

1 Introduction
Macromonomers (defined as polymers with polymerisable entities at one or both chain ends and generally low molar masses) were shown to copolymerise with low molar mass monomers for the first time more than 40 years ago 〚1〛. They were utilised for some applications in the late 60s 〚2, 3〛. It was only since the work of Milkovich in the 70s 〚4, 5〛 that macromonomers have been increasingly employed for macromolecular engineering. Macromonomers with a wide range of chemical structures are now accessible 〚6〛. They can either be homopolymerised to yield comb polymers, or copolymerised with a large number of comonomers to yield graft copolymers. The macromonomer technique was applied efficiently to the synthesis of well-defined branched polymers with controlled length of the grafts, controlled degree of polymerisation of the main chain and controlled composition of the backbone and the grafts. Depending on the chemical nature, graft density, graft length and degree of polymerisation, these polymacromonomers can adopt several conformations in solution (Fig. 1), leading to various properties. Many physico-chemical studies were performed in order to learn more about their structure-properties relationships 〚7–15〛.
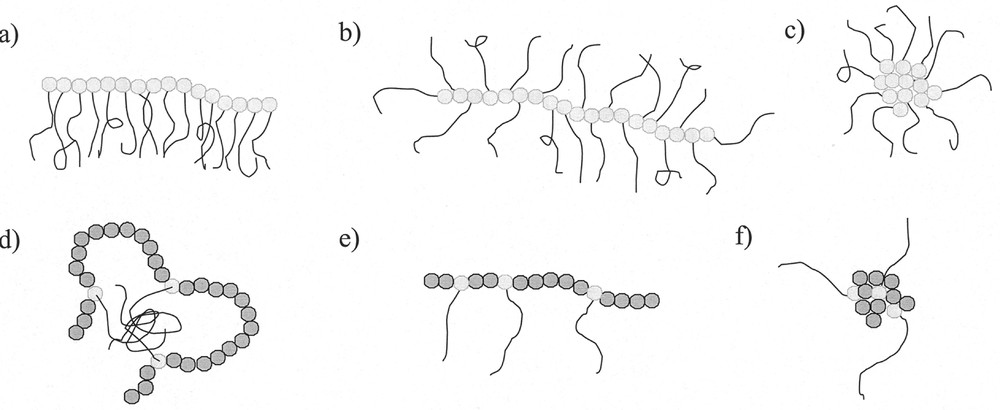
Architectures obtained for the (co)polymerisation of macromonomers (a, e: comb-like; b: bottlebrush; c, f: star-like ; d: flower-like) 〚12〛.
Over the years, macromonomers have been (co)polymerised using different polymerisation processes. Anionic and free radical processes were first employed 〚16〛. Without being exhaustive, one can mention the works of Rempp and co-workers 〚17〛, Lutz and co-workers 〚18〛, and Hirao and co-workers 〚19〛 for the anionic polymerisation of macromonomers. A lot of work was also done based on free radical polymerisation procedures 〚20–29〛. The review published by Capek 〚29〛 provides a good overview for free radical polymerisations of macromonomers of different chemical nature as well as kinetic data for these processes. It is only in the past 10 years that the (co)polymerisation of macromonomers with transition-metal based processes has been investigated. In this paper, we intend to extensively review published contributions dealing with the homopolymerisation of macromonomers in the presence of transition metal complexes. Their homopolymerisation by Ring Opening Metathesis Polymerisation (ROMP) 〚30–45〛, Atom Transfer Radical Polymerisation (ATRP) 〚46–49〛 and Ziegler–Natta type polymerisation (ZN) 〚50–52〛 will be successively discussed. Finally, the particular case of isocyanide-terminated macromonomers polymerised in the presence of NiCl2 will be presented 〚53〛.
2 Results and discussion
2.1 Ring opening metathesis polymerisation (ROMP) 〚30–45〛
Among the different polymerisation processes mentioned above, ROMP is probably the one that has received the most attention in the last years for the homopolymerisation of macromonomers. In particular, several groups described the synthesis of polymacromonomers with ruthenium- or molybdenum-based catalysts with variable efficiency (Fig. 2).
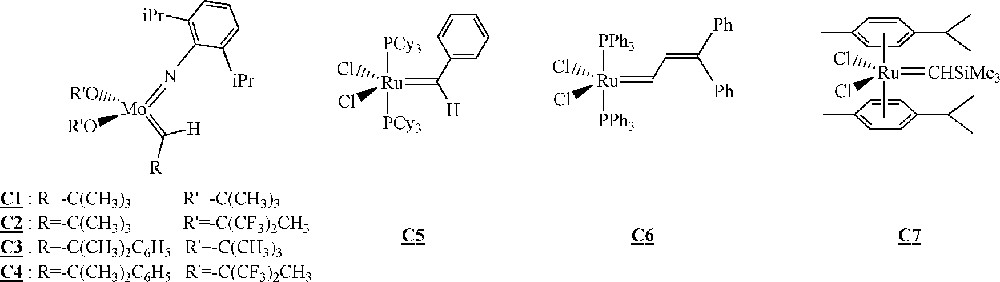
Catalysts for the homopolymerisation of macromonomers by ROMP 〚30–45〛.
Feast and co-workers were among the first to study the homopolymerisation of polystyrene macromonomers by ROMP in 1994 〚30〛. The macromonomers were synthesised by living anionic polymerisation, leading to species bearing 1 or 2 pendant polystyrene chains (Fig. 3, macromonomers and ).
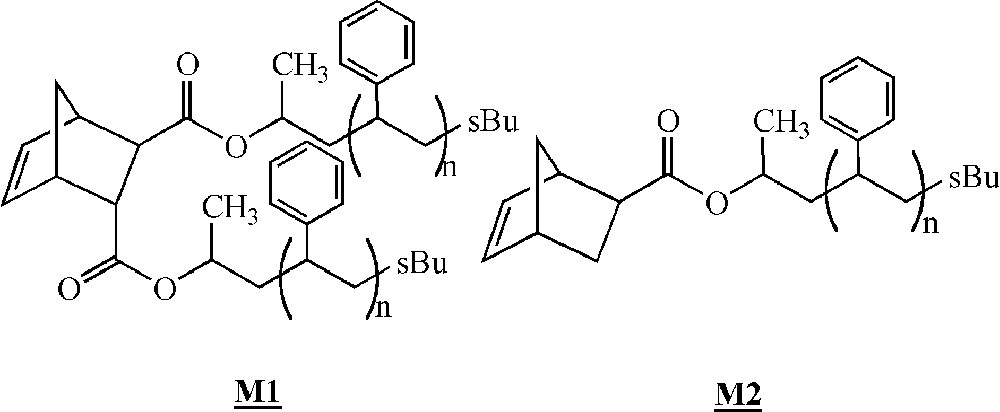
Macromonomers synthesised by Feast and co-workers 〚30–32〛.
The ROMP of these macromonomers with PS branches having (on average) 4, 7 or 9 styrene units was then studied with Schrock catalysts (Fig. 2, complexes and ). For macromonomers with such low molar masses, the polymerisation was shown to be living. Oligomacromonomers with measured molar masses in good agreement with the calculated ones as well as low polymolecularity indices were obtained for degrees of polymerisation up to 15. No unreacted macromonomer was detected. On the contrary, attempts with macromonomers bearing longer PS chains (14, 24 or 46 styrene units) led to a mixture of polymacromonomer and unreacted macromonomer. Despite this observation, it was shown by 1H NMR spectroscopy that the reactive organometallic chain ends remained present, even if there was no more incorporation of the macromonomer. The presence of unreacted macromonomer after very long reaction time was attributed by the authors to steric hindrance (thermodynamic inhibition). Moreover, it was evidenced that the maximum length of the polynorbornene backbone was dependent on the PS branch length 〚31〛 (Fig. 4).
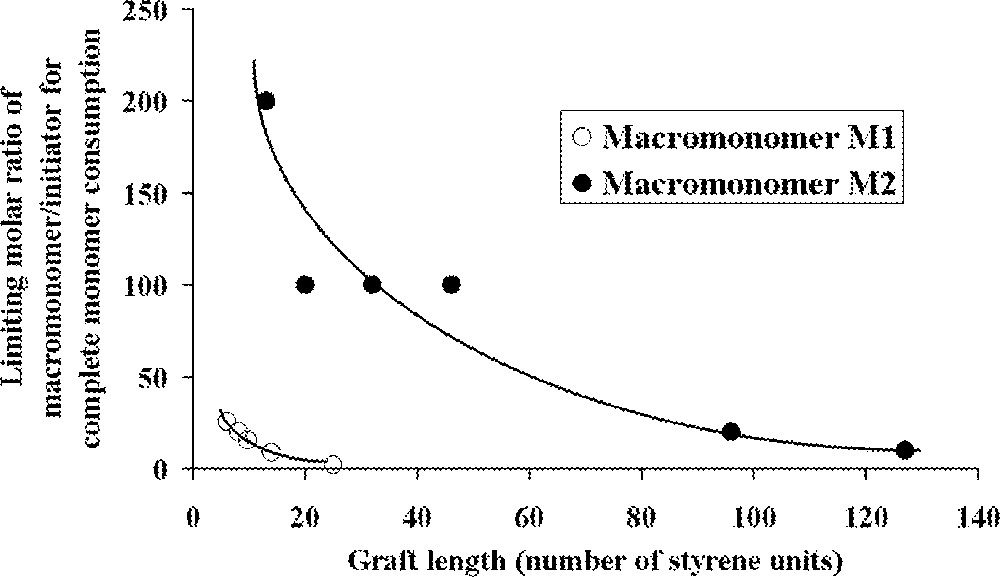
Relationship between the length of polystyrene graft in the macromonomer and limiting molar ratio of macromonomer to initiator that results in complete monomer consumption 〚31, 32〛.
By using macromonomer (one chain per macromonomer unit) instead of macromonomer (two chains per macromonomer unit) (Fig. 3), the authors were able to obtain polymacromonomers with much higher degrees of polymerisation (up to 200), and complete consumption of the macromonomer was observed, the maximum length of the norbornene backbone depending again on the PS branch length 〚32〛 (Fig. 4). Similar results were observed for polymacromonomers obtained by anionic polymerisation 〚18〛.
At about the same time, Gnanou and co-workers described the ROMP of ω-norbornenyl PS macromonomers 〚33〛. In subsequent contributions, they described the behaviour of macromonomers of various chemical natures (Fig. 5) 〚33–40〛. All these macromonomers were synthesised by living anionic polymerisation, the norbornene unit being introduced either during the initiation step (macromonomers , , , , ) or during the deactivation step (macromonomers , , , ). Macromonomers with block copolymers as branches (macromonomers to ) were also synthesised by anionic living polymerisation, the two comonomers being added sequentially.
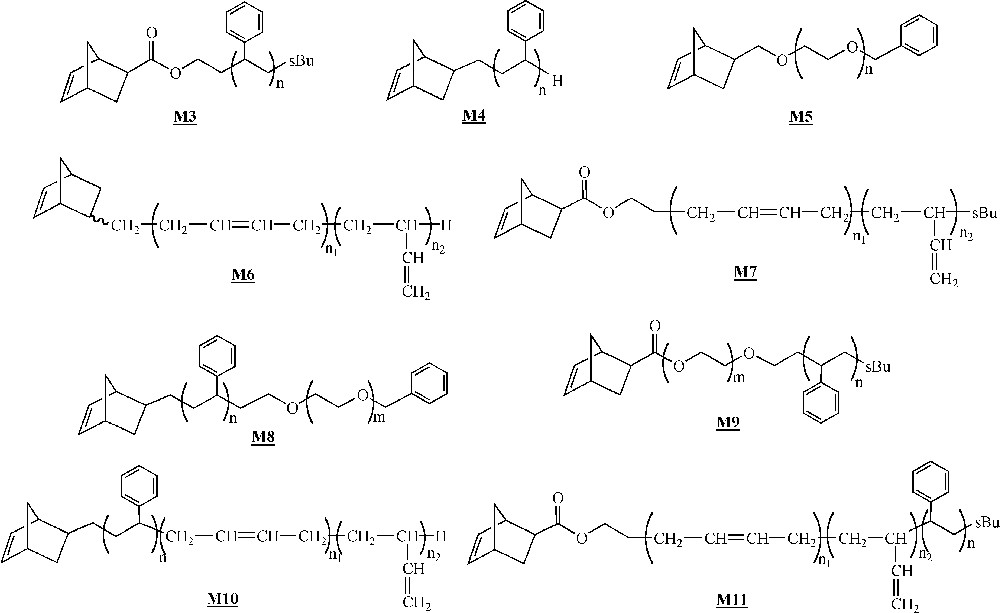
Macromonomers synthesised by Gnanou and co-workers 〚33–40〛.
The homopolymerisation of ω-norbornenyl PS macromonomers (Fig. 5, macromonomer ) was first investigated with the classical ROMP initiating system WCl6/SnPh4. This catalyst was rapidly abandoned because only dimers and trimers were obtained 〚34〛. It was replaced by one of the Schrock catalysts (Fig. 2, complex ). Under these conditions, polymacromonomers with degrees of polymerisation up to 100 were obtained, and a complete consumption of the macromonomer was observed. Moreover, the measured molar masses were in very good agreement with the expected ones for branches up to 5000 g mol–1. For longer branches (11 000 g mol–1), a difference appeared that was attributed by the authors to the difficulty of controlling accurately the initiator concentration.
The homopolymerisation of α-norbornenyl PS macromonomers (Fig. 5, macromonomer , = 2600 g mol–1) was also studied with a Schrock catalyst (Fig. 2, complex ) 〚35〛. Again, a very good control of the polymerisation was observed with measured molar masses very close to expected ones and full macromonomer conversion.
The homopolymerisation of α-norbornenyl poly(ethylene oxide) macromonomers (Fig. 5, macromonomer ) led to unexpected results 〚36〛. It was first observed that with catalyst (Fig. 2), only a 20% macromonomer conversion could be obtained. With catalyst (Fig. 2), the polymerisation went to completion, but measured molar masses were always well above the expected ones, while polymolecularity indices remained quite low (< 1.3). The authors demonstrated that this peculiar behaviour was due to a competition between the oxygen atoms of the poly(ethylene oxide) branches and the double bond of the norbornenyl unit for the complexation to the catalyst. As a result of this competition, quantitative initiation was impossible.
The homopolymerisation of α- and ω-norbornenyl polybutadiene macromonomers (Fig. 5, macromonomers and ) suffered from the presence of acyclic C=C double bonds on the branches that can also react with the organometallic species and interfere with the polymerisation 〚37〛. As the number of 1,2- and 1,4-butadiene units is dependent on the synthetic conditions used during the macromonomer synthesis, the synthesis of macromonomer , which was achieved in a mixture of diethyl ether and toluene as solvents, led to 40–50% 1,2-units, whereas the synthesis of macromonomer , which was performed in toluene alone, led to 10–15% 1,2-units. The authors used several catalysts (Fig. 2, complexes , , and ). Catalysts and led to complete degradation of the macromonomers because of their very high reactivity towards acyclic double bonds. With catalyst , no polymerisation was observed for macromonomer and poor results (16% conversion) were obtained for macromonomer . Catalyst gave the best results. For the macromonomer containing 40–50% 1,2-butadiene units (macromonomer ), the polymerisation did not proceed to completion (70%), but for macromonomers containing 10–15% 1,2-butadiene units, the polymerisation went to completion. As expected by the authors, the presence of many 1,2-units led to considerable problems: these double bonds are indeed more liable to participate in the metathesis process than their 1,4-analogues. As a result, the maximum yield in polymacromonomer was directly correlated to the number of 1,2-units (Fig. 6). It was also evidenced that a degradation of the formed polymacromonomer occurred after long reaction times.
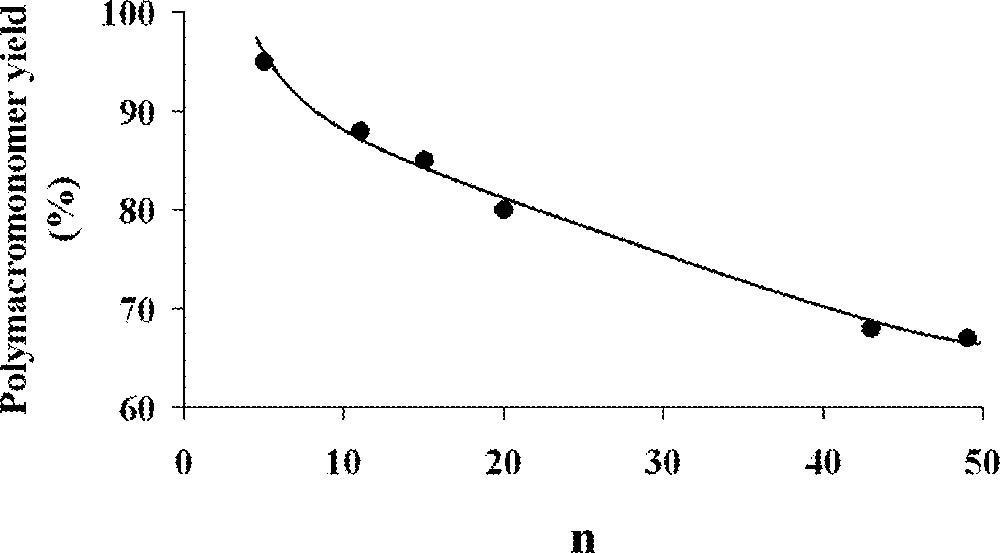
Maximum polymacromonomer yield as a function of 1,2-units contents 〚37〛.
The authors also studied the homopolymerisation of macromonomers constituted of block copolymer branches (Fig. 5, macromonomers to ) 〚38–40〛.
For poly(styrene-b-ethylene oxide) macromonomers ( and ), the objective was to build amphiphilic architectures 〚38〛. The degree of polymerisation of the polymacromonomers was limited to 20 and 10, respectively, and a very good control of the molar masses was obtained with complete consumption of the macromonomers (catalyst ). The presence of a poly(ethylene oxide) block did not affect the initiation efficiency as previously observed for poly(ethylene oxide) macromonomers.
The same authors also studied the homopolymerisation of poly(styrene-b-butadiene) macromonomers 〚40〛. For macromonomer with low content in 1,2-units, homopolymerisation occurred in 90% yield with very good control for degrees of polymerisation up to 50 (catalyst ). For macromonomer , with higher content in 1,2-units, the authors evidenced that the presence of a polystyrene spacer separating the norbornenyl chain end from the polybutadiene block prevents side reactions due to 1,2-units, even if the PS block is very short (5 units). It was possible to obtain polymacromonomers in 90% yield with good control for degrees of polymerisation up to 50. The same authors also synthesised Janus-type polymacromonomers 〚37–39〛 by sequential polymerisation of macromonomers of different chemical natures.
More recently, Dubois and co-workers described the ROMP of α-norbornenyl poly(ϵ-caprolactone) macromonomers (Fig. 7) 〚41〛, which were synthesised by ring opening polymerisation of ϵ-caprolactone in the presence of hydroxymethylnorbornene and triethyl aluminum. Their homopolymerisation was conducted in the presence of 〚RuCl2(p-cymene)〛2/PCy3/trimethylsilyl diazomethane as the initiating system. Polymacromonomers with yields up to 90% were obtained. The living and controlled characters of the polymerisation are not entirely clear.
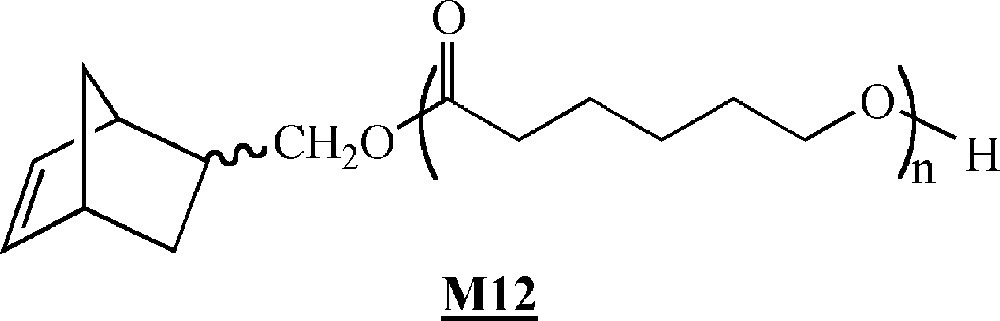
Macromonomer synthesised by Dubois and co-workers 〚41〛.
Nomura and co-workers studied the homopolymerisation of ω-norbornenyl polynorbornene macromonomers and of some derivatives (Fig. 8) 〚42–44〛. The macromonomers were synthesised via ring opening metathesis polymerisation of norbornene (or some derivatives) with catalyst (Fig. 2) and end-capped with p-Me3SiC6H4CHO. After hydrolysis of the trimethylsilyl group with NaOH followed by a coupling with norbornenecarbonyl chloride, macromonomers – were obtained.
Macromonomers synthesised by Nomura and co-workers 〚42–44〛.
Catalysts and (Fig. 2) were used to homopolymerise macromonomer . In both cases, yields up to 90% were obtained for degrees of polymerisation of 10. It was shown that the polymolecularity of the polymacromonomer obtained with catalyst was broader due to a control of the polymerisation that was worse than with catalyst . The homopolymerisation of macromonomers or with catalyst led to oligomacromonomers in good yields and with a degree of polymerisation of 5.
Macromonomer was synthesised in order to build amphiphilic polymacromonomer-based architectures. The authors hydrolysed the silyl ether functions, but unfortunately the resulting polymacromonomers were poorly soluble in all common solvents.
Finally, in that area of the homopolymerisation of macromonomers by ROMP, one has to mention the work published very recently by Allcock and co-workers on the homopolymerisation of polyphosphazenes macromonomers (Fig. 9) 〚45〛. The macromonomers were obtained via living cationic polymerisation of Cl3P=NSiMe3 quenched with a norbornenyl phosphoranimine, followed by a replacement of the chlorine atoms with CF3CH2O– units. The macromonomer was homopolymerised with catalyst (Fig. 2), leading to polymacromonomers with molar masses lower than expected and high polymolecularity indices. The homopolymerisation of macromonomer with catalyst led to highly cross-linked materials that were not characterised.
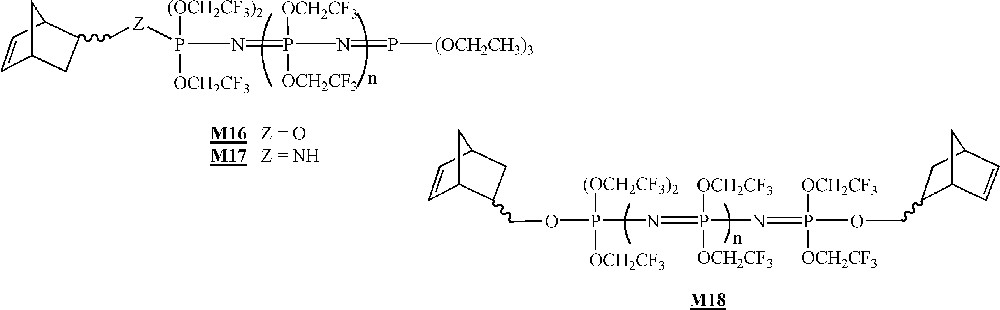
Macromonomers synthesised by Allcock and co-workers 〚45〛.
As described above, ROMP of macromonomers represents a nice way to synthesise various polymacromonomers of different chemical natures, provided appropriate polymerisation conditions are applied. Control of the molar masses and architectures is thus possible, which represents an important improvement compared to the free radical and anionic polymerisations, which were first employed. Nevertheless, the maximum degree of polymerisation attainable from large macromonomers seems to be somehow limited.
2.2 Atom Transfer Radical Polymerisation (ATRP) 〚46–49〛
Although ATRP has benefited from a huge development over the last 5–7 years 〚54–57〛, only few examples of its use for the homopolymerisation of macromonomers have been described in the literature. Thus far, ATRP has mostly been used for the synthesis of macromonomers and their copolymerisation with small monomers 〚58〛. The ‘grafting-from’ approach is also widely used 〚58〛.
In studies dealing with the homopolymerisation of macromonomers, the metal utilised is always copper(I) associated to several ligands (Fig. 10, ligands –). Several initiators were also employed (Fig. 10, initiators –).
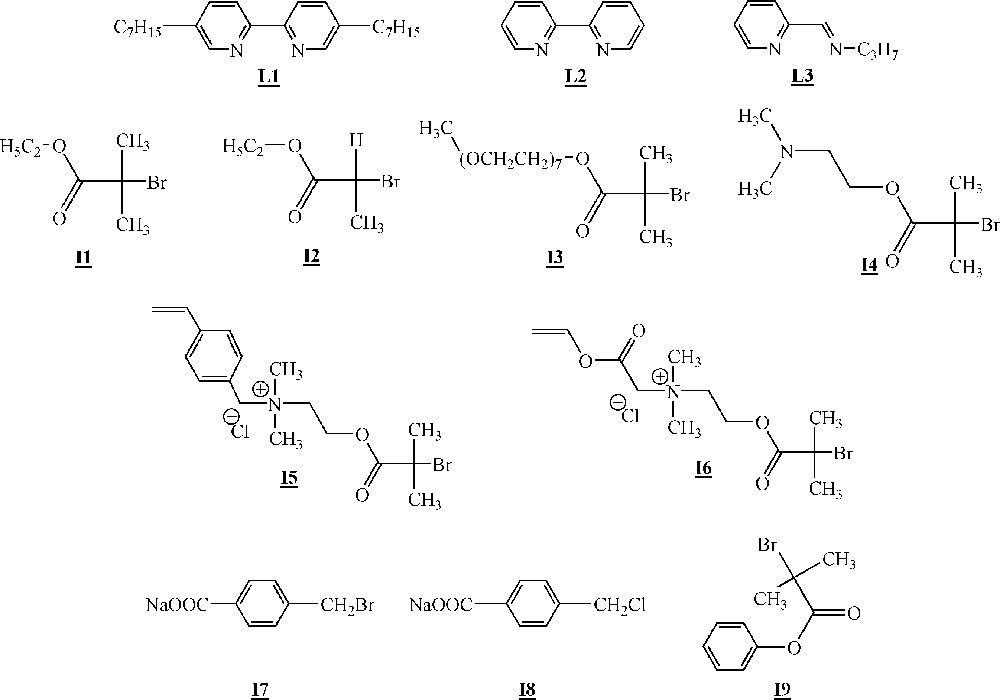
Ligands and initiators used for the ATRP of macromonomers 〚46–49〛.
Minoda and co-workers homopolymerised α-methacryloyl poly(isobutyl vinyl ether) macromonomers (Fig. 11) via ATRP 〚46〛. The macromonomers were synthesised by living cationic polymerisation. ATRP of the macromonomers was carried out using ligand (Fig. 10), Cu(I)Br and initiator (Fig. 10). All the evidences of a controlled polymerisation process were observed. Polymacromonomers with a degree of polymerisation of up to 200 were obtained for the shortest branches. The authors also observed that the polymerisation rate decreased with the length of the macromonomers.
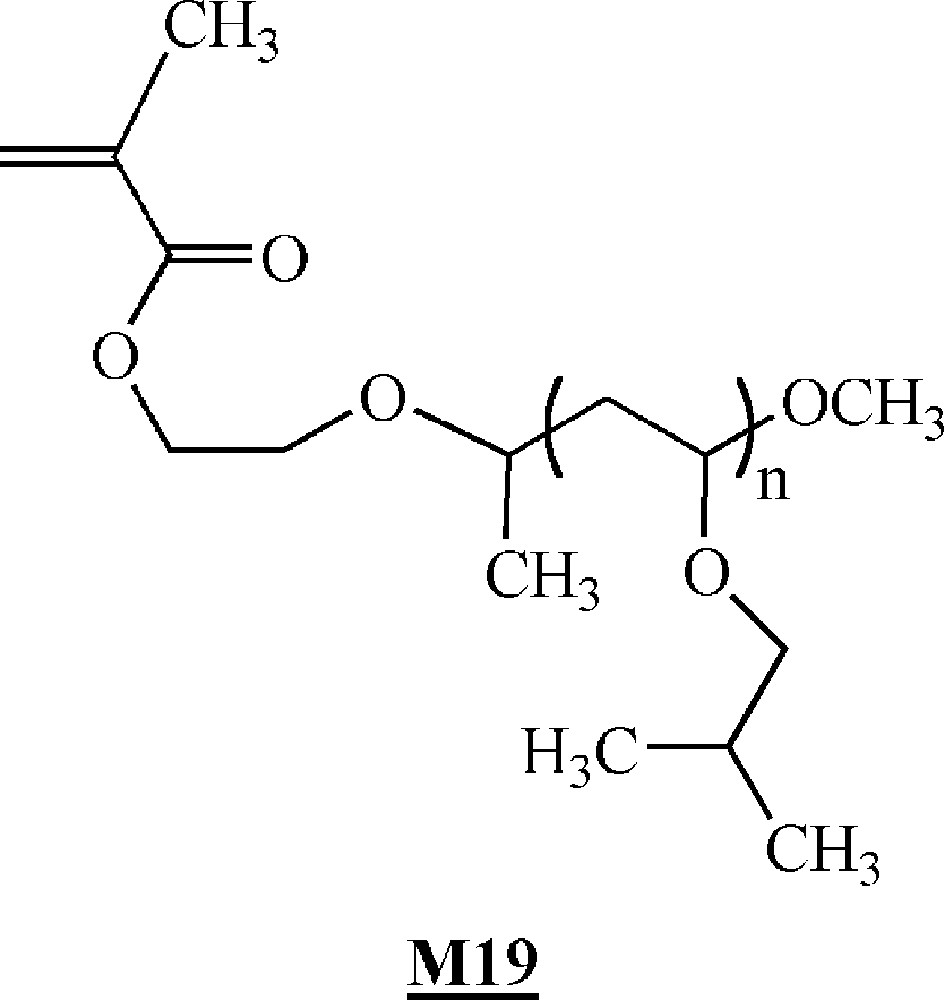
Macromonomers synthesised by Minoda and co-workers 〚46〛.
Some examples of ATRP of poly(ethylene oxide) (PEO) macromonomers are also mentioned in the literature 〚47–49〛. The macromonomers were obtained from commercial PEOs.
Armes and co-workers studied extensively the homopolymerisation of macromonomer (Fig. 12) using ligand , CuCl and several brominated initiators (initiators -, Fig. 10) 〚47, 48〛. The polymerisations were performed either in bulk or in aqueous solution at room temperature. It appeared clearly that the homopolymerisation was faster in water than in bulk. This was attributed to a change in the catalyst structure in more polar reaction medium. For the shortest branches (7–8 ethylene oxide units), quantitative yields were observed with measured molar masses in good agreement with the expected ones. For the longest branches (45 ethylene oxide units), quantitative yields were observed again, but shoulders appeared on the size exclusion chromatograms at high theoretical degrees of polymerisation (100).
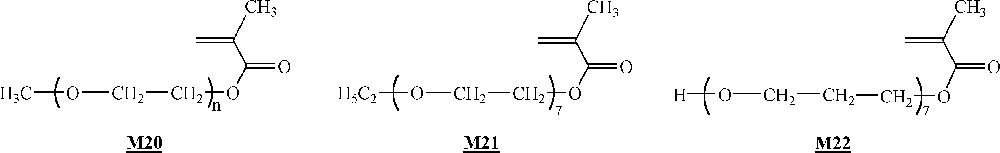
Macromonomers used by Armes and co-workers and Haddleton and co-workers, respectively 〚47–49〛.
Haddleton and co-workers have studied the homopolymerisation of methoxy end-capped poly(ethylene oxide) methacrylate (macromonomer , Fig. 12) using ligand (Fig. 10), Cu(I)Br and initiator (Fig. 10) in toluene 〚49〛. High yield (> 80%) and molar masses in good agreement with expected ones were observed for degrees of polymerisation up to 40. It also appeared that the rate of polymerisation was higher than foreseen. Like Armes and co-workers, they postulated that a change in the catalyst structure upon variation of the reaction medium polarity was responsible for the increased rate.
Like ROMP, ATRP provides a good control of the molar masses for the polymacromonomers, which is an important improvement compared to ‘classical’ free radical polymerisations. The maximum degree of polymerisation being attainable is also dependent on the macromonomer length. An additional advantage of ATRP and other living/controlled free radical processes results from the possibility to run polymerisation in water, which could be of great interest from an industrial point of view.
2.3 Ziegler–Natta type polymerisation 〚50–52〛
Metallocenes and late transition metal catalysts have been extensively utilised over the last few years for the (co)polymerisation of olefins 〚59–61〛, but only a few examples dealing with the homopolymerisation of macromonomers have been described in the literature.
Lutz and co-workers have studied the homopolymerisation of ω-terminated polystyrene macromonomers (Fig. 13) using early or late transition metal catalysts (Fig. 14) 〚50, 51〛. The macromonomers were synthesised via living anionic polymerisation.
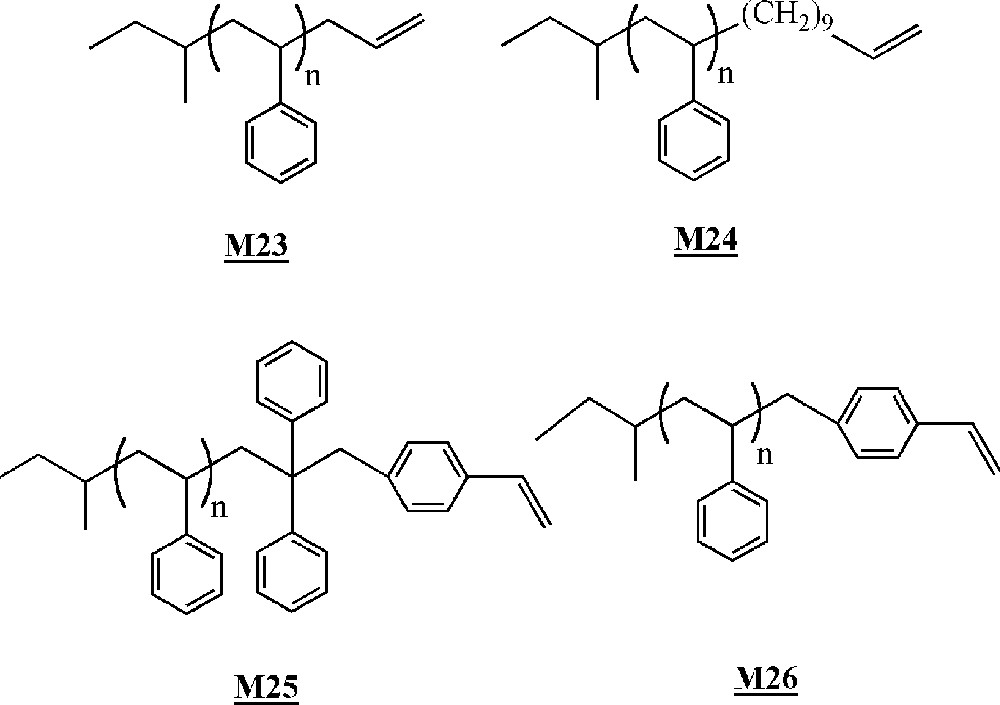
Macromonomers synthesised by Lutz and co-workers 〚50, 51〛.
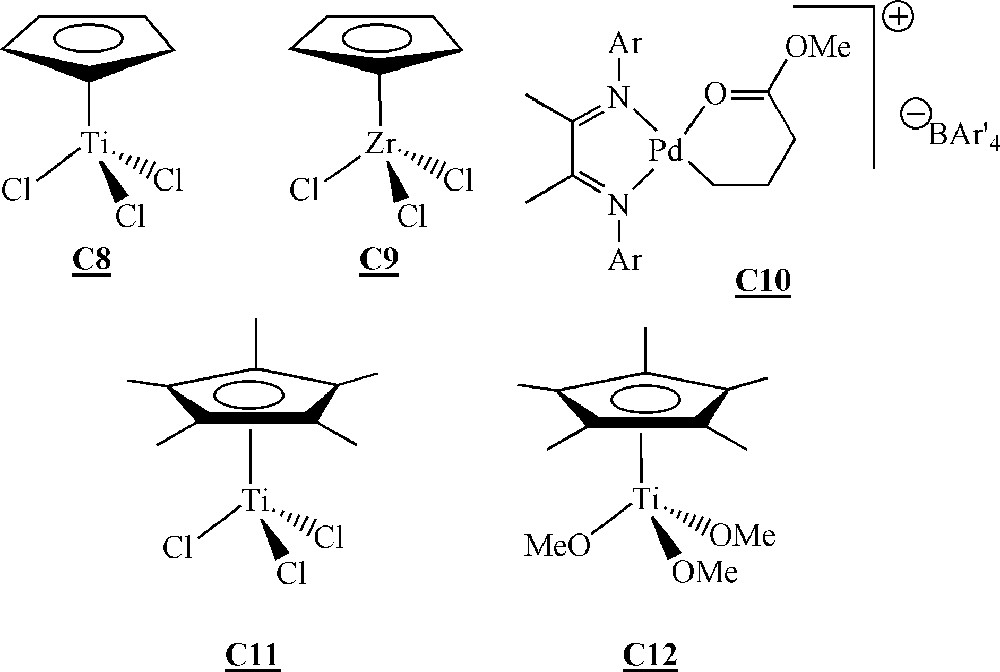
Catalysts for the homopolymerisation of macromonomers by Ziegler–Natta type polymerisation 〚50–52〛.
The homopolymerisation of macromonomers and was performed with catalysts , and . No homopolymerisation was detected with catalyst . With catalysts and , only dimerisation of the macromonomers occurred with low yields (< 10%).
Macromonomers and were homopolymerised with catalysts and activated by methylaluminoxane (MAO). The best results were obtained with catalyst and macromonomer (up to 60% conversion, degrees of polymerisation of up to 20). The better results obtained with macromonomer compared to macromonomer were explained by a decrease in the steric hindrance around the terminal reactive double bond, resulting in a facilitated coordination to the metal. The authors showed that even if there was no real control of the polymerisation, yields and molar masses were influenced by a couple of reaction conditions. The yield increased with the Al/Ti ratio, initiator concentration and polymerisation time. On the contrary, molar masses decreased with the Al/Ti ratio and initiator concentration, but remained constant with polymerisation time. Moreover, a lower polymerisation temperature led to higher molar masses but lower yields.
Endo and co-workers have investigated the homopolymerisation of ω-styryl polyisoprene macromonomers (Fig. 15) with catalyst , and (Fig. 14) activated with MAO 〚52〛. The macromonomers were synthesised via living anionic polymerisation. A degree of polymerisation of 10 with catalyst and a conversion of 30% with catalyst constitute the best-observed results. Like Lutz and co-workers, these authors observed an increase of the yield as well as a decrease of the molar masses at increasing Al/Ti ratio. They also obtained higher molar masses but lower yields by decreasing the polymerisation temperature.
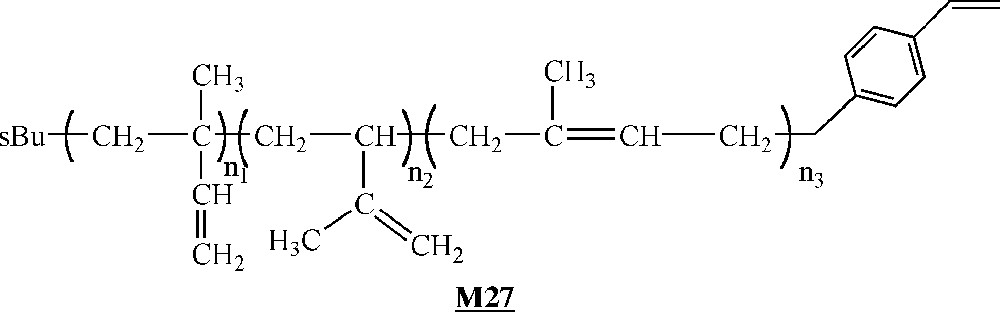
Macromonomers synthesised by Endo and co-workers 〚52〛.
As it was well known that the polymerisation of styrene catalysed by leads to highly syndiotactic polystyrene 〚62〛, the authors checked if the presence of polyisoprene side-chains influenced the expected stereoregularity. To this end, they removed the PI side chains by ozonolysis. Structural analysis by 13C NMR spectroscopy demonstrated that the remaining polystyrene backbone is highly syndiotactic, as expected.
The main advantage of Ziegler–Natta type macromonomer homopolymerisation lies in the fact that controlling the tacticity of the backbone is possible, leading to new comb-shaped polymer topologies.
2.4 Polymerisation of isocyanide-terminated macromonomers 〚53〛
Galin and co-workers have described the homopolymerisation of isocyanide-terminated macromonomers (Fig. 16) obtained by living cationic polymerisation of tetrahydrofurane, the living polymers being end-capped by 3-(dimethyl amino)propyl isocyanide.
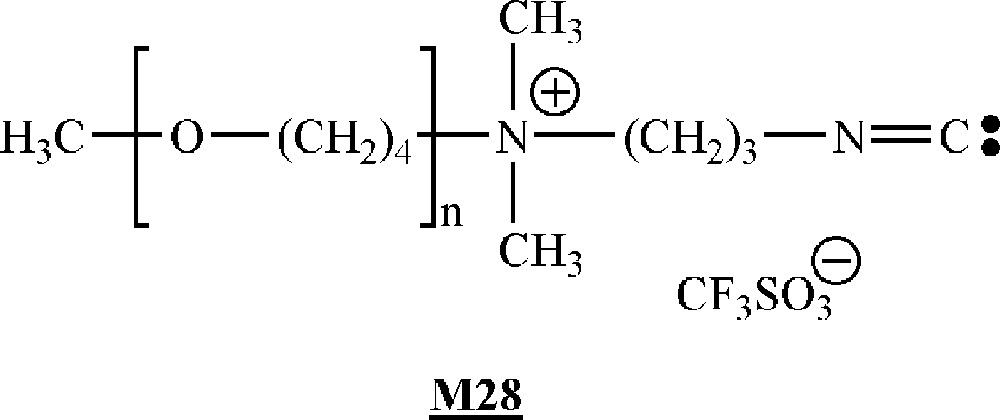
Macromonomers synthesised by Galin and co-workers 〚53〛.
The homopolymerisation was initiated by NiCl2. Very high molar mass () polymacromonomers were obtained. The molar masses increased with increasing macromonomer/initiator ratios. The degree of polymerisation of the polymacromonomer was also dependent on the macromonomer length, but not monotonously. The most important feature of these polymacromonomers is their very high degree of branching, each carbon atom of the backbone bearing a branch. This specific placements of the side substituents leads to super-hairy semi-rigid polymers.
3 Conclusions
In summary, transition metal-based polymerisation processes applied to macromonomers appears to represent an important breakthrough in the field. It is now possible, for example, to control the molar masses of polymacromonomers of different chemical nature (polystyrene, poly(ethylene oxide), polybutadiene...) via ROMP or ATRP over a wide range of molar masses. It is also possible to control the tacticity of the polymacromonomer backbone, via metallocene catalysis, opening a route to entirely new topologies.