conversion des acides gras avec des acides gras à longue chaîne [3]. Ces résultats confirment que la lipase de C. antarctica est plus spécifique avec les acides gras à longue chaîne. Une explication pourrait être que les courtes et moyennes chaînes d’acide gras (C8-C14) sont relativement plus polaires, ce qui entraînerait un bas taux d’élimination de l’eau pendant la synthèse [3].
3.3.4 Influence du ration fructose/acide laurique
En revanche, le ratio molaire fructose:acide laurique a une plus grande influence sur la conversion. En effet, plus la quantité d’acide est élevée moins la conversion est importante, allant de 76 % pour un ratio 1:1 à seulement 33,6 % pour un ratio 1:5 en passant par 59,0 % pour un ratio 1:2. Nos résultas sont en accord avec ceux de Soultani [3]. Se basant sur le taux de conversion du sucre, Arcos [8] dans l’acétone, a trouvé que le taux de conversion est plus élevé dans les cas d’un ratio 1:3 et 1:5 (100 %) que pour un ratio de 1:1 (55 %). Ces résultas sont également en accord avec ceux de Sekeroglu [4]. Dans notre situation, la lipase de C. antarctica est probablement inhibée par le donneur d’acyle ou bien par une limitation de la concentration en fructose dans le milieu, ce qui concorderait avec les travaux de Soultani et al. [3].
3.3.5 Influence de la teneur initiale en eau [28]
Le dernier paramètre qui a été étudié est la quantité initiale d’eau dans le milieu réactionnel. Le pourcentage d’eau a été déterminé par la méthode de Karl Fisher.
Six expériences ont été réalisées : la première en présence de tamis moléculaire, la deuxième en l’absence de tamis moléculaire et enfin en ajoutant 50, 100, 150 et 200 μl d’eau au début de la réaction, ce qui donne, respectivement, 0,02 %, 0,21 %, 0,42 %, 0,61 %, 0,80 % et 0,96 % d’eau dans le milieu après 48 h de réaction (Tableau 1). La teneur en eau influence grandement le taux de conversion, celui-ci passe de 72 % pour 0,02 % d’eau à 4,5 % à 0,96 % d’eau (Fig. 4 et Tableau 1). En l’absence de tamis moléculaire mais sans ajout d’eau (ce qui correspond à 0,21 %), le rendement est alors de 21 %. Chamouleau observe le même phénomène pour la synthèse du palmitate de fructose [6] et Yoo aussi pour l’oléate de xylitol [26]. Il est possible de modéliser la courbe obtenue. Celle-ci suit en effet une loi en puissance à trois paramètres d’Herschel-Bulkley (Éq. (3) et Fig. 4), déterminée par le module Two variables statistic Version 1.31 [20] qui fait parti du programme Gabriel Data Analysis [19].
Taux de conversion et ratio polyester sur monoester en fonction de la teneur en eau.
| Avec tamis moléculaire | Sans tamis moléculaire | Avec de l’eau ajoutée | ||||
| 50 μl | 100 μl | 150 μl | 200 μl | |||
| Teneur en eau (%) | 0,14 ± 0,01a | 0,150 ± 0,005a | 0,27 ± 0,02a | 0,434 ± 0,006a | 0,65 ± 0,04a | 0,87 ± 0,02a |
| 0,02 ± 0,01b | 0,207 ± 0,006b | 0,424 ± 0,009b | 0,608 ± 0,004b | 0,80 ± 0,02b | 0,96 ± 0,02b | |
| Taux de conversion (%)c | 72 ± 6 | 21 ± 3 | 10,1 ± 0,8 | 7,2 ± 0,8 | 5,3 ± 0,7 | 4,5 ± 0,9 |
| Polyester/Monoester (%/%)d | 30,2/69,8 | 2,6/97,4 | 1,4/98,6 | nd/– | nd/– | nd/– |
a Teneur initiale en eau déterminée par la méthode de Karl Fisher. Les résultats sont la moyenne de deux réactions différentes et de six données au total.
b Teneur en eau après 48 h de réaction déterminée par la méthode de Karl Fisher. Les résultats sont la moyenne de deux réactions différentes et de six données au total.
c Basée sur la conversion de l’acide laurique après 48 h. Les résultats sont la moyenne de deux réactions différentes et de quatre données au total.
d Déterminée par HPLC – deux répétitions – voir figure S 3.

Taux de conversion en fonction de la teneur initiale en eau (notre étude et celle de Chamouleau et al. [6]) ou de la quantité d’eau ajoutée (étude de Mutua et Akoh [29]).
En 1993, Akoh [29] publie les résultats de l’incorporation d’acide oléique au cours de la transestérification de glucoside de méthyle par l’oléate de méthyle dans le benzène/pyridine catalysée par la lipase de Candida sp. (SP382) lorsque de l’eau est ajoutée. Plus récemment, les mêmes informations ont été publiées par Chamouleau et al. lors de l’estérification du fructose par l’acide palmitique en 2-méthyl-2-butanol catalysée par la Novozym 435, l’incorporation de l’eau dans le milieu réactionnel étant réalisée par utilisation d’atmosphères de solutions salines saturées [6]. De plus, en 2007, Yoo contrôlait la teneur en eau par l’addition de différentes quantités de tamis moléculaires lors de l’estérification du xylitol par l’acide oléique [26].
La modélisation de courbes de points expérimentaux est importante pour les procès industriels. Pour cela, il a semblé important de modéliser aussi les travaux de Chamouleau et d’Akoh. La modélisation a fait appel au module Two variables statistic Version 1.31 [20] du programme Gabriel Data Analysis [19]. Comme la réaction de Chamouleau est la même que la nôtre (estérification), l’équation de la conversion en fonction de la teneur initiale en eau peut être décrite par la même équation de loi en puissance à trois paramètres (Éq. (4) et Fig. 4).
Comme la réaction d’Akoh est différente, puisqu’il s’agit d’une transestérification, le meilleur modèle correspond à une loi exponentielle donnée à l’Éq. (5) et sur la Fig. 4.
| (3) |
| (4) |
| (5) |
En comparant les coefficients de l’équation de notre étude à ceux déterminés à partir des résultats de l’étude de Chamouleau, il est possible de conclure que le premier coefficient est en relation avec les conditions expérimentales si l’eau n’est pas présente et que le troisième coefficient (l’exposant) correspond au « pouvoir d’action » de l’eau (pour faire décroître le taux de conversion). Dans ce cas, le premier coefficient indique que nos conditions sont meilleures que celles des travaux de Chamouleau, mais le troisième terme montre clairement que la diminution du taux de conversion est plus forte dans nos conditions que dans les conditions de Chamouleau, cela étant dû au fait que nous introduisons l’eau directement dans le milieu réactionnel contrairement à Chamouleau qui agit avec des conditions plus douce par diffusion en utilisant une atmosphère de solutions saturées en sels.
Par ailleurs, l’étude quantitative par HPLC montre que la concentration en produits formés est différente (figure S 3 et Fig. 1) en fonction de la teneur en eau. La synthèse de polyesters diminue de 30,7 % à 1,4 %, respectivement avec des tamis moléculaires jusqu’à l’addition de 50 μl d’eau (0,14 à 0,27 % de teneur en eau initiale). Un résultat similaire dans l’acétone avec de l’acide oléique a été trouvé par Arcos et al. [8]. Selon Chamouleau, la perte de la sélectivité de l’enzyme est due à la réduction de la couche d’hydratation des protéines ; le manque d’eau proche de l’enzyme augmentant l’hydrophobie de l’environnement et donc une diminution de la solubilité du fructose. Dans ces conditions, le monoester devient un meilleur substrat que le fructose [6]. Contrairement à nous, Chamouleau rapporte les cinétiques pour deux points seulement, avec et sans tamis moléculaires, alors que rapportons ici une étude complète de l’addition de plusieurs quantités d’eau (Tableau 1 et figure S 3).
Il est impossible de conclure si le phénomène de diminution de la synthèse des polyesters est le même avec une quantité d’eau ajoutée supérieure à 50 μl. En termes de cinétique, la synthèse des polyesters est le carré de la vitesse de la synthèse du monoester. Or le taux de conversion diminue avec l’ajout d’eau, la détection des polyesters par HPLC n’étant plus assurée (figure S 3).
Dans les mêmes conditions, quelle que soit la quantité d’eau dans la milieu, la lipase de C. antarctica ne produit que des monoesters avec le glucose comme sucre au lieu de fructose (données non montrées).
3.4 Purification des esters
Le mélange réactionnel est d’abord filtré pour récupérer l’enzyme et le tamis moléculaire. Le filtrat est alors dilué par de l’eau, puis extrait par du dichlorométhane. La phase organique est évaporée sous vide et les produits de la synthèse sont repris soit par du chloroforme (méthode 1), soit par du dichlorométane (méthode 2).
La première séparation (méthode 1) a été réalisée par colonne chromatographique préparative, dont le système de solvant est basé sur le chloroforme, d’abord pur, puis un mélange chloroforme/méthanol (90/10, v/v) et pour finir par un mélange chloroforme/méthanol/eau (64/10/1, v/v/v). Les esters obtenus ont alors été utilisés comme témoins pour l’analyse HPLC et ont servi à l’établissement de droite d’étalonnage mais aussi à la caractérisation physicochimique. La pureté des esters a été contrôlée par HPLC (voir Section 3.2.3) et est supérieure dans tous les cas à 99 % (figure S 4). Dans la littérature, plusieurs hypothèses sont données pour expliquer les deux pics visibles sur la figure S 4 (a) dans le cas du monolaurate de fructose : pour Arcos [8], Chamouleau [6] et Soultani [3], il pourrait s’agir soit des anomères α et β, soit d’une acylation en 1 et en 6, tandis que pour Scheckermann [14] et Coulon [30], il s’agirait plutôt des formes furanose et pyranose du fructose.
La seconde version (méthode 2) est basée sur le gradient d’un mélange dichlorométhane et éthanol, passant d’une fraction 19/1 v/v à une fraction 17/3 v/v. Dans ce cas, la pureté des produits fut contrôlée par CCM.
3.5 Résultats sur les propriétés interfaciales
3.5.1 Propriétés de surface
Cette étape est la plus importante pour la caractérisation des propriétés intrinsèques des molécules synthétisées. En effet, celles-ci gouvernent les propriétés technofonctionnelles (propriétés moussantes, émulsionnantes – voir Section 3.5.3). De plus, ce type de mesures met en œuvre de faibles quantités.
Nous avons en premier déterminé la tension de surface du mono- et du polylaurate de fructose à 60 g.L−1. À la lecture du Tableau 2, nous constatons alors que le monolaurate de fructose est plus actif que le polylaurate.
Tension de surface des surfactants synthétisés.
| Tension de surface de solution à 60 g.L−1 (mN.m−1) | Tension de surface de solution non filtrée 1 % (mN.m−1) | Tension de surface de solution filtrée 1 % (mN.m−1) | |
| Monolaurate de fructose | 26,3 ± 0,3 | ||
| Polylaurate de fructose | 32,5 ± 0,2 | 40,8 | 46,7 |
| Monolaurate + polylaurate de fructose | 28,4 | 27,7 |
Dans la littérature, différentes valeurs de concentration micellaire critique (CMC) ont été trouvées pour le laurate de fructose. Il y a parfois un facteur dix entre elles : 2,8 × 10−4 M pour Scheckermann et al. [14] à 2,76 × 10−5 M pour Sin [15] en passant par 7,20 × 10−5 M pour Sekeroglu [4]. Il nous a semblé opportun alors de déterminer cette valeur. Pour le monolaurate de fructose, il a été obtenu une valeur de 8 × 10−5 M (soit 29 mg.L−1) et un γ = 32,18 mN.m−1 (Fig. 5(a)).

(a) détermination de la CMC du monolaurate de fructose, (b) détermination de la CMC du polylaurate de fructose, (c) détermination de la CMC du polylaurate de fructose à partir d’une solution filtrée et diluée, (d) détermination de la CMC sur un mélange réactionnel et (e) détermination de la CMC sur un mélange de monolaurate et polylaurate de fructose 50/50.
La Fig. 5(b) montre qu’il n’est pas possible de déterminer la CMC du polylaurate de fructose. Après d’autres essais, nous nous sommes rendus compte d’une solubilité limitée (vide infra).
Lors de l’étude à 60 g.L−1, il avait été constaté qu’il est difficile de dissoudre complètement le polylaurate de fructose. Afin de voir son influence, une étude sur une solution de polylaurate pur à 1 % a été entreprise aussi bien sur des solutions brutes et sur des solutions filtrées et les résultats sont présentés dans le Tableau 2. Une variation de presque 20 % est constatée entre la solution non filtrée et la solution filtrée et aussi que la nouvelle tension de surface est nettement supérieure à la valeur trouvée dans l’étude à 60 g.L−1 (Tableau 2).
La Fig. 5(c) montre qu’il n’est toujours pas possible, malgré la dilution et la filtration, de déterminer la CMC du polylaurate de fructose.
Un des buts de cette étude est aussi l’utilisation des bruts récationnels à des fins industrielles. Pour cela, le mélange réactionnel, débarrassé du fructose et de l’acide laurique n’ayant pas réagi, a été l’objet de travaux, dont les résultats sont présentés dans le Tableau 2 et sur la Fig. 5(d). Le mélange « naturel » a une CMC de 27 mg.L−1 avec γCMC = 28,55 mN.m−1. En comparant ces résultats avec ceux du monolaurate de fructose, il est constaté que les valeurs obtenues sont proches : comme le polylaurate est peu soluble, les résultats sont dus au monolaurate qui est soluble totalement.
Comme, il peut être intéressant industriellement d’obtenir de nouvelle caractéristique, la détermination de la CMC d’un mélange « artificiel reconstitué » (qui consiste à prendre du monolaurate et du polylaurate de fructose pur et à les mélanger) 50/50 a donné 41 mg.L−1 avec un γCMC = 28,64 mN.m−1 (Fig. 5(e)).
3.5.2 DSC
Pour avoir une meilleure connaissance des molécules mais aussi pour pouvoir éventuellement poser les bases d’une purification par une méthode douce comme la (re)cristallisation, de la DSC (ou ACD) a été utilisée dont les thermogrammes sont présentés sur la Fig. 6.
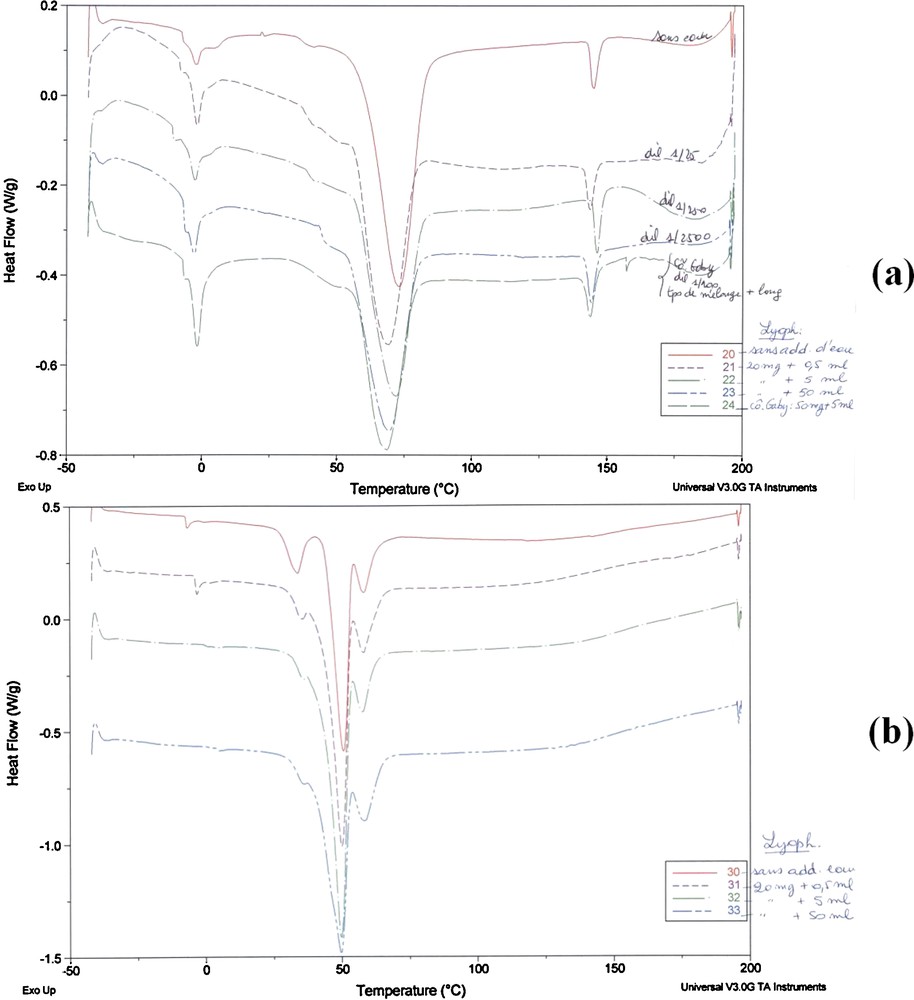
DSC (a) monolaurate de fructofuranosyle, (b) polylaurate de fructofuranosyle.
Le thermogramme obtenu pour le monolaurate présente deux pics exothermiques. Le premier entre 55 et 80 °C correspond vraisemblablement à une transition à l’état de cristaux liquides. Le second peu avant 150 °C correspond à la fusion totale et l’obtention d’un liquide anisotrope.
Le polylaurate ne semble pas présenter d’organisation en cristaux liquides.
Dans les deux cas, les conditions de préparation (quantité d’eau ajoutée avant lyophilisation) ont peu d’impact sur l’allure des thermogrammes mais occasionnent un léger « shift » des valeurs de température.
3.5.3 Propriétés technofonctionnelles
Elles correspondent aux propriétés moussantes et émulsionnantes. Seuls des tests orientatifs ont été réalisés et ont pu mettre en évidence que le laurate de fructose est peu moussant.
Grâce au « Group Number » de Davies [31], nous avons calculé la Balance Hydrophile-Lipophile (HLB). Le HLB calculé pour le laurate de fructose est de 8,8, soit émulsionnant huile dans eau, agent mouillant, légèrement soluble dans l’eau (ni hydrophile, ni lipophile) et peu moussant.
4 Conclusion
Grâce à la littérature, nous savions que la synthèse enzymatique à partir du fructose était possible. Nous avons néanmoins repris un certain nombre de paramètres afin d’optimiser les conditions opératoires, cela dans une démarche plus industrielle. Nos conditions expérimentales sont maintenant supérieures à celles de la littérature et nos rendements en esters le montrent : nous obtenons 78 % de laurate de fructose où le meilleur de la littérature (Soultani) annonce 57 %. De plus, nous avons passé en revue différents paramètres tels que le temps de réaction, l’influence de la longueur de chaîne de l’acide gras, le solvant, etc. et la réaction est bien maîtrisée maintenant.
De nombreuses difficultés d’ordre analytique ont été résolues et nous avons développé des méthodes rapides (basées sur différentes conditions CCM en fonction des informations désirées), des méthodes précises (basées sur l’HPLC) et plusieurs méthodes de purification.
Remerciements
Ce travail a reçu le soutien financier de la Direction générale de l’agriculture de la région Wallonne (no D31/1143). Ce travail est dédié à M. Benoit Dispa, Bourgmestre de Gembloux.
1 En accord avec Sekeroglu, un tensioactifs ou surfactant est une molécule amphiphile qui tend à diminuer les tensions interfaciales aux interphases [1].
2 Les spots CCM identifiés par une flèche sur la Fig. 2 seront nommés par la suite « polylaurates ». Il est en fait impossible d’identifier s’il s’agit de différents conformères du dilaurate ou bien de tri-, tétra-esters sans analyses complémentaires poussées.