1 Introduction
Les strigolactones sont des hormones végétales dérivées du métabolisme des caroténoïdes [1]. Leur nom est lié à la découverte, au milieu des années 1960, de leur action dans l’induction de la germination des graines de Striga. Si ces composés sont présents dans environ 80 % des plantes terrestres, leur véritable étude ne s’est développée que très récemment, en raison des quantités infimes présentes dans les plantes et des difficultés d’isolement et de caractérisation. Depuis, cette découverte suscite un engouement extrêmement important et de nombreux travaux concernant l’étude de leur mode d’action sont apparus dans la littérature [2].
On reconnaît deux fonctions essentielles aux strigolactones. Ces hormones sont impliquées dans la germination de certaines plantes. En effet, elles provoquent la médiation chimique dans les interactions entre plantes et leurs champignons mycorhiziens. Ces derniers sont guidés vers les racines de la plante et stimulés dans leur croissance, avec pour conséquence une augmentation des chances de mycorhization rapide. Il y a alors création d’une association symbiotique entre la plante et le champignon, les deux espèces bénéficiant de l’augmentation globale de la surface racinaire.
Les strigolactones sont également connues pour contrôler la ramification des plantes. Ce type d’hormone agit en inhibant la croissance des bourgeons situés à l’aisselle des feuilles. Les plantes mutantes qui n’en produisent pas sont « hyper-ramifiées ».
Du point de vue de la structure, les strigolactones sont constituées d’un noyau tricyclique octahydroindénofuranone ABC et d’un cycle buténolide D lié au tricycle par un bras méthylèneoxy (Schéma 1). En raison de ces faibles quantités présentes naturellement dans les plantes et de la synthèse assez délicate de ces composés [3], les préparations d’analogues simplifiés se sont multipliées au cours des dernières années [4]. On peut par exemple citer le GR24 [5], analogue de synthèse possédant un cycle A aromatique, qui est particulièrement intéressant car il est extrêmement actif et par conséquent couramment utilisé comme référence dans des tests de germination [6].
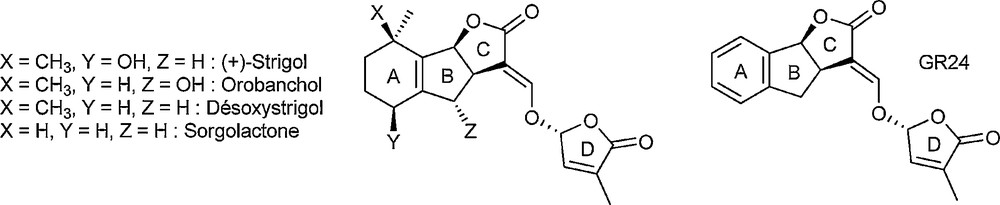
Strigolactones naturelles et analogue GR24.
Plusieurs voies de synthèse multi-étapes de strigolactones ou d’analogues, basées sur la préparation du noyau tricyclique ABC, puis la mise en place subséquente du bras méthylèneoxy et du buténolide D, ont été décrites dans la littérature [7–9]. Néanmoins, plusieurs de ces études ont montré que la mise en place du cycle D ne posait pas de problème particulier. Il apparait donc évident que l’une des difficultés principales est la préparation rapide et efficace du noyau tricyclique ABC. Dans ce cadre, il n’existait pas, à notre connaissance, de voie de synthèse domino du squelette ABC de strigolactones ou d’analogues à partir de réactifs simples et/ou commerciaux.
Ces dernières années, notre groupe s’est tourné vers le développement de réactions multicomposants impliquant des réactifs organométalliques générés in situ, [10] en particulier par l’intermédiaire du système Zn(0)/Co(II) [11]. Il a ainsi été montré, en particulier, que des γ-butyrolactones multi-substituées pouvaient être obtenues par une réaction domino [12] addition conjuguée/aldolisation/lactonisation entre des halogénures aromatiques, des dérivés carbonylés, et l’itaconate de diméthyle [10c]. Dans la continuité de ces travaux, nous présentons ici une voie de synthèse du noyau tricyclique ABC des strigolactones et d’analogues tels que le GR24 par réaction domino entre dérivés carbonylés ortho-halogénés et l’itaconate de diméthyle.
2 Résultats et discussion
Si l’on regarde la préparation de l’analogue GR24 en série racémique, plusieurs voies faisant appel à la préparation commune de l’indanone substituée 1 ont été publiées, en particulier par Zwanenburg et al. (Schéma 2) [7].
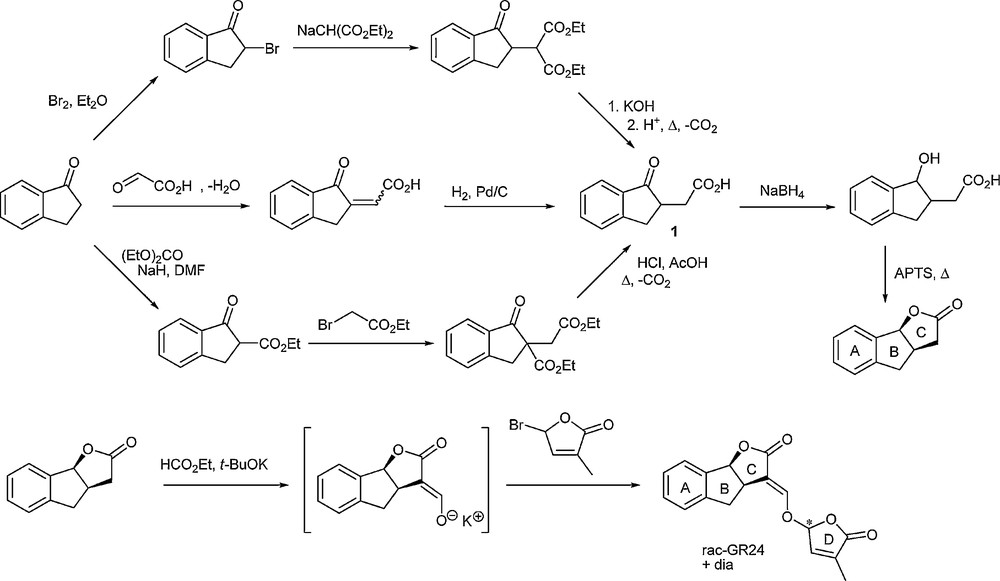
Synthèses multi-étapes du GR24.
Une fois formé par des méthodes classiques, cet intermédiaire est réduit puis cyclisé pour donner le motif tricyclique ABC du GR24. Par la suite, le cycle D est ajouté par formation d’un énolate intermédiaire et piégeage par un bromobuténolide. Les diastéréoisomères, épimères au niveau du cycle D, sont ensuite séparés par chromatographie. D’un point de vue synthétique, il apparaît que ce bras n’est pas très compliqué à mettre en place. En effet, il peut être introduit par déprotonation en α du carbonyle lactonique et piégeage de l’énolate par du formiate d’éthyle. Le nouvel énolate alors formé s’additionne sur la bromofuranone correctement substituée pour donner le composé final attendu.
Zwanenburg et al. ont publié récemment une autre voie de synthèse passant par une condensation de Stobbe, une hydrogénation catalytique, une formation de chlorure d’acyle et une réaction de Friedel et Crafts afin de former l’indanone substituée 1 sous sa forme ester, qui mène au noyau tricyclique ABC dans les réactions décrites précédemment [8].
Une voie de synthèse très élégante du GR24 a été publiée très récemment par De Mesmaeker et al. (Schéma 3) [9].
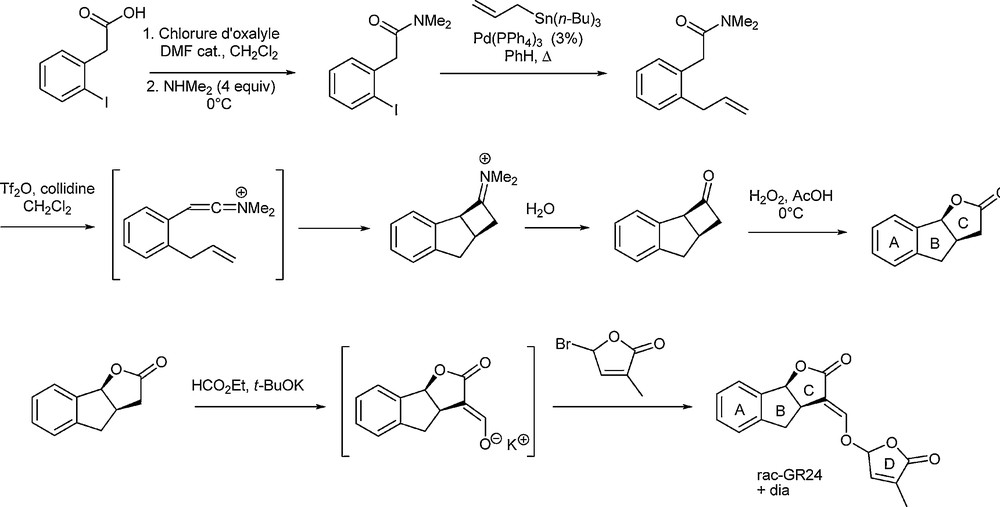
Synthèse du GR24 par De Mesmaeker et al.
Elle met en jeu la formation de cétène iminium permettant d’accéder à une cyclobutanone tricyclique qui subit une réaction de Baeyer–Villiger afin de former le cycle lactonique. La suite de la synthèse est classique avec la mise en place du cycle buténolide D. Un avantage important de cette méthode est l’utilisation possible d’amine chirale lors de la formation de la cyclobutanone afin d’obtenir une induction asymétrique qui permet, au final, de récupérer un seul des énantiomères optiquement pur.
Au travers de ces trois exemples, et compte- tenu de la facilité d’accès au bras méthylèneoxybuténolide, on se rend compte que l’un des challenges, au-delà du contrôle de la stéréochimie, est donc la préparation rapide, efficace, et permettant une diversification fonctionnelle du noyau tricyclique ABC.
Au cours de travaux précédents effectués au laboratoire, nous avions montré qu’il était possible de préparer des γ-butyrolactones multi-substituées par réaction multicomposants entre un dérivé carbonylé, un bromure aromatique et l’itaconate de diméthyle (Schéma 4).
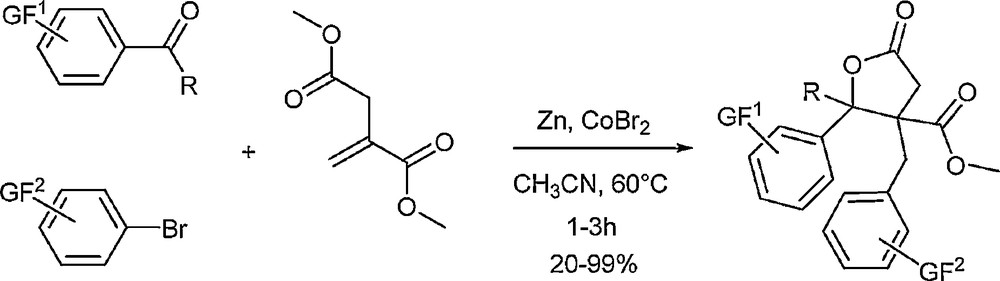
Synthèse multicomposants de γ-butyrolactones multi-substituées.
Le mécanisme réactionnel présumé de cette réaction implique un processus domino addition conjuguée/aldolisation/lactonisation qui permet la création de trois liaisons carbone-carbone en une seule étape expérimentale. Lorsque l’on regarde la structure des composés formés, il est possible d’imaginer une « fusion » des deux cycles aromatiques afin d’avoir accès lactones tricycliques. Une analyse rétrosynthétique conduit à envisager l’utilisation de substrats carbonylés ortho-halogénés afin d’avoir accès au noyau tricyclique ABC du GR24, celui-ci pouvant être, en outre, habillé avec une chaîne R éventuelle, une fonctionnalité sur le cycle benzénique et une fonction ester supplémentaire en jonction de cycle (Schéma 5).
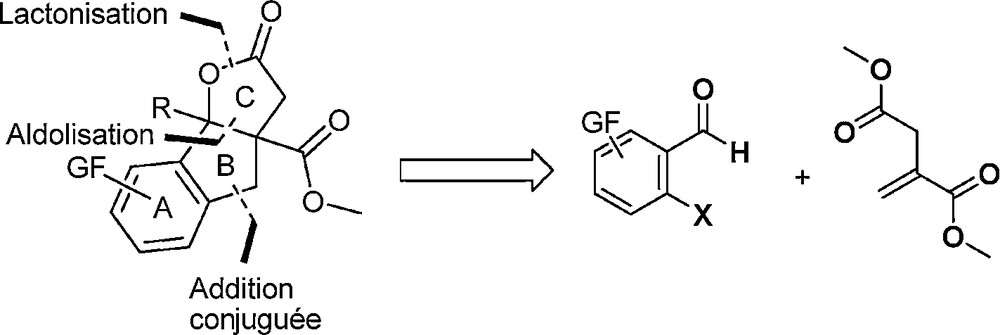
Analyse rétrosynthétique.
La stratégie de synthèse a donc été la suivante : en utilisant un aldéhyde aromatique halogéné en ortho et l’itaconate de diméthyle, l’addition conjuguée de l’organométallique correspondant sur l’itaconate permettrait la formation d’un énolate qui s’additionnerait sur l’aldéhyde. L’alcoolate formé réagirait ensuite sur la deuxième fonction ester pour permettre la formation de la lactone. Malgré les résultats obtenus lors des synthèses multicomposants de lactones, un doute subsistait malgré tout sur la faisabilité de cette approche. En effet, des travaux antérieurs par Cho et al. avaient montré que les substrats utilisés menaient à des composés totalement différents lorsque la réaction était catalysée au palladium. En effet, dans ce cas, des naphtalènes ortho-disubstitués étaient obtenus par réaction tandem Heck–Aldol [13].
Un essai préliminaire, réalisé dans les conditions utilisées pour la synthèse multicomposants de lactones [10c] a rapidement confirmé que le système réactionnel permettait bien la formation de lactones tricycliques. Nous avons donc, dans un premier temps, tenté de mettre au point les conditions réactionnelles optimales. Ainsi, nous avons pu montrer au cours de ces essais que les meilleures conditions étaient les suivantes : le zinc est activé par l’utilisation conjointe d’une faible quantité d’acide trifluoroacétique et de 1,2-dibromoéthane, la quantité de cobalt est de 8 % molaire, l’itaconate de diméthyle est utilisé en excès (deux équivalents), la réaction est conduite à 60 °C pendant une à trois heures. Enfin, le milieu réactionnel n’est pas hydrolysé mais simplement filtré sur célite avant chromatographie afin de préserver la lactone formée d’une dégradation éventuelle.
Après cette phase de mise au point, nous avons testé des benzaldéhydes ortho-halogénés ou -pseudo-halogénés dans la réaction (Tableau 1).
Influence de l’halogénure ou du pseudo-halogénure sur la réactiona.
| Entrée | X | Rendement (%)b |
| 1 | Br | 99 |
| 2 | I | 87 |
| 3 | I | 73c |
| 4 | Cl | 20 |
| 5 | OTf | 69d |
a Les expériences ont été typiquement réalisées à 60 °C avec 10 mL d’acétonitrile, 3,95 g (25 mmol) d’itaconate de diméthyle, 12,5 mmol du benzaldéhyde ortho-substitué, 1,50 g (23 mmol) de poudre de zinc et 0,23 g (1 mmol) de CoBr2.
b Rendement isolé.
c Réaction conduite à température ambiante.
d 2 mmol de CoBr2 ont été utilisées.
Ces expériences ont montré que la réaction fonctionne avec divers halogénures aromatiques comme les bromures (Tableau 1, entrée 1), les iodures, à chaud (Tableau 1, entrée 2) ou à température ambiante (Tableau 1, entrée 3), voire même les chlorures mais avec des rendements plus limités (Tableau 1, entrée 4). Des pseudo-halogénures comme les triflates aromatiques fonctionnent également dans la réaction (Tableau 1, entrée 5) et constituent une alternative à l’utilisation d’halogénures aromatiques. Néanmoins, les bromures aromatiques constituant les meilleurs compromis entre rendement, disponibilité commerciale et coût, nous avons ensuite évalué la portée des bromoaldéhydes aromatiques dans la réaction (Schéma 6).
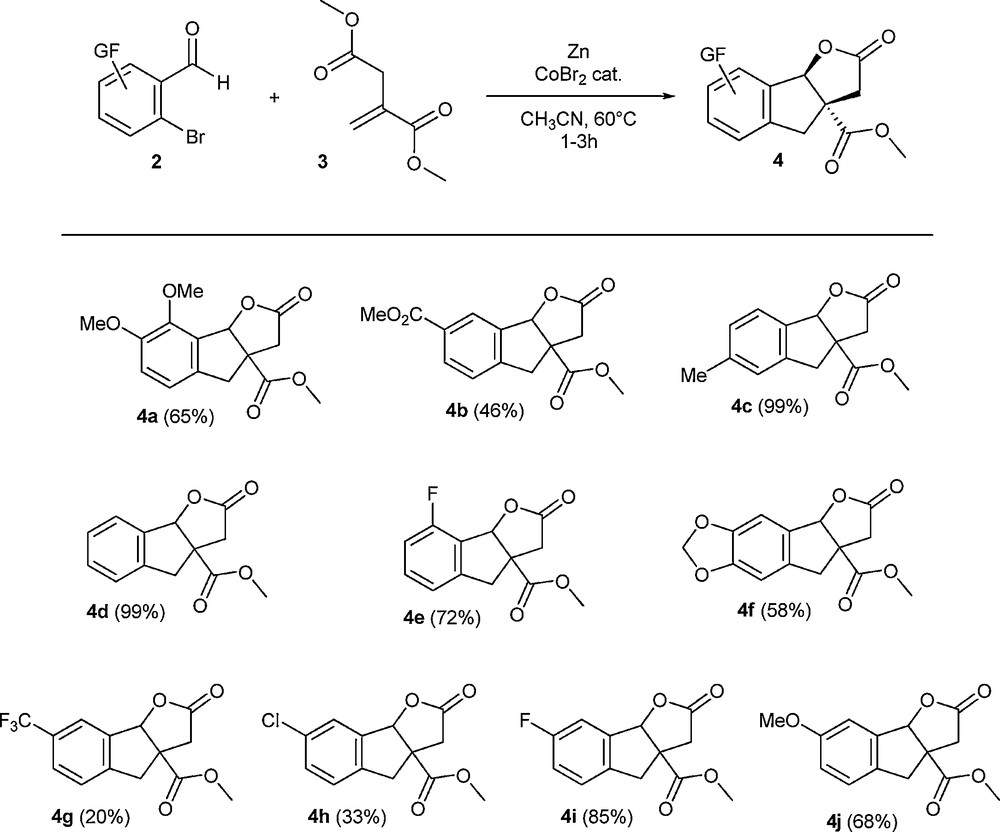
Réaction de l’itaconate de diméthyle avec des bromoaldéhydes aromatiques substitués. Les rendements indiqués sont des rendements en produit isolé. Les expériences ont été typiquement effectuées à 60 °C avec 10 mL d’acétonitrile, 3,95 g (25 mmol) d’itaconate de diméthyle, 12,5 mmol d’ortho-bromobenzaldéhyde, 1,50 g (23 mmol) de poudre de zinc et 0,23 g (1 mmol) de dibromure de cobalt.
Les résultats présentés indiquent que la réaction fonctionne avec plusieurs types de substituants puisque des groupements électrodonneurs ou électroattracteurs peuvent être utilisés. Néanmoins, des résultats curieux ont été observés avec un groupement trifluorométhyle en position 5 (formation de 4g), avec un rendement limité à 20 % sans qu’une explication évidente puisse être trouvée. En revanche, le rendement de seulement 33 % observé avec le composé chloré en position 5 (formation de 4h) provient d’une métallation additionnelle du cycle aromatique au niveau de la liaison carbone-chlore, ce qui mène à la perte d’une partie du produit final lors du traitement.
Nous avons ensuite cherché à déterminer si une extension aux cétones ortho-halogénées était envisageable. Dans ce but, nous avons réalisé un essai préliminaire à partir de 2-chloroacétophénone 2k et d’itaconate de diméthyle 3 (Schéma 7). Après trois heures de réaction, le produit de couplage 4k est obtenu avec un rendement isolé de 54 %, ce qui constitue un résultat intéressant, permettant d’envisager des travaux additionnels à l’avenir.
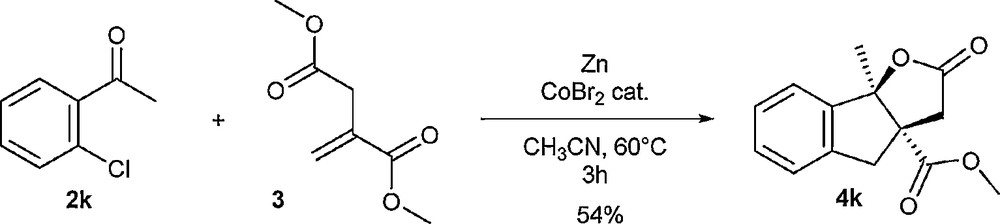
Réaction de la 2-chloroacétophénone avec l’itaconate de diméthyle.
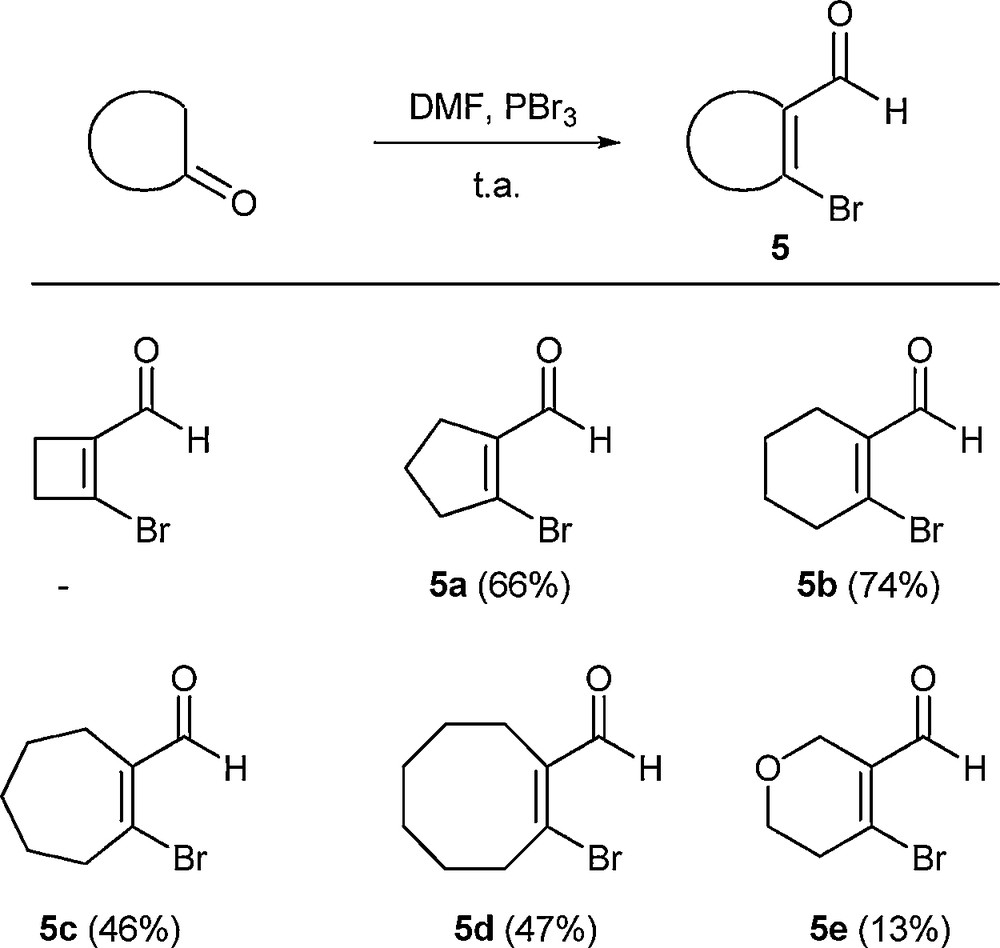
Dans la mesure où toutes les strigolactones naturelles possèdent un noyau A cyclohexénique, nous avons ensuite tenté d’utiliser des bromoaldéhydes vinyliques cycliques dans la réaction. Nous avons, bien entendu, considéré les composés cycliques à six chaînons, mais également envisagé d’étendre la réaction à des composés vinyliques de tailles différentes afin d’étudier leur réactivité. Pour la préparation des substrats vinyliques, plusieurs voies avaient été initialement testées. Néanmoins, les meilleurs résultats ont été obtenus en utilisant une réaction de Vilsmeier–Haack à partir des cétones cycliques correspondantes [14]. Ainsi, différentes cétones ont été soumises à l’action de PBr3 et de DMF [15] (Schéma 8) et nous avons pu obtenir les composés attendus avec des rendements qui sont malheureusement restés généralement limités. Cela est vraisemblablement dû à la faible stabilité des composés formés, qui pour l’un d’entre eux, s’est dégradé de manière violente1 après isolement.
En conséquence, bien que les réactions sur les bromoaldéhydes aromatiques aient été réalisées à 60 °C, nous avons choisi, pour ces essais, de rester à température ambiante afin de limiter la dégradation de ces substrats dans le milieu réactionnel. Les essais de couplage réalisés ont montré que la réaction était possible avec certains bromoaldéhydes vinyliques, bien que des rendements moins élevés soient obtenus (Schéma 9).
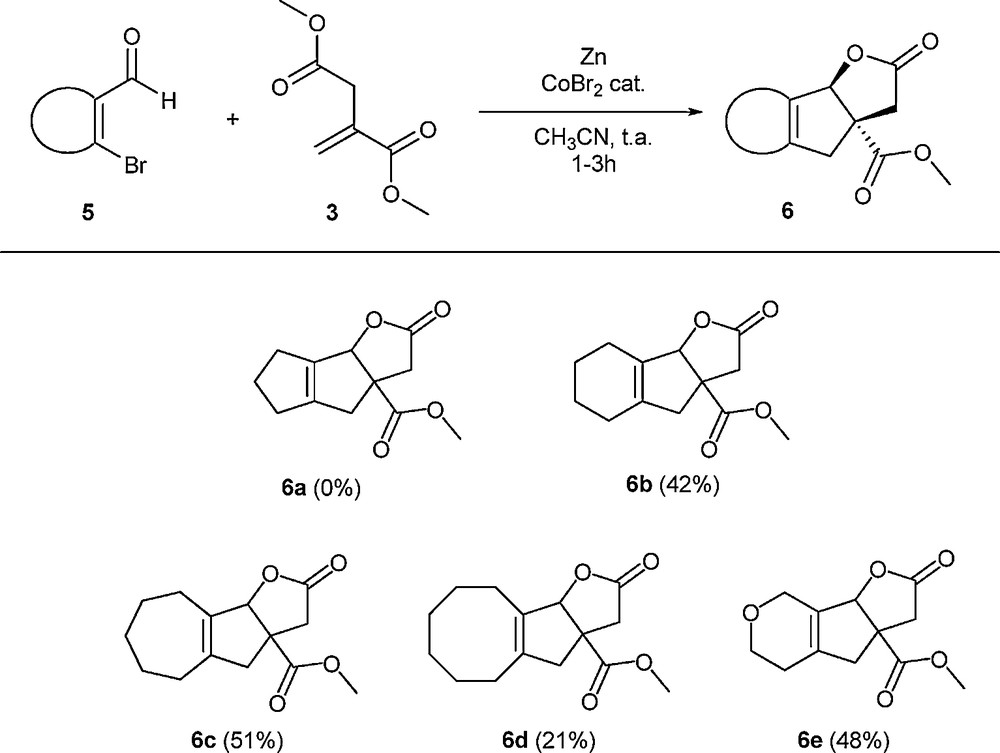
Réaction de l’itaconate de diméthyle avec des aldéhydes vinyliques bromés. Les rendements indiqués sont des rendements en produit isolé. Les expériences ont été typiquement effectuées à température ambiante avec 5 mL d’acétonitrile, 4,74 g (30 mmol) d’itaconate de diméthyle, 6 mmol de composé vinylique disubstitué 5, 0,75 g (11,5 mmol) de poudre de zinc et 0,11 g (0,5 mmol) de dibromure de cobalt.
Dans le cas de l’essai réalisé à partir du composé vinylique à cinq chaînons 5a, nous avons attribué l’absence de réaction à la dégradation trop rapide du composé de départ. Dans les autres cas, nous ne savons pas si l’instabilité des substrats limite les rendements ou si l’obligation de travailler à température ambiante génère un manque de réactivité de la part de ceux-ci.
En ce qui concerne le mécanisme de la réaction, nous envisageons, en relation avec des travaux précédents [16], qu’un organocobalt soit l’organométallique impliqué dans l’étape d’addition conjuguée et le processus en cascade qui suit (Schéma 10).
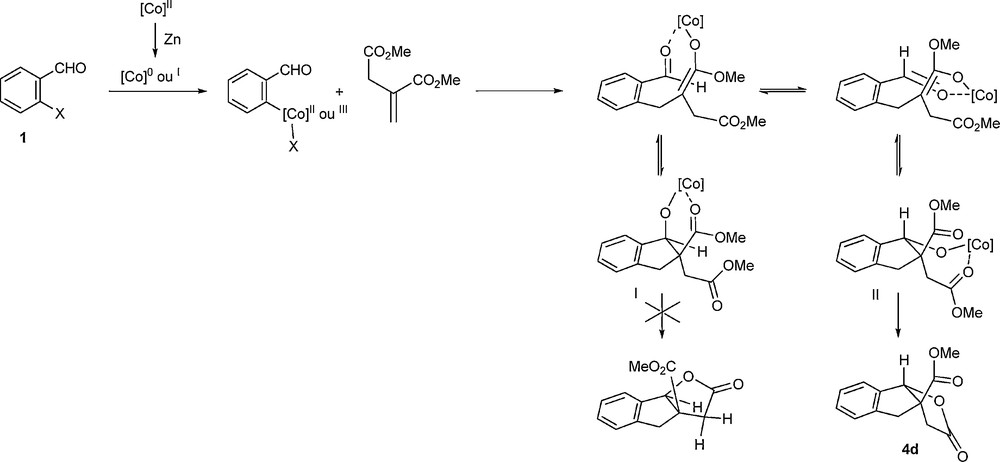
Mécanisme réactionnel possible.
Ainsi, le cobalt(II), après réduction par le zinc métallique en cobalt(0), s’insèrerait dans la liaison C–Br de l’halogénure organique et c’est l’intermédiaire organométallique correspondant qui réaliserait l’addition conjuguée sur l’accepteur de Michael. Les énolates Z ou E formés s’additionneraient sur l’aldéhyde pour donner des alcoolates qui ne permettraient la cyclisation que dans le cas de l’alcoolate II, menant à la formation de la lactone à jonction de cycle cis. Cela est d’ailleurs le cas général dans ce type de cyclisations à cinq chaînons.
Compte tenu de la présence sur le noyau tricyclique ABC d’une fonction ester supplémentaire en comparaison des strigolactones ou de leurs analogues, nous avons tenté la décarboxyméthylation par réaction de Krapcho [17], après oxydation sélective en position benzylique (Schéma 11). La fonction carbonyle supplémentaire en position benzylique pouvant ensuite être potentiellement réduite partiellement ou totalement pour avoir accès à des structures apparentées aux strigolactones naturelles.
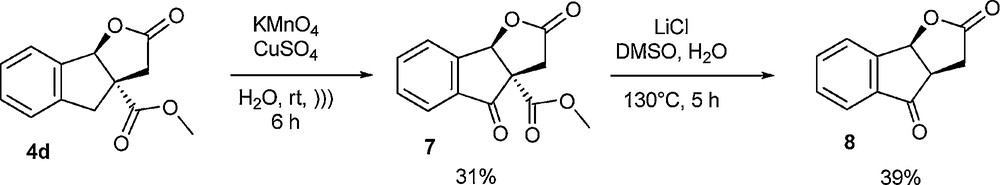
Essai de décarboxyméthylation après oxydation benzylique.
Les résultats préliminaires se sont avérés intéressants, bien que non optimisés. En effet, l’oxydation s’est montrée très sélective et a pu être menée dans des conditions douces, selon une méthode inédite. En effet, les conditions hétérogènes décrites dans la littérature [18] n’avaient pas donné les résultats escomptés en termes de rendement et de rapidité de réaction et nous avons été amenés à développer une variante des oxydations classiques, cette fois en milieu aqueux. La réaction de Krapcho subséquente s’est montrée relativement efficace, et contrairement à ce que nous redoutions, l’ouverture du cycle lactonique n’a pas été observée à la température fixée. Néanmoins, une étude portant sur la modification de la température du milieu réactionnel s’avérerait nécessaire pour tenter d’optimiser cette étape, dans la mesure ou il s’agirait de trouver un bon compromis entre temps de réaction et absence de dégradation du cycle lactonique.
3 Conclusion
En conclusion, nous avons pu montrer au cours de ce travail que le système Zn(0)/Co(II) constitue un outil intéressant pour la synthèse domino de quelques lactones tricycliques originales, possédant le noyau tricyclique ABC des strigolactones et d’analogues, dont le GR24. Des perspectives possibles seraient les suivantes : il pourrait être envisagé :
- • d’ajouter le cycle buténolide (cycle D) ;
- • de réaliser des tests biologiques afin d’évaluer l’importance du CO2Me sur l’activité germinatrice ;
- • de préparer des analogues azotés de ces molécules et/ou d’augmenter la taille du cycle lactonique en partant d’accepteurs de Michael homologues.
C’est l’objet d’un travail qui est actuellement en cours et qui sera rapporté dès que possible.
4 Partie expérimentale
4.1 Conditions générales
Toutes les réactions ont été effectuées sous atmosphère d’argon. Les réactif et solvants commerciaux ont été utilisés sans purification supplémentaire. Toutes les réactions ont été suivies par chromatographie en phase gazeuse (CPG) en utilisant une colonne BP1 de 5 m. Les points de fusion (PF) ont été déterminés sur un appareil de fusion capillaire et ne sont pas corrigés. Les spectres infrarouges ont été enregistrés sur un spectromètre infra rouge à transformée de Fourrier en mode ATR. Les spectres RMN ont été réalisés dans CDCl3 ou C3D6O, à 400 MHz (1H), 100 MHz (13C) et 376 MHz (19F). Les déplacements chimiques (δ) sont donnés en parties par million (ppm) par rapport au signal du solvant non deutéré résiduel. Les valeurs des constants de couplage (J) sont données en Hertz (Hz). Les multiplicités apparentes sont notées comme suit : s (singlet) ; br s (singlet large) ; d (doublet) ; t (triplet) ; q (quadruplet) ; m (multiplet) ; dd (doublet de doublets). La connectivité des atomes est établie grâce à l’analyse des spectres COSY, DEPT, HMBC et HSQC. Les chromatographies flash ont été réalisées sur silice (particules de 35 à 70 μm, 550 mesh). Les spectres de masse ont été obtenus sur un appareil CPG/SM en mode ESI+.
4.2 Synthèse des bromoaldéhydes aromatiques substitués
Le 2-iodobenzaldéhyde a été préparé en une étape à partir d’alcool 2-iodobenzylique selon un mode opératoire décrit par Corey et al. [19].
Le 2-formylphényl trifluorométhanesulfonate a été préparé en une étape à partir de 2-hydroxybenzaldéhyde selon un mode opératoire décrit par Frantz et al. [20].
Le 2-bromo-5-chlorobenzaldéhyde a été préparé en deux étapes à partir d’acide 2-bromo-5-chlorobenzoïque. L’acide 2-bromo-5-chlorobenzoïque a été réduit selon un mode opératoire décrit par Ray et al. [21] pour obtenir l’alcool 2-bromo-5-chlorobenzylique qui a été oxydé selon un mode opératoire décrit par Corey et al. pour obtenir le 2-bromo-5-chlorobenzaldéhyde.
Le 4-bromo-3-formylbenzoate de méthyle a été préparé en deux étapes à partir de 4-bromo-3-méthylbenzoate de méthyle selon un mode opératoire décrit par Burrows et al. [22].
4.3 Synthèse des bromoaldéhydes vinyliques
Les bromoaldéhydes vinyliques 5 ont été préparés selon un mode opératoire décrit par Bekele et al. [15] et utilisés sans purification supplémentaire, en raison de l’instabilité de ces composés.
4.4 Modes opératoires pour la réaction domino
4.4.1 Mode opératoire général pour les substrats aromatiques
De l’acétonitrile (10 mL) est ajouté à un ballon sec de 50 mL sous courant d’argon. Du dodécane (0,1 mL), de la poudre de zinc (1,5 g, 23 mmol, 1,8 equiv), de l’itaconate de diméthyle (3,55 mL, 25 mmol, 2 equiv) et le dérivé carbonylé disubstitué (12,5 mmol, 1 equiv) sont ajoutés sous agitation. Du dibromure de cobalt (0,22 g, 1 mmol, 8 %), de l’acide trifluoroacétique (0,05 mL) et du 1,2-dibromoéthane (0,1 mL) sont ajoutés au mélange réactionnel porté à 60 °C et agité jusqu’à la consommation totale de l’aldéhyde halogéné (suivi par CPG). Le mélange réactionnel est alors filtré sur Célite. La Célite est lavée plusieurs fois à l’éther diéthylique et les fractions organiques combinées sont concentrées sous vide. Le résidu est purifié par chromatographie flash sur silice en utilisant comme éluant un mélange pentane/éther diéthylique de (1:0) à (0:1) pour obtenir la lactone tricyclique, en tant que diastéréoisomère cis.
4.4.2 Mode opératoire général pour les substrats vinyliques
De l’acétonitrile (5 mL) est ajouté à un ballon sec de 25 mL sous courant d’argon. Du dodécane (0,05 mL), de la poudre de zinc (0,75 g, 11,5 mmol, 1,8 equiv), l’itaconate de diméthyle (4,27 mL, 30 mmol, 5 equiv) et l’aldéhyde ortho-halogéné (6 mmol, 1 equiv) sont ajoutés sous agitation. Du dibromure de cobalt (0,11 g, 0,5 mmol, 8 %), de l’acide trifluoroacétique (0,025 mL) et du 1,2-dibromoéthane (0,05 mL) sont ajoutés au mélange réactionnel à température ambiante et agité jusqu’à la consommation totale de l’aldéhyde halogéné (suivi par CPG). Le mélange réactionnel est alors filtré sur Célite. La Célite est lavée plusieurs fois à l’éther diéthylique et les fractions organiques combinées sont concentrées sous vide. Le résidu est purifié par chromatographie flash sur silice en utilisant comme éluant un mélange pentane/éther diéthylique de (1:0) à (0:1) pour obtenir la lactone tricyclique, en tant que diastéréoisomère cis.
4.5 Données analytiques
6,7-Diméthoxy-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4a)
Préparé selon le mode opératoire général, à partir de 6-bromovératraldéhyde (2,45 g, 12,5 mmol). Le composé (4a) est obtenu sous la forme d’une huile brune (1,89 g, 65 %).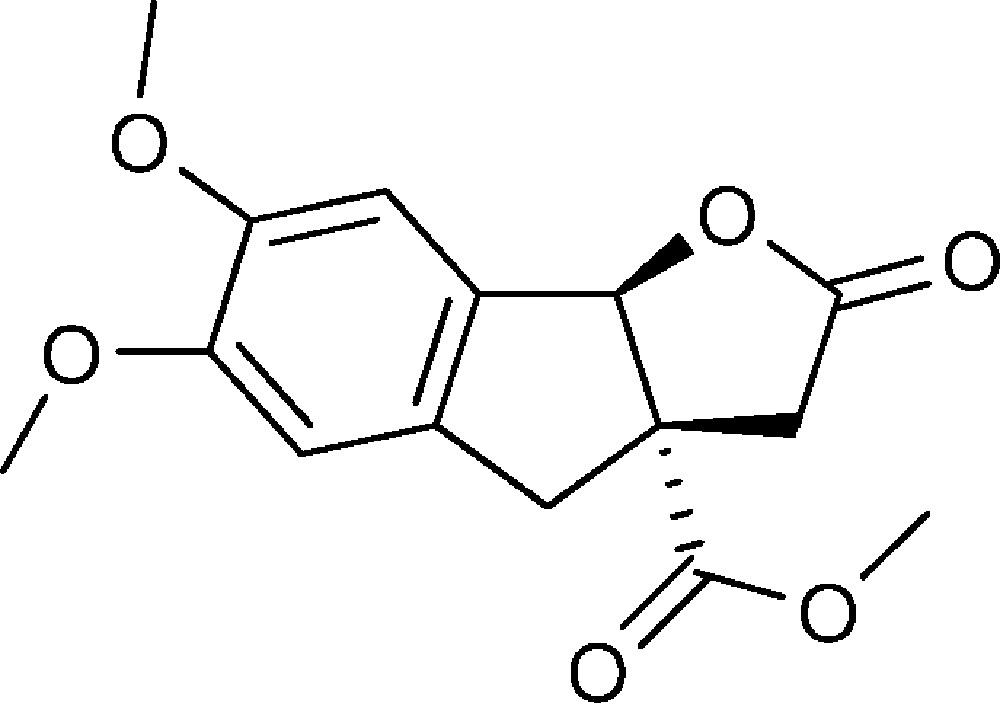
RMN 1H (CDCl3) : δ = 2,59 (1H, d, J 18,2), 3,05 (1H, d, J 16,2), 3,30 (1H, d, J 18,2), 3,61 (1H, d, J 16,2), 3,79 (3H, s), 3,86 (6H, s), 5,98 (1H, s), 6,70 (1H, s), 6,89 (1H, s) ;
RMN 13C (CDCl3) : δ = 39,2, 41,6, 53,2, 55,4, 56,0, 90,2, 107,1, 107,9, 129,3, 132,7, 149,5, 151,3, 173,6, 175,0 ;
IR : ν = 641, 703, 870, 983, 1219, 1267, 1334, 1506, 1613, 1719, 2361, 3396 cm−1 ;
SM (IE) : m/z = 293 (13), 292 (80), 248 (19), 233 (10), 232 (7), 215 (8), 205 (7), 190 (17), 189 (100), 188 (6), 175 (6), 174 (24), 158 (7), 145 (10), 115 (9) ;
m/z calculé pour C15H16O6 (M+1,0078) : 293,1020 ; trouvé : 293,1023.
2-Oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a,6-dicarboxylate de diméthyle (4b)
Préparé selon le mode opératoire général, à partir de 4-bromo-3-formyl benzoate de méthyle (0,94 g, 4 mmol). Le composé (4b) est obtenu sous la forme d’une huile (0,53 g, 46 %).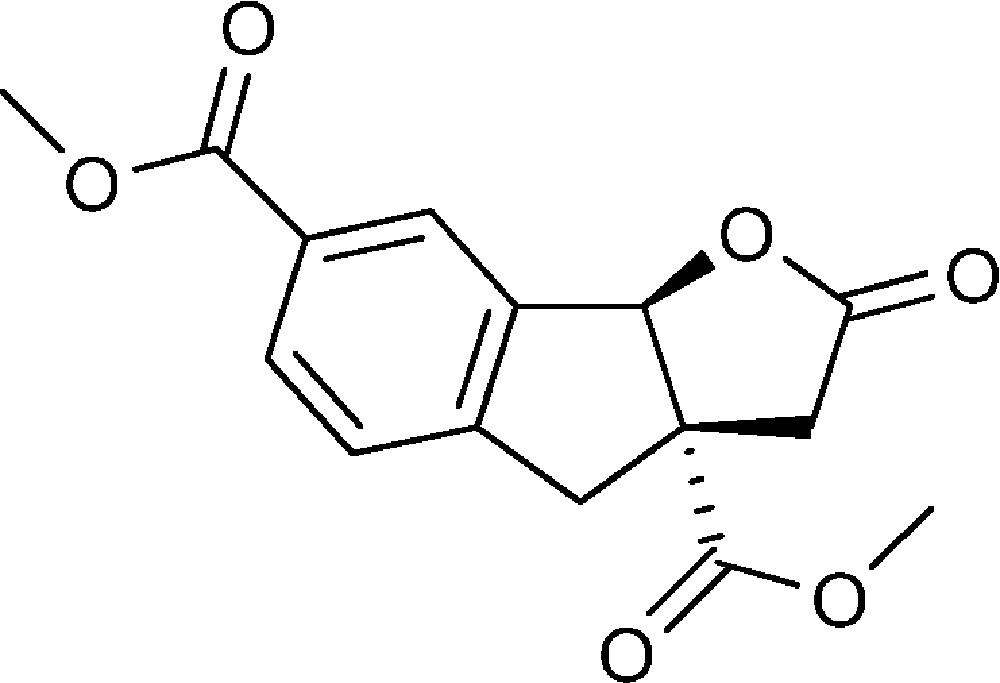
RMN 1H (CDCl3) : δ = 2,57 (1H, d, J 18,3), 3,17 (1H, d, J 17,3), 3,31 (1H, d, J 18,3), 3,74 (1H, d, J 17,3), 3,80 (3H, s), 3,90 (3H, s), 6,02 (1H, s), 7,33 (1H, d, J 7,3), 8,05 (1H, d, J 7,3), 8,12 (1H, s) ;
RMN 13C (CDCl3) : δ = 30,9, 38,7, 41,5, 52,3, 53,3, 55,2, 88,9, 125,3, 127,8, 131,8, 138,1, 166,3, 173,1, 174,2 ;
IR : ν = 696, 758, 1016, 1172, 1198, 1276, 1435, 1715, 2954, 3402 cm−1 ;
SM (IE) : m/z = 272 (59), 259 (41), 258 (44), 248 (38), 245 (19), 244 (100), 231 (22), 230 (97), 216 (17), 203 (25), 188 (45), 187 (34), 186 (18), 171 (25), 157 (22), 155 (18), 143 (20), 128 (40), 127 (20) ;
m/z calculé pour C15H13O6 (M-1,0078) : 289,0712 ; trouvé : 289,0726.
6-Méthyl-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4c)
Préparé selon le mode opératoire général, à partir de 4-méthyl-2-bromobenzaldéhyde (0,61 g, 3,1 mmol). Le composé (4c) est obtenu sous la forme d’une huile orange (0,88 g, 99 %).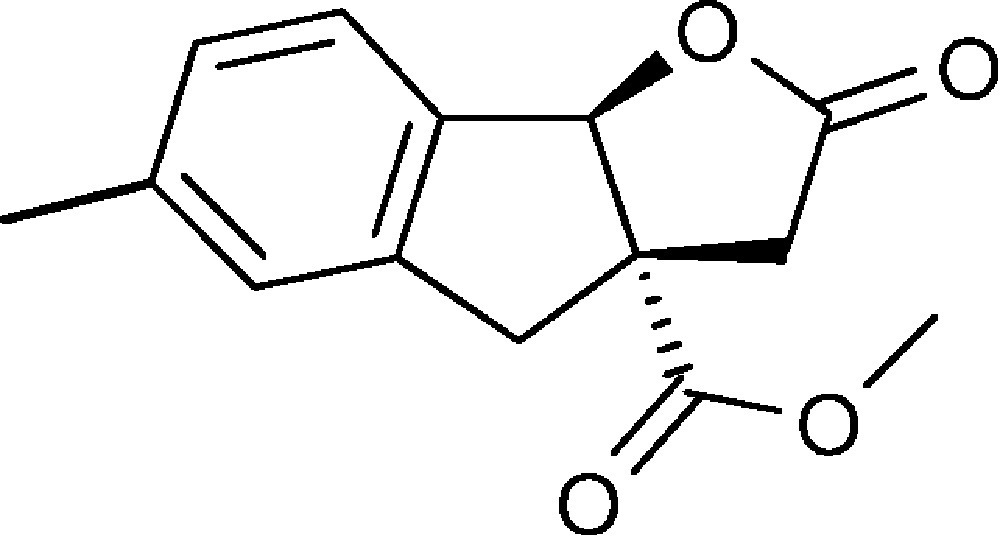
RMN 1H (CDCl3) : δ = 2,34 (3H, s), 2,63 (1H, d, J 18,3), 3,09 (1H, d, J 16,7), 3,35 (1H, d, J 18,3 Hz), 3,63 (1H, d, J 16,7), 3,78 (3H, s), 6,04 (1H, s), 7,06 (1H, s), 7,11 (1H, d, J 7,9), 7,32 (1H, d, J 7,8) ;
RMN 13C (CDCl3) : δ = 21,5, 39,3, 41,4, 53,3, 55,2, 90,4, 125,7, 126,0, 129,1, 134,5, 140,7, 140,9, 173,5, 176,3 ;
IR : ν = 689, 810, 1005, 1180, 1200, 1234, 1437, 1613, 1722 cm−1 ;
SM (IE) : m/z = 228 (35), 214 (31), 204 (30), 200 (50), 186 (53), 172 (17), 159 (48), 158 (15), 144 (46), 143 (81), 142 (29), 141 (38), 131 (23), 130 (13), 129 (39), 128 (100), 127 (20), 116 (17), 115 (69), 91 (14) ;
m/z calculé pour C14H14O4Na (M+22,9898) : 269,0790 ; trouvé : 269,0788.
2-Oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4d)
Préparé selon le mode opératoire général, à partir de 2-bromobenzaldéhyde (1,46 mL, 12,5 mmol). Le composé (4d) est obtenu sous la forme d’une huile brunâtre (2,90 g, 99 %).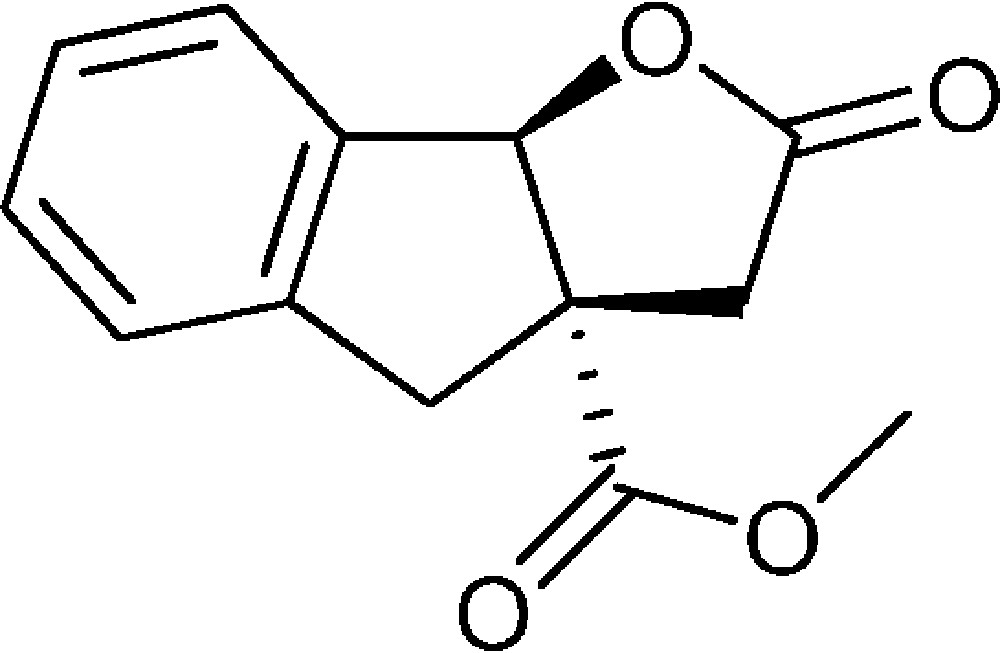
RMN 1H (CDCl3) : δ = 2,60 (1H, d, J 18,2), 3,15 (1H, d, J 16,7), 3,32 (1H, d, J 18,2), 3,69 (1H, d, J 16,7), 3,80 (3H, s), 6,06 (1H, s), 7,26-7,37 (4H, m) ;
RMN 13C (CDCl3) : δ = 37,8, 41,4, 53,2, 54,9, 89,8, 125,3, 126,8, 127,9, 129,8, 140,7, 153,4, 173,6, 175,0 ;
IR : ν = 690, 719, 809, 1006, 1179, 1234, 1268, 1437, 1608, 1723, 3391 cm−1 ;
SM (IE) : m/z = 214 (52), 200 (35), 190 (40), 187 (15), 186 (100), 173 (13), 172 (88), 158 (23), 145 (48), 144 (17), 143 (12), 131 (11), 130 (51), 129 (63), 128 (77), 127 (23), 117 (24), 116 (22), 115 (49), 102 (11) ;
m/z calculé pour C13H12O4Na (M+22,9898) : 255,0633 ; trouvé : 255,0634.
8-Fluoro-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4e)
Préparé selon le mode opératoire général, à partir de 2-bromo-6-fluorobenzaldéhyde (0,81 g, 4,0 mmol). Le composé (4e) est obtenu sous la forme d’une huile brune (0,72 g, 72 %).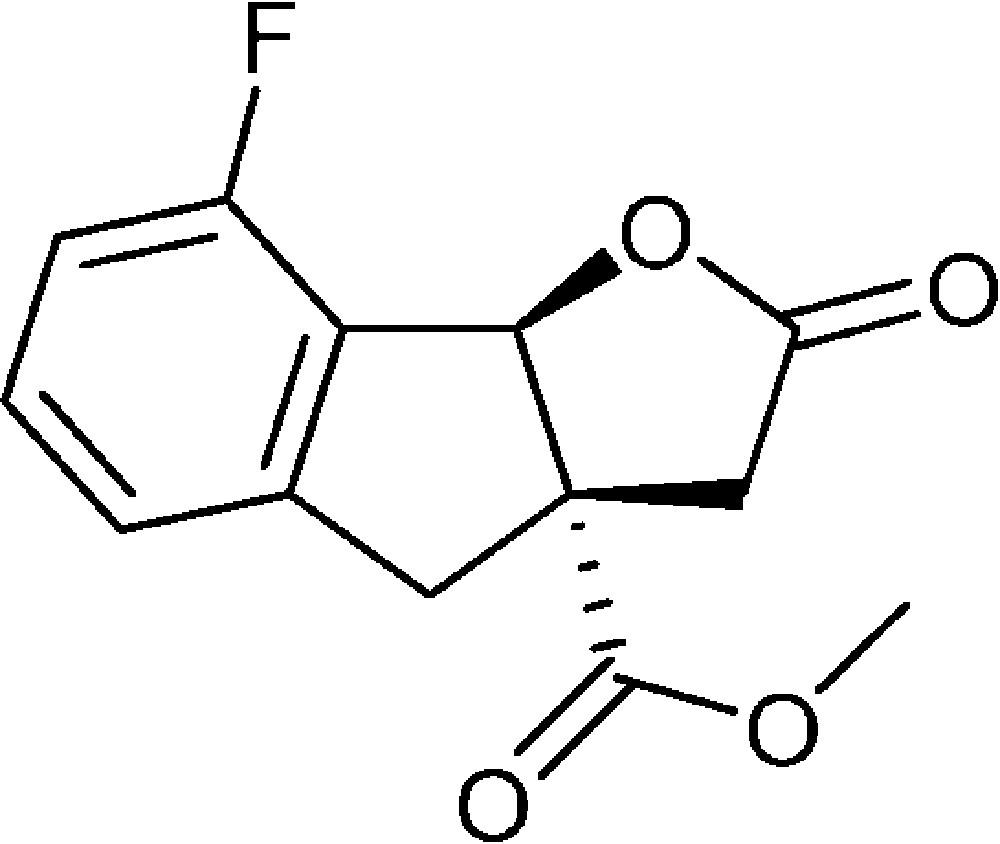
RMN 1H (CDCl3) : δ = 2,66 (1H, d, J 18,3), 3,16 (1H, d, J 17,0), 3,34 (1H, d, J 18,3) 3,67 (1H, d, J 17,0), 3,78 (3H, s), 6,15 (1H, s), 6,94–7,62 (3H, m) ;
RMN 13C (CDCl3) : δ = 39,0, 42,1, 53,4, 55,2, 86,8, 114,6 (d, J 19,4), 115,7 (d, J 21,8), 118,7 (d, J 2), 119,5 (d, J 3,5), 120,9 (d, J 3,8), 131,7 (d, J 254), 173,0, 174,4 ;
RMN 19F (CDCl3) : δ = –115,9 ;
IR : ν = 681, 782, 990, 1235, 1361, 1624, 1693, 3380 cm−1 ;
SM (IE) : m/z = 249 (15), 248 (100), 231 (15), 230 (78), 218 (11), 217 (69), 214 (22), 203 (12), 202 (66), 201 (88), 200 (6), 199 (14), 188 (11), 183 (12), 182 (15), 181(18), 171 (7), 170 (14), 169 (12), 51 (6) ;
m/z calculé pour C13H11FO4Na (M+22,9898) : 273,0539 ; trouvé : 273,0543.
6-Oxo-6,7,7a,8-tétrahydro-4bH-furo[2′,3′:1,2]indéno[5,6-d][1,3]dioxole-7a-carboxylate de méthyle (4f)
Préparé selon le mode opératoire général, à partir de 6-bromo-1,3-benzodioxole-5-carbaldéhyde (2,86 g, 12,5 mmol). Le composé (4f) est obtenu sous la forme d’une huile brunâtre (2,01 g, 58 %).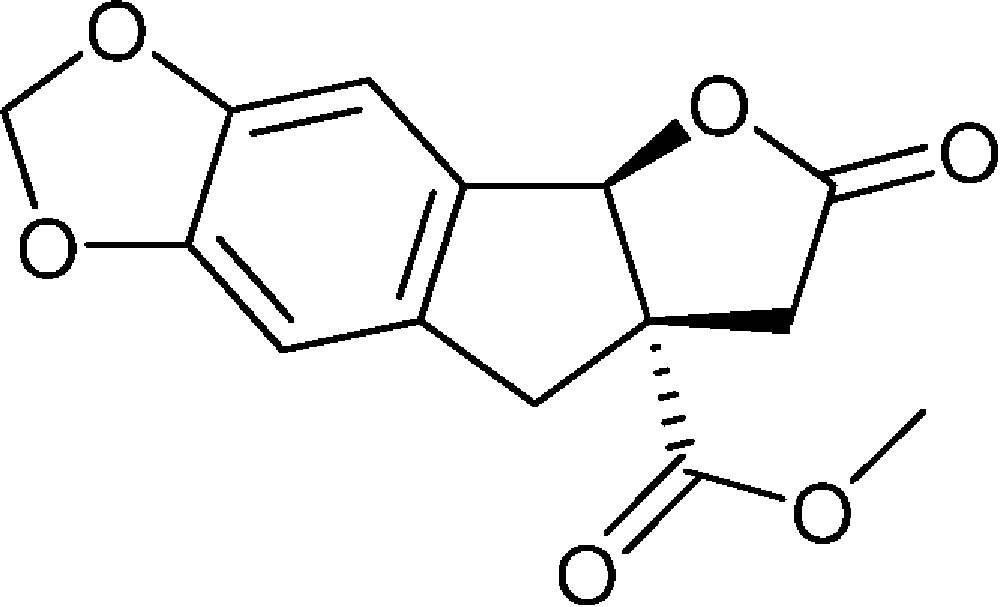
RMN 1H (CDCl3) : δ = 2,60 (1H, d, J 18,2), 3,00 (1H, d, J 16,4), 3,29 (1H, d, J 18,2), 3,56 (1H, d, J 16,4), 3,78 (3H, s), 5,92 (1H, s), 5,92 (2H, s), 6,64 (1H, s), 6,82 (1H, s) ;
RMN 13C (CDCl3) : δ = 39,5, 41,8, 53,3, 56,1, 90,1, 102,7, 105,9, 106,2, 131,9, 136,2, 148,8, 150,7, 174,3, 175,4 ;
IR : ν = 560, 850, 1035, 1254, 1476, 1612, 1718, 3408 cm−1 ;
SM (IE) : m/z = 277 (16), 276 (97), 234 (6), 233 (5), 232 (16), 230 (6), 217 (10), 216 (14), 202
(6), 199 (8), 189 (11), 174 (19), 173 (100), 172 (12), 171 (7), 143 (15), 116 (6), 115 (36), 103 (6) ;
m/z calculé pour C14H12O6Na (M+22,9898) : 299,0532 ; trouvé : 299,0540.
7-(Trifluorométhyl)-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4g)
Préparé selon le mode opératoire général, à partir de 2-bromo-5-trifluorométhylbenzaldéhyde (1,01 g, 4 mmol). Le composé (4g) est obtenu sous la forme d’huile (0,23 g, 20 %).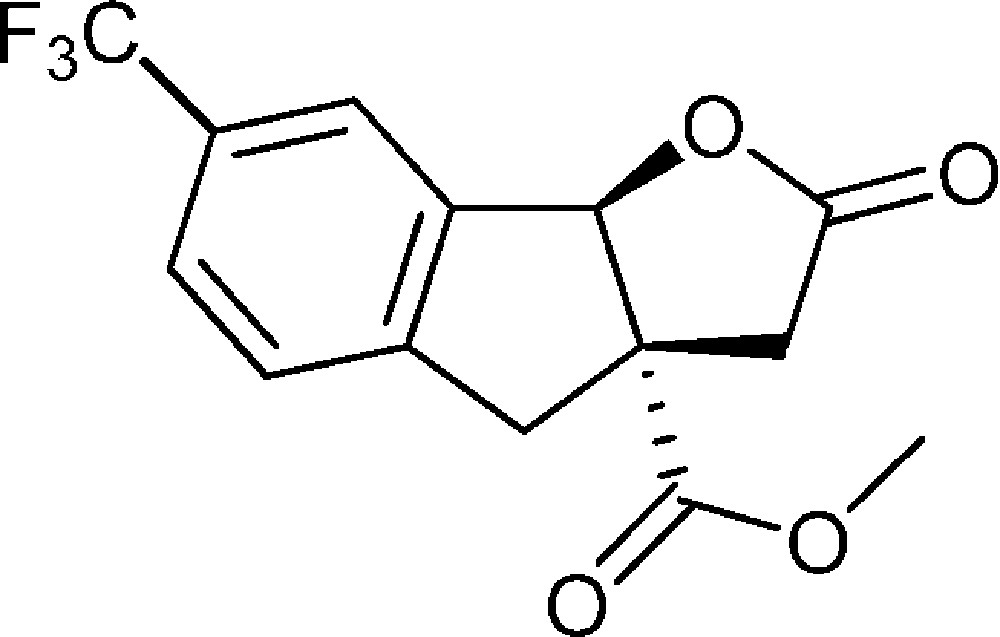
RMN 1H (CDCl3) : δ = 2,58 (1H, d, J 18,2), 3,18 (1H, d, J 17,3), 3,33 (1H, d, J 18,2), 3,69 (1H, d, J 17,3), 3,80 (3H, s), 6,03 (1H, s), 7,38 (1H, d, J 8,5), 7,62 (1H, d, J 8,5), 7,71 (1H, s) ;
RMN 13C (CDCl3) : δ = 38,6, 41,4, 53,3, 55,1, 88,6, 123,5 (q, J 4,1), 123,8 (q, J 272,0), 125,8, 127,5 (q J 4,2), 130,8 (q, J 32,4), 138,5, 144,8, 173,0, 174,0 ;
RMN 19F (CDCl3) : δ = –62,7 ;
IR : ν = 662, 703, 806, 901, 1017, 1062, 1118, 1161, 1285, 1326, 1439, 1625, 1733 cm−1 ;
SM (IE) : m/z = 222 (7), 221 (42), 207 (16), 206 (100), 190 (6), 178 (38), 162 (8), 150 (8), 146 (13), 136 (6), 130 (6), 122 (10), 120 (6), 116 (6), 91 (12), 77 (5), 57 (9) ;
m/z calculé pour C14H11F3O4Na (M+22,9898) : 323,05016 ; trouvé : 323,05006.
7-Chloro-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4h)
Préparé selon le mode opératoire général, à partir de 2-bromo-5-chlorobenzaldéhyde (0,70 g, 3 mmol). Le composé (4h) est obtenu sous la forme d’une huile (0,27 g, 33 %).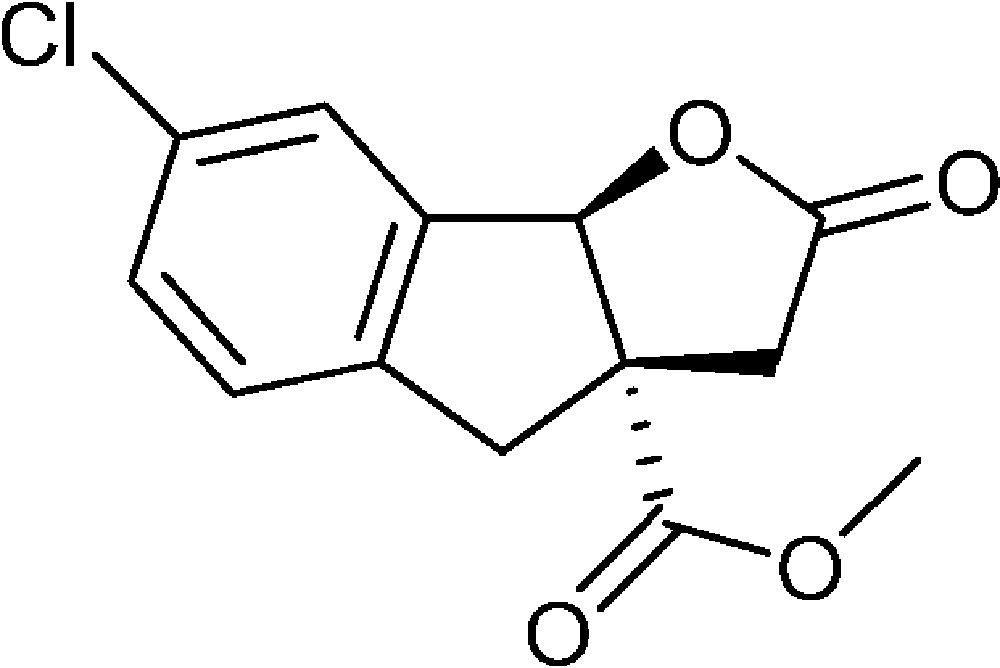
RMN 1H (CDCl3) : δ = 2,58 (1H, d, J 18,2), 3,09 (1H, d, J 16,8), 3,30 (1H, d, J 18,2), 3,63 (1H, d, J 16,8), 3,79 (3H, s), 5,98 (1H, s), 7,18 (1H, d, J 8,2), 7,32 (1H, dd, J 1,7 ; 8,2), 7,41 (1H, s) ;
RMN 13C (CDCl3) : δ = 31,1, 38,7, 40,9, 53,3, 55,3, 89,0, 126,4, 130,7, 134,0, 139,0, 139,3, 173,1, 174,6 ;
IR : ν = 564, 616, 692, 806, 1014, 1073, 1177, 1418, 1730, 1770, 2955 cm−1 ;
SM (IE) : m/z = 250 (20), 248 (59), 234 (55), 224 (32), 222 (33), 221 (16), 220 (87), 208 (32), 207 (17), 206 (100), 189 (36), 179 (35), 166 (17), 165 (23), 164 (64), 163 (34), 162 (31), 128 (43), 127 (35), 115 (47) ;
m/z calculé pour C13H11O4ClNa (M+22,9898) : 289,0244 ; trouvé : 289,0235.
7-Fluoro-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4i)
Préparé selon le mode opératoire général, à partir de 2-bromo-5-fluorobenzaldéhyde (1,95 g, 9,6 mmol). Le composé (4i) est obtenu sous la forme d’une huile brune (2,05 g, 85 %).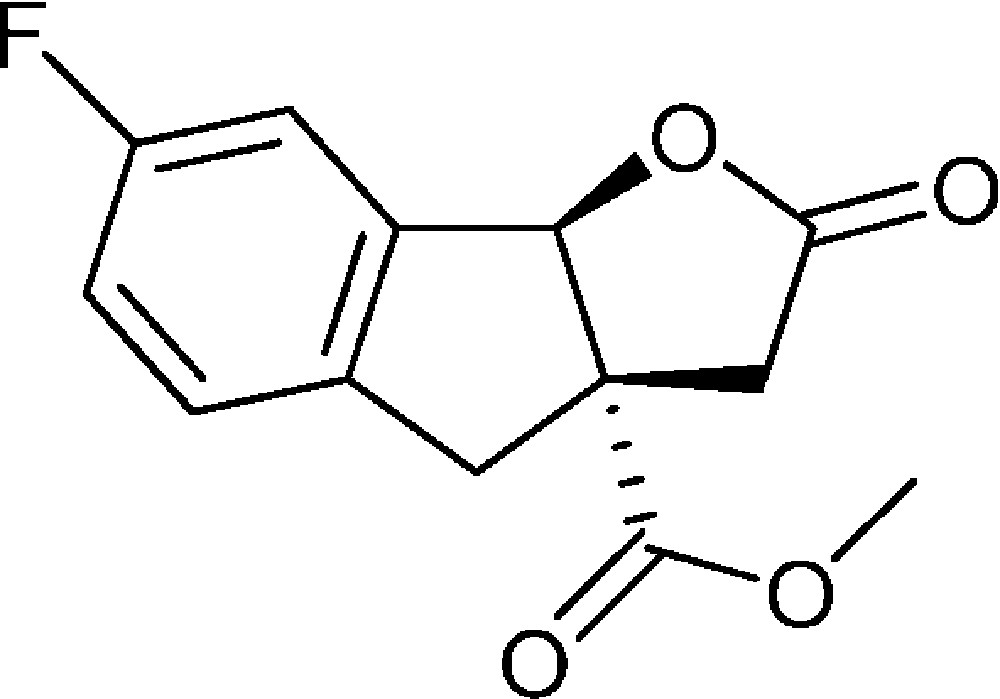
RMN 1H (CDCl3) : δ = 2,58 (1H, d, J 18,3), 3,09 (1H, d, J 16,5), 3,30 (1H, d, J 18,3), 3,61 (1H, d, J 16,5), 3,78 (3H, s), 5,99 (1H, s), 7,02-7,11 (3H, m) ;
RMN 13C (CDCl3) : δ = 38,8, 40,7, 53,3, 55,6, 89,2, 113,0 (d, J 23), 117,9 (d, J 23), 126,7 (d, J 9), 136,0 (d, J 3), 139,4 (d, J 8), 162,7 (d, J 247), 173,2, 174,9 ;
RMN 19F (CDCl3) : δ = –113,0 ;
IR : ν = 878, 956, 1009, 1196, 1319, 1439, 1490, 1614, 1718, 2400 cm−1 ;
SM (IE) : m/z = 232 (57), 218 (45), 208 (44), 205 (12), 204 (100), 191 (12), 190 (91), 163 (40), 162 (13), 161 (15), 149 (12), 148 (62), 147 (51), 146 (69), 145 (10), 135 (19), 134 (16), 133 (36), 127 (28), 115 (14) ;
m/z calculé pour C13H11O4FNa (M+22,9898) : 273,0539 ; trouvé : 273,0543.
7-Méthoxy-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4j)
Préparé selon le mode opératoire général, à partir de 2-bromo-5-méthoxybenzaldéhyde (2,70 g, 12,5 mmol). Le composé (4j) est obtenu sous la forme d’une huile jaune (2,25 g, 68 %).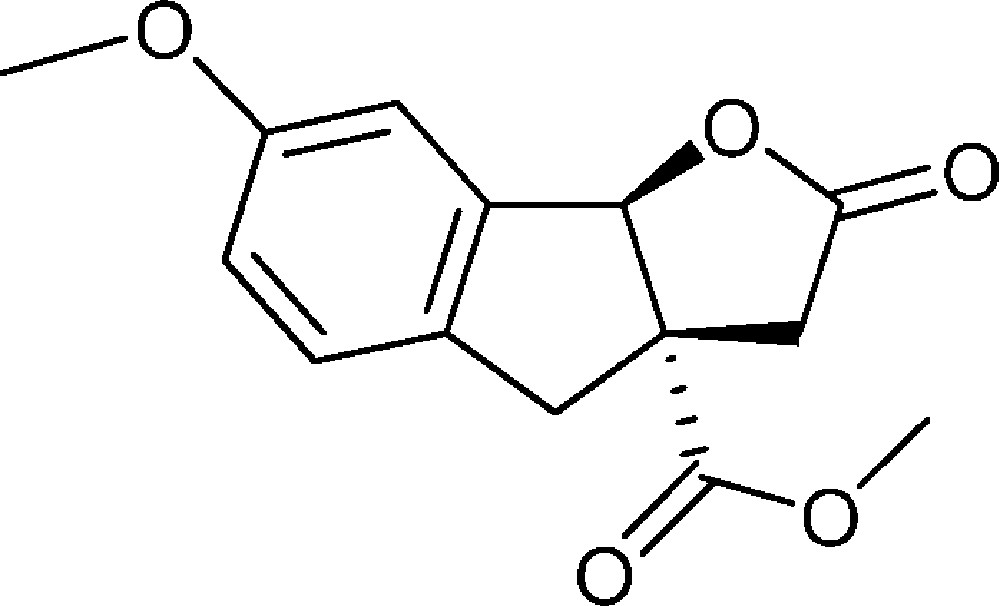
RMN 1H (CDCl3) : δ = 2,56 (1H, d, J 18,2), 3,05 (1H, d, J 16,2), 3,28 (1H, d, J 18,2), 3,58 (1H, d, J 16,2), 3,78 (3H, s), 3,79 (3H, s), 6,00 (1H, s), 6,89-6,93 (2H, m), 7,13 (1H, d, J 8,2) ;
RMN 13C (CDCl3) : δ = 38,8, 40,7, 53,2, 55,5, 55,6, 89,7, 109,9, 117,8, 126,0, 132,1, 138,9, 159,9, 173,6, 174,8 ;
IR : ν = 814, 1005, 1201, 1495, 1614, 1718, 3400 cm−1 ;
SM (IE) : m/z = 264 (16), 263 (100), 230 (17), 220 (15), 218 (9), 216 (50), 203 (21), 202 (90), 188 (13), 175 (24), 174 (20), 173 (8), 161 (13), 160 (73), 159 (23), 158 (17), 145 (10), 144 (17), 116 (11), 115 (26) ;
m/z calculé pour C14H14O5Na (M+22,9898) : 285,0739 ; trouvé : 285,0732.
8b-Méthyl-2-oxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (4k)
Préparé selon le mode opératoire général, à partir de 2-chloroacétophénone (1,94 g, 12,5 mmol). Le composé (4k) est obtenu sous la forme d’une huile (1,65 g, 54 %).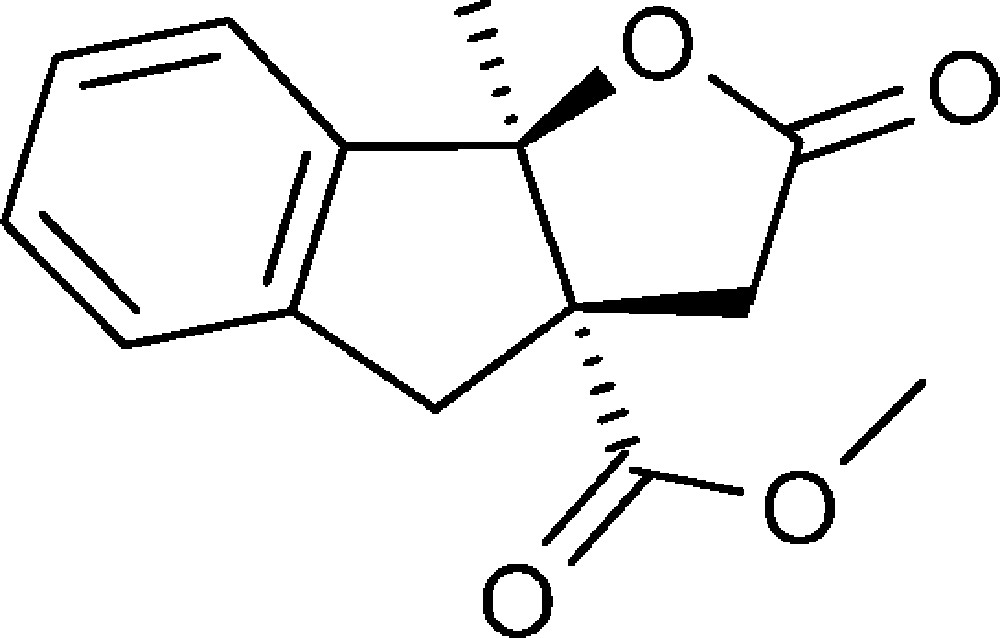
RMN 1H (CDCl3) : δ = 1,62 (3H, s), 2,55 (1H, d, J 17,7), 3,04 (1H, d, J 16,5), 3,29 (1H, d, J 17,7), 3,15 (1H, d, J 18,4), 3,75–3,85 (4H, m), 7,25–7,45 (4H, m) ;
RMN 13C (CDCl3) : δ = 22,3, 39,0, 40,0, 52,8, 59,2, 96,1, 124,3, 125,3, 128,1, 130,0, 138,9, 141,7, 172,5, 174,2 ;
SM (IE) : m/z = 248 (10), 229 (62), 215 (55), 201 (94), 187 (70), 146 (63), 145 (35), 144 (100) ; 129 (48), 116 (36).
2-Oxo-3,3a,4,5,6,7,8,8b-octahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (6b)
Préparé selon le mode opératoire général, à partir de 2-bromo-cyclohex-1-ène-carbaldéhyde (1,13 g, 6 mmol). Le composé (6b) est obtenu sous la forme d’une huile (0,60 g, 42 %).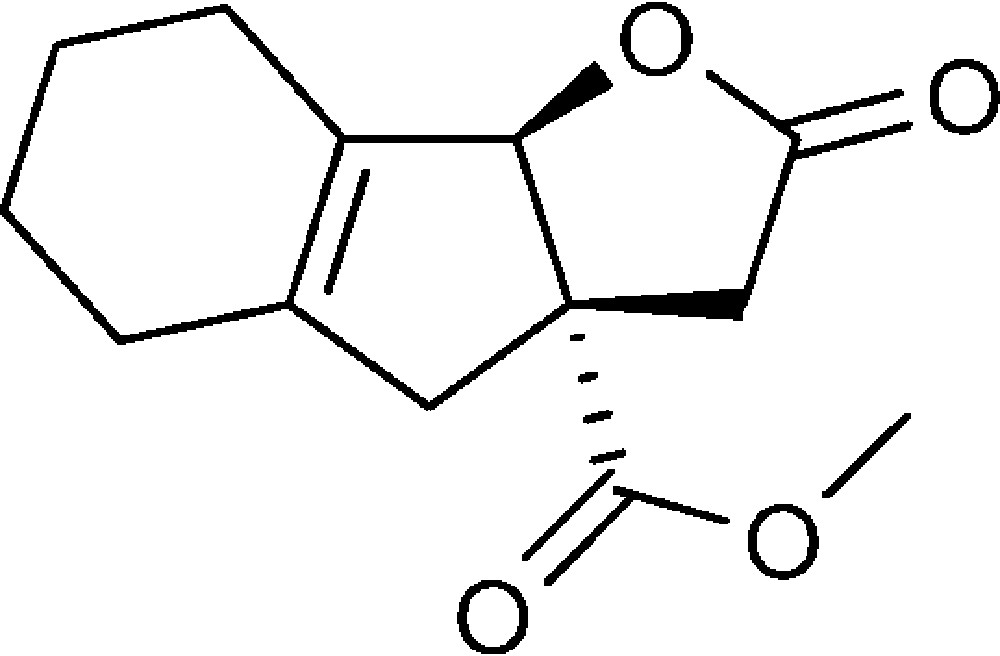
RMN 1H (CDCl3) : δ = 1,53-1,59 (4H, m), 1,93-2,02 (4H, m), 2,37 (1H, d, J 16,7), 2,45 (1H, d, J 18,4), 2,93 (1H, d, J 16,4), 3,18 (1H, d, J 18,4), 3,70 (3H, s), 5,35 (1H, s) ;
RMN 13C (CDCl3) : δ = 21,7, 22,8, 25,4, 39,4, 46,0, 52,1, 52,7, 93,9, 132,3, 139,8, 174,2, 175,2 ;
IR : ν = 667, 770, 993, 1166, 1277, 1732, 1776, 2929 cm−1 ;
SM (IE) : m/z = 236 (19), 218 (22), 204 (87), 194 (26), 192 (19), 190 (56), 177 (71), 176 (91), 162 (22), 149 (48), 148 (81), 132 (22), 131 (55), 120 (23), 117 (30), 105 (26), 91 (100), 79 (27), 77 (20) ;
m/z calculé pour C13H16O4Na (M+22,9898) : 259,0946 ; trouvé : 259,0951.
2-Oxo-2,3,3a,4,5,6,7,8,9,9b-décahydroazuléno[1,2-b]furan-3a-carboxylate de méthyle (6c)
Préparé selon le mode opératoire général, à partir de 2-bromo-cyclohept-1-ène-carbaldéhyde (1,30 g, 6 mmol). Le composé (6c) est obtenu sous la forme d’une huile (0,80 g, 51 %).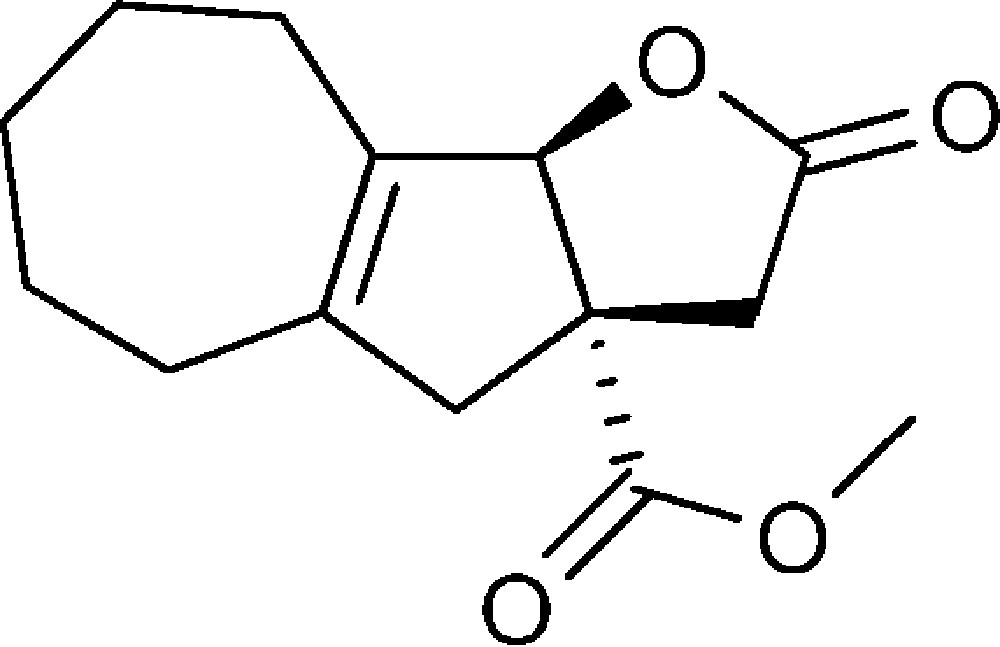
RMN 1H (CDCl3) : δ = 1,50–2,14 (10H, m), 2,35 (1H, d, J 16,7), 2,42 (1H, d, J 18,3), 2,95 (1H, d, J 16,7), 3,10 (1H, d, J 18,3), 3,63 (3H, s), 5,28 (1H, s) ;
RMN 13C (CDCl3) : δ = 26,6, 27,1, 29,9, 30,3, 39,6, 48,2, 52,1, 52,8, 96,0, 135,3, 142,9, 174,1, 175,4 ;
IR : ν = 855, 886, 1168, 1277, 1437, 1732, 1777, 2853, 2924 cm−1 ;
SM (IE) : m/z = 250 (21), 232 (22), 219 (17), 218 (89), 208 (16), 204 (37), 192 (14), 191 (100), 190 (86), 176 (21), 163 (41), 162 (77), 147 (29), 145 (45), 131 (34), 119 (18), 105 (30), 91 (72), 79 (16), 77 (18) ;
m/z calculé pour C14H18O4Na (M+22,9898) : 273,1103 ; trouvé : 273,1100.
2-Oxo-3,3a,4,5,6,7,8,9,10,10b-decahydro-2H-cycloocta[4,5]cyclopenta[1,2b]furan-3a-carboxylate de méthyle (6d)
Préparé selon le mode opératoire général, à partir de 2-bromo-cyclooct-1-ène-carbaldéhyde (1,30 g, 6 mmol). Le composé (6d) est obtenu sous la forme d’une huile (0,305 g, 19 %).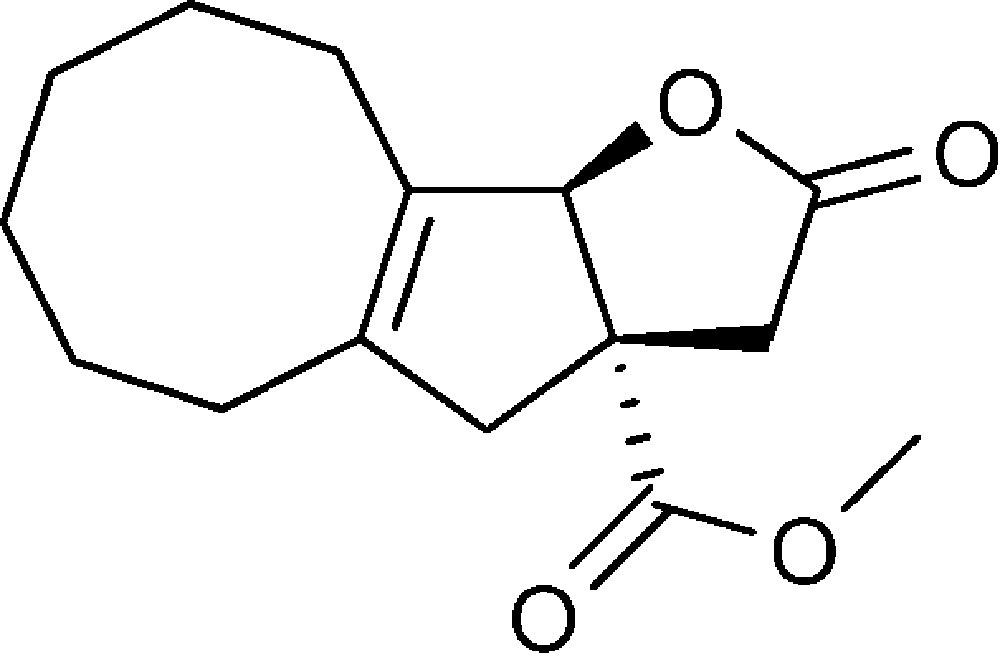
RMN 1H (CDCl3) : δ = 1,35–1,50 (8H, m), 2,05–2,14 (4H, m), 2,35 (1H, d, J 16,7), 2,41 (1H, d, J 18,6), 2,95 (1H, d, J 16,7), 3,15 (1H, d, J 18,4), 3,66 (3H, s), 5,35 (1H, s) ;
RMN 13C (CDCl3) : δ = 25,5, 26,0, 26,8, 27,4, 28,4, 39,5, 46,1, 52,1, 52,8, 94,6, 133,8, 141,7, 174,1, 175,2 ;
IR : ν = 751, 852, 886, 996, 1165, 1283, 1334, 1733, 1776, 2854, 2924 cm−1 ;
SM (IE) : m/z = 264 (35), 232 (93), 222 (22), 218 (49), 205 (100), 204 (98), 203 (22), 190 (38), 177 (48), 176 (87), 175 (22), 162 (21), 161 (30), 159 (30), 145 (30), 117 (25), 105 (32), 91 (70), 79 (22), 77 (21) ;
m/z calculé pour C15H20O4Na (M+22,9898) : 287,1259 ; trouvé : 287,1256.
2-Oxo-2,3,3a,4,5,6,8,8b-octahydrofuro[3′,2′:4,5]cyclopenta[1,2-c]pyran-3a-carboxylate de méthyle(6e)
Préparé selon le mode opératoire général, à partir de 2-bromo-5,6-dihydro-2H-pyran-3-carbaldéhyde (0,44 g, 2,3 mmol). Le composé (6e) est obtenu sous la forme d’une huile (0,265 g, 48 %).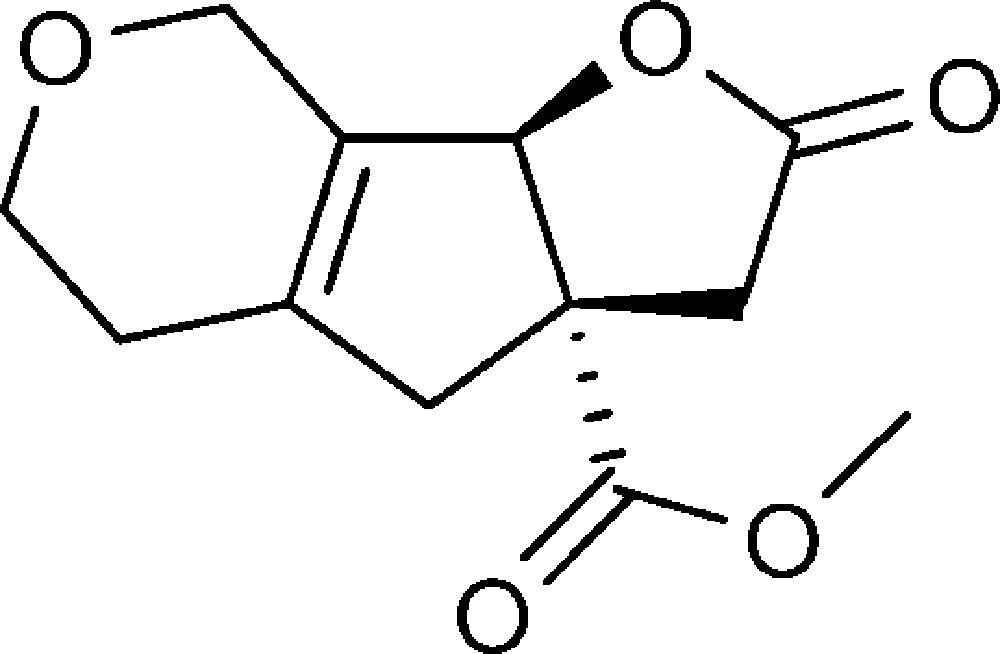
RMN 1H (CDCl3) : δ = 2,17 (2H, m), 2,52 (1H, d, J 15,7), 2,55 (1H, d, J 18,5), 3,09 (1H, d, J 15,7), 3,30 (1H, d, J 18,5), 3,67–3,84 (5H, m), 4,19–4,23 (2H, m), 5,51 (1H, s) ;
RMN 13C (CDCl3) : δ = 26,2, 39,4, 46,0, 52,7, 53,2, 63,6, 64,2, 91,5, 131,9, 138,5, 173,8, 174,8 ;
SM (IE) : m/z = 208 (51), 206 (46), 190 (37), 179 (100), 178 (20), 177 (24), 176 (75), 164 (16), 149 (18), 148 (15), 135 (36), 133 (46), 121 (19), 105 (49), 96 (34), 95 (17), 91 (37), 81 (27), 79 (25), 77 (32) ;
m/z calculé pour C12H14O5Na (M+22,9898) : 261,0733 ; trouvé : 261,0734.
(3aS,8bR)-2,4-Dioxo-3,3a,4,8b-tétrahydro-2H-indéno[1,2-b]furan-3a-carboxylate de méthyle (7)
Un mélange oxydant est préparé par broyage de KMnO4 et CuSO4, 5 H2O en proportion identique (m/m). Le composé 4d (1,5 g, 6,5 mmol) est introduit dans un ballon contenant 15 g du mélange oxydant. De l’eau est ajoutée puis le ballon est placé dans la cuve d’un bain ultrasons, sous air, à température ambiante durant six heures. Le mélange est filtré sur Büchner, le filtrat extrait au dichlorométhane (3 × 50 mL) et la phase organique séchée sur MgSO4, puis évaporée. Le produit est chromatographié sur SiO2, en utilisant comme éluant un mélange pentane/éther diéthylique de (8:2) à (3:7). Le composé est obtenu sous la forme d’un solide blanc (0,5 g, 31 %).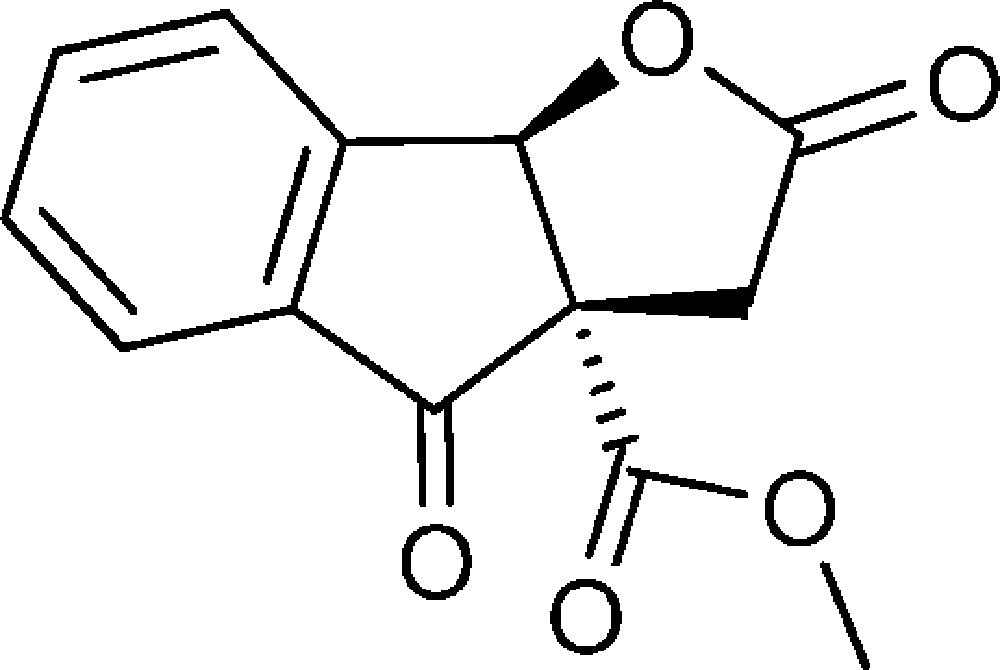
RMN 1H (CDCl3) : δ = 2,92 (1H, d, J 19,3), 3,65 (1H, d, J 19,3), 3,80 (3H, s, OCH3), 6,09 (1H, s), 6,64–6,71 (1H, m), 7,80–7,94 (3H, m) ;
RMN 13C (CDCl3) : δ = 34,4, 53,8, 61,4, 81,8, 125,4, 127,6, 131,5, 135,1, 136,8, 149,2, 168,3, 173,1, 197,2 ;
SM (IE) : m/z = 247 (2), 220 (12), 219 (98), 188 (28), 187 (100), 175 (8), 160 (20), 144 (8), 134 (24), 116 (16).
(3aR,8bS)-3,3a-Dihydro-2H-indéno[1,2-b]furan-2,4(8bH)-dione (8)
Dans un ballon équipé d’un réfrigérant et contenant la lactone 7 (0,2 g, 0,8 mmol) est ajouté du DMSO (5 mL), LiCl (0,035 g, 0,8 mmol) et H2O (0,25 mL). Le mélange est chauffé à 130 °C sous agitation pendant 5 h. Après refroidissement, de l’eau (20 mL) est ajoutée et les produits organiques sont extraits au dichlorométhane (3 × 15 mL). La phase organique est lavée avec NaCl sat., séchée sur MgSO4, puis évaporée. Le produit est chromatographié sur SiO2, en utilisant comme éluant un mélange pentane/éther diéthylique de (7:3) à (4:6). Le composé est obtenu sous la forme d’un solide jaune (0,06 g, 39 %).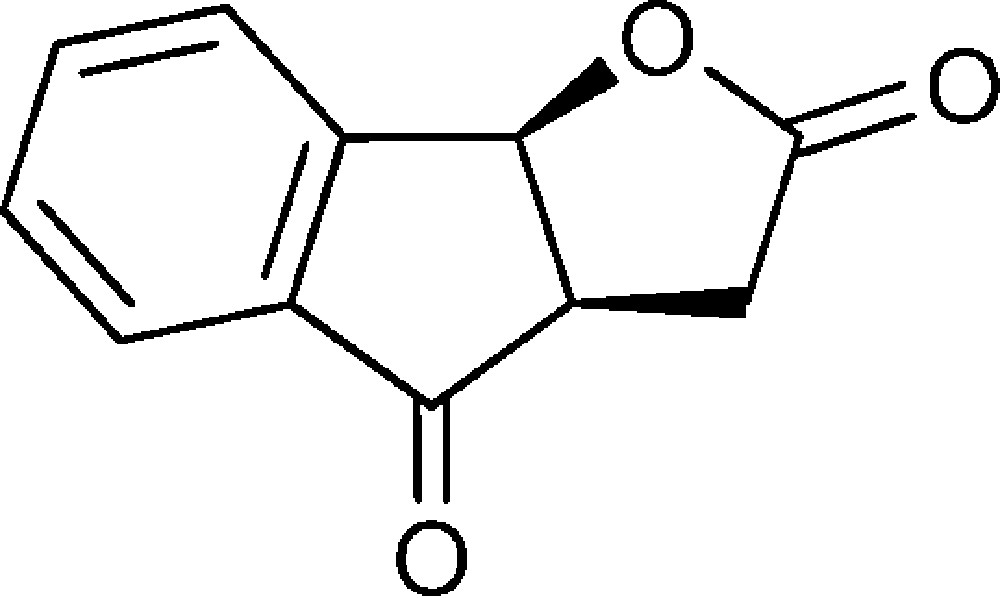
RMN 1H (CDCl3) : δ = 2,81 (1H, dd, J 19,0, 4,5), 3,07 (1H, dd, J 19,0, 12,5), 3,54–3,65 (1H, m), 6,02 (1H, d, J 6,8), 6,60–6,67 (1H, m), 6,77–6,85 (2H, m), 7,86 (1H, d, 7,7) ;
RMN 13C (CDCl3) : δ = 31,2, 45,8, 79,1, 124,5, 127,6, 131,2, 136,1, 136,3, 149,7, 174,7, 202,4 ;
SM (IE) : m/z = 208 (12), 189 (10), 161 (16), 147 (100), 132 (24), 116 (54), 105 (28).
1 Un ballon contenant le produit 5a brut a éclaté pendant la nuit, avec formation d’un solide noir, alors qu’il était stocké à température ambiante sous la hotte.