

1 Introduction
If ten years ago somebody asked for covalently constructed, cylindrically shaped molecules with dimensions of a few to several hundred nanometres (diameter and length, respectively), he or she would presumably have been referred to some biological systems with helical conformation or to bucky tubes. No structure of this kind made by controlled synthetic procedures was known, although, depending on concrete structural conditions, interesting properties were to be expected. By merging both dendrimer [1,2] and polymer concepts, the situation has meanwhile changed and been developed to the point where one can choose between various molecular cylinders which differ in chemical constitution, stiffness, surface decoration, backbone properties, etc. [3–6]. After considerable effort in straightening and simplifying synthetic procedures, some of these cylinders can now even be provided on the 10 g scale with high and quantifiable structure perfection [7,8].
How have these two concepts been combined? Dendrimers consist of a few dendrons that are connected to a small core molecule. These dendrons have regularly and repeatedly branched structures that can reach respectable molar masses and quite densely fill volumes on the order of cubic nanometres, because of their branched nature. The idea was to attach dendrons of increasing size (generation) to a polymer instead of to a small core and to exploit the steric repulsion between them as a means to stretch the backbone. For that purpose they needed to have the largest realizable steric demand and be attached with the tightest possible spacing to the backbone. In the extreme case, a basically linear chain would result, which was tightly wrapped by a highly branched dendritic layer, or it would become a (filled) cylindrical object. Fig. 1 illustrates in cartoon form this transition of a coiled polymer into a cylindrical object by the attachment of increasingly more demanding dendrons.
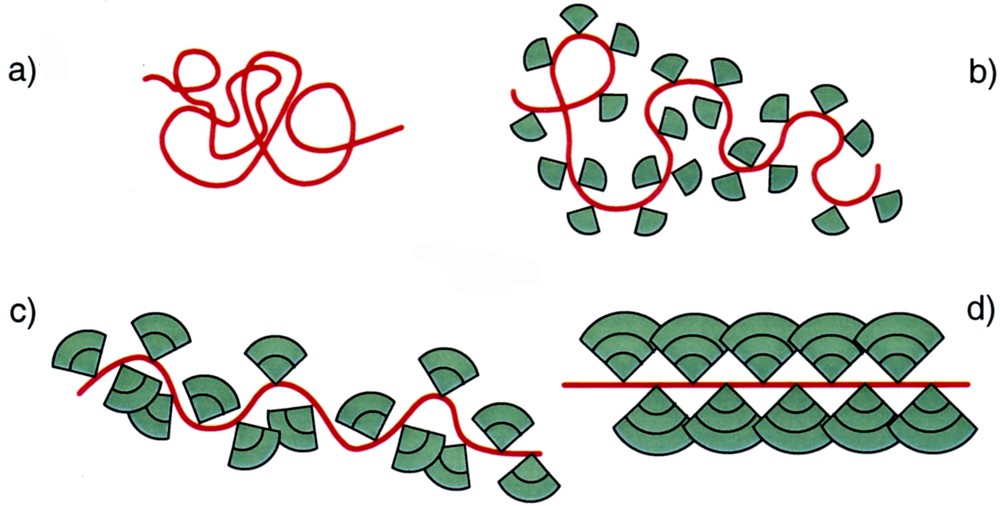
Cartoon representation of a coiled polymer backbone’s stretching through the attachment of increasingly sterically demanding dendrons. Polymer backbone with (a) no dendrons, (b) dendrons of the first, (c) second, and (d) third generation.
Higher generation dendronized polymers have a set of features which make them unique macromolecules and interesting candidates for a variety of applications, some of which were not foreseen at the beginning of this research. They are not only huge molecular objects with a cylindrical or near-cylindrical shape but their surface and backbone’s chemical nature are engineerable as well. Fig. 2 gives an overview of areas in which dendronized polymers were and are presently being put to work. This helps illustrate their versatility and explain why a steadily increasing number of research groups worldwide are now entering this fascinating field of research. For a more detailed discussion of their potential applications the reader is referred to the pertinent literature [3–6,9–22].
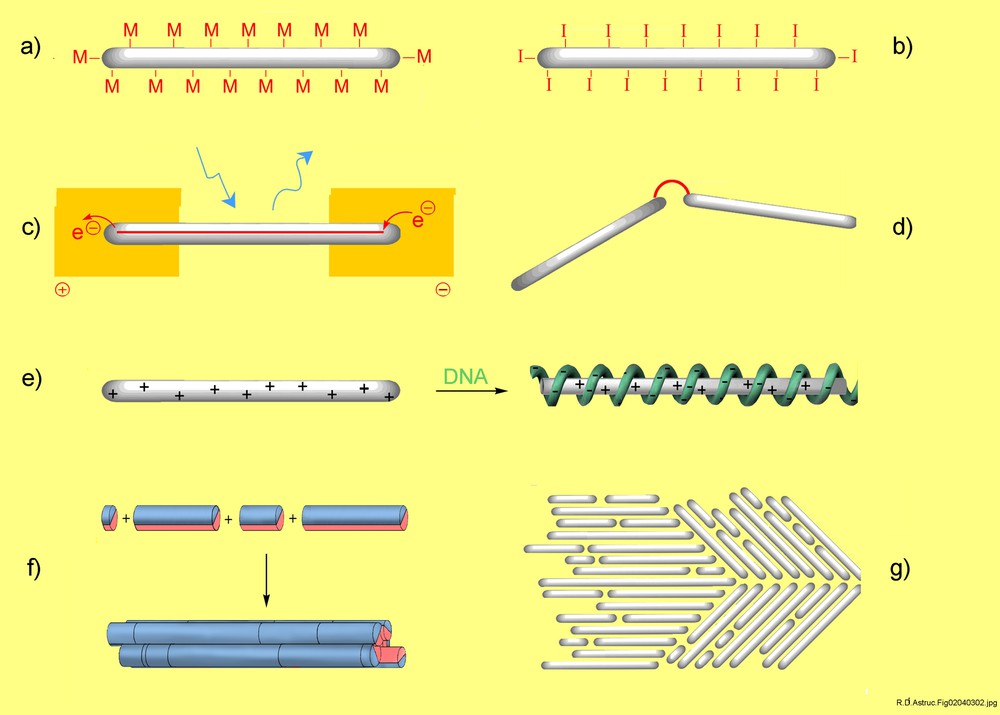
Dendronized polymers, represented as white/grey (or blue/red) cylinders, put to work as (a) catalyst (M) supports in nanodimensions [9], (b) polyinitiator for the synthesis of ‘hairy’ functional derivatives, energy transfer, light harvesting, and/or electrically conducting materials [10–15], objects for covalent attachment by the move–connect–prove strategy between individualized molecules on solid surfaces [16], novel, ultra-highly charged polyelectrolytes for, e.g., wrapping with DNA and subsequent gene transfection studies [17], lengthwise segregated polar/non-polar constituents of novel ‘supercylinders’ [18,19], nanoobjects for surface patterning and to induce periodicity changes from the Angstrom to nanometre scale [20,21].
Dendronized polymers can be approached by different strategies that have recently been compared in order to provide an overall feasibility picture [23]. This article focuses on the easiest of these approaches, the so-called macromonomer route. Here monomers are used which already carry the entire dendron, which ends up after polymerisation as a substituent on each repeat unit. This way the tedious divergent built-up of the dendritic layer using polymers as starting material is avoided. For space reasons we chose to concentrate here on the polymers rather than their precursors. There is no uniformly accepted definition of a dendron generation. This article does not make an attempt to remedy this problem and uses the nomenclature suggested by the respective authors instead.
2 Synthesis
The macromonomer route starts from monomers already carrying the final dendron (Fig. 3, shown for G4 dendrons). They are polymerised either according to (a) step– or (b) chain-growth mechanisms with all the enormous consequences this difference in mechanism has on the achievable molar mass and molar mass distribution, the applicability of controlled procedures, and the importance of monomer purity and stoichiometric balance, etc. This route has the advantage that the dendronized products are intrinsically structurally perfect regarding their dendrons, which is sometimes a critical aspect for the alternative routes in which the pendant dendrons are synthesized after polymerisation (not described here). On the other hand, the price paid for this advantage is sterical hindrance, which can cause polymerisation, especially of monomers with high generation dendrons, to furnish only low molar mass products or even no polymer at all. Because of the considerable molar mass of dendronized monomers, this route is normally referred to as the macromonomer approach. Synthetic aspects include finding out the macromonomer’s compatibility with the polymerisations conditions (no chain transfer, no loss of end groups, etc.) and the mechanism tolerating large dendron sizes. Also it is interesting to investigate the applicability of the so-called controlled methods to macromonomers in order to achieve more narrowly distributed dendronized polymers.
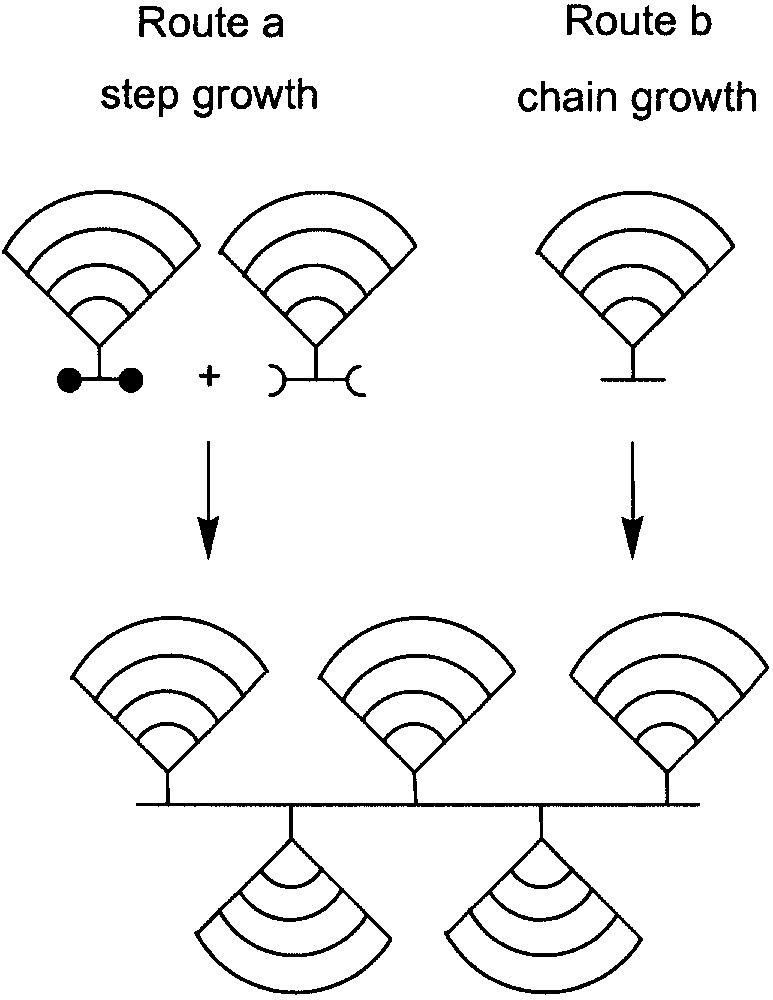
Cartoon-like illustration of the step-growth (route a) and chain growth (route b) macromonomer routes to dendronized polymers.
The steric repulsion intentionally implemented in many but not all of the final products sooner or later cause problems during synthesis in the sense that reaction centres (chain end and polymerisable unit of incoming monomer) cannot easily reach each other anymore. Consequently, growth reactions turn too slowly and side channels come into play. Thus, synthesis of densely packed dendronized polymers always has an element of unpredictability and seeming subtleties in the reaction conditions can have an enormous effect on the outcome. There are also more specific, structure related aspects that need to be considered whenever the synthesis of a dendronized polymer with a certain function or an application is the goal. The choice of reactions and conditions then depends on their compatibility with the respective requirement. This may pose considerable limitations to the tools available to a synthetic chemist and cause almost insurmountable hurdles.
2.1 Step-growth mechanisms
Dendronized polymers offer the unique opportunity to surround an electro-optically active (conjugated) backbone by its own solubilizing and/or shielding/insulating (dendritic) layer. It was, thus, a natural development to synthesize corresponding representatives and test for energy transfer and conductivity properties. Most work with conjugated backbones used step-growth mechanisms. For a recent report from Percec’s laboratory using chain-growth procedures to polyacetylenes with pendant dendronized carbozole units see below [24].
Aida [10] and Bao [11,12] were the first to report work along these lines. Aida et al. described the light-harvesting properties of 1(G2–4) and provided, i.e., evidence that the dendritic layer prevents collisional quenching of the excited state. Bao et al. reported on the poly(phenylenevinylene)s 2(G1,2) initially for their liquid crystalline properties but then also for their electro-luminescent ones. Recently Müllen also contributed interesting examples based on the polyfluorenes 3(n = 0) and 3(n = 1) (Fig. 4) [13,14]. Specifically the former turned out a useful material for organic light emitting diodes with blue emission. Their G1 dendrons do not interfere with the charge transport or the material’s emission properties and help the colour stability of the devices. Both polymers were synthesized according to the Yamamoto procedure, which is basically a Ni-catalysed reductive homocoupling of aromatic dibromides (here: dendronized 4,4'–dibromofluorene macromonomers) [25]. The GPC molar masses obtained are approximately on the order of 50–60 000 Da and, thus, within the normal limits for this method, even if one takes into account that the molar masses of rod-like polymers are normally overestimated by GPC versus polystyrene standard. This overestimation will be compensated to some unknown degree by the dendritic substitution’s opposite effect.
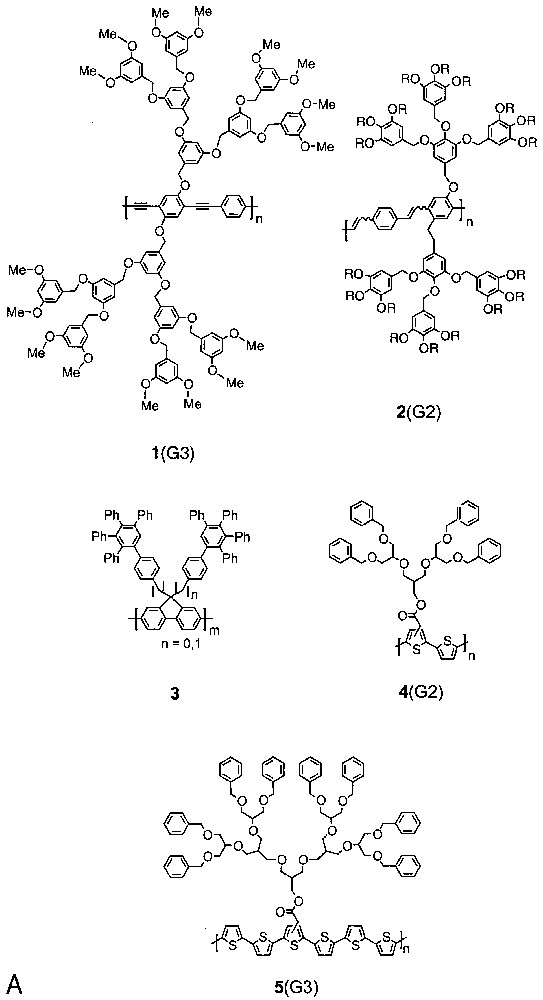
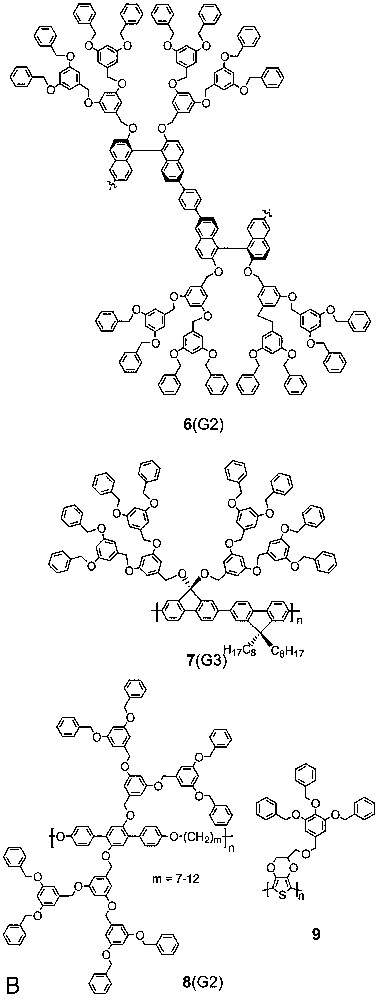
Dendronized polymers prepared according to step-growth procedures (route a).
Along similar lines, Fréchet reported first results on the synthesis of the dendronized polythiophenes 4(G2), 4(G3), 5(G2), and 5(G3) using the Stille cross-coupling procedure of AA and BB-type monomers [15]. They employed 2,5-bis(trimethylstannyl)thiophene as AA-monomer in all cases and coupled it with the dendronized dibromothiophene and dibromopentathiophene as the corresponding BB-type macromonomers. The dendron size was not sufficient for 4(G2) to mediate solubility in organic solvents. All other products were soluble. Stille cross coupling is normally accompanied by homocoupling and methyl group transfer reactions and, thus, is a problematical reaction for polymerization purposes [26]. This can be seen from the GPC elution curve of polymer 4(G3) which is rather complex. Nevertheless, high molar mass fractions could be obtained by preparative GPC and the electrical properties successfully tested.
Both these works are intentionally exploratory and preliminary. Synthesis was purposely only developed to the point where it could be seen whether the product actually shows the expected property. Since the properties in both cases look promising, a further optimisation of the synthetic procedures is justified.
Suzuki polycondensation (SPC) is perhaps the most powerful tool for synthesizing polyarylenes [27–29]. It was shown that even for macromonomers with enormous steric load (G4 Fréchet-type dendrons) high molar mass polymers could be obtained after some optimisation work [30]. It is, therefore, not surprising that this method has recently also been used for the synthesis of novel dendronized polyarylenes. Xi reported the chiral main chain representatives 6(G1) and 6(G2), whose chirality is based on the known atropisomeric dinaphthyl units [31]. 1,4-Benzene bisboronic acid served as AA-monomer and the G1 and G2 dendronized 6,6′-dibromo-1,1′-dinaphthyls, respectively, as the BB macromonomer counterparts. For 6(G1) a weight average molar mass was stated as 240 000 (corresponding to a weight average DPw = 250) and for 6(G2) as 11 000 (corresponding to DPw = 6). Although these values have to be considered with care, it is not easy to understand why the molar mass of the higher generation polymer seems to be drastically lower than for 6(G1). A problem here may have been the difficulty to precisely match the required 1:1 stoichiometry. Boronic acids tend to contain water and the weighed quantities have to be corrected accordingly, which is not always easy to do. SPC was also used by Shu et al. who reported on the dendronized polyfluorenes 7(G1–G3) [32]. As expected, they observed the photoluminescence efficiency to increase with increasing dendron size. This again underlines the potential importance of a dendritic layer’s shielding abilities for luminescent backbones in device technology.
The macromonomer has not only been followed by transition-metal mediated step-growth procedures. Kallitsis reported a successful polyether formation between G1 and G2 dendronized terphenyls with terminal phenolic groups and α,ω-dibromoalkanes as coupling partners to give 8(G1) (structure not shown) and 8(G2) [33]. The polymerisations were carried out in the two-phase system ortho–dichlorobenzene/NaOH (10 N) in the presence of a phase transfer catalyst. Depending on reaction time very respectable molar masses as high as, e.g., Mw = 280 000 (GPC calibrated by light scattering) for 8(G2) were obtained. It may have been helpful in this regard that the steric repulsion of consecutive dendrons is small. The polymers’ thermal and dynamic mechanical behaviour reveals a tendency to phase separation of the dendritic fragments and the main chain. Kumar reported the electrochemical synthesis of G1 polymer 9, which was prepared for its electrochromic properties [34].
2.2 Chain-growth mechanisms
As pointed out above and in the earlier reviews, a main concern with the macromonomer route is the achievement of high molar mass polymers for macromonomers with generation three and higher [4]. In most cases, irrespective of polymerisation mechanism, only relatively low molar mass products were obtained. This general observation was further substantiated by a work of Scrivante and Chessa [35]. They tried to synthesize polymers 10(G1–G3) that carry rather interesting pyridine containing dendrons for complexation studies (Fig. 5). Reasonable molar masses could only be obtained for 10(G1) by standard radical techniques. Already for the next higher homologue, 10(G2), the product contains oligomers, and dimers and trimers form the majority of the material for 10(G3).
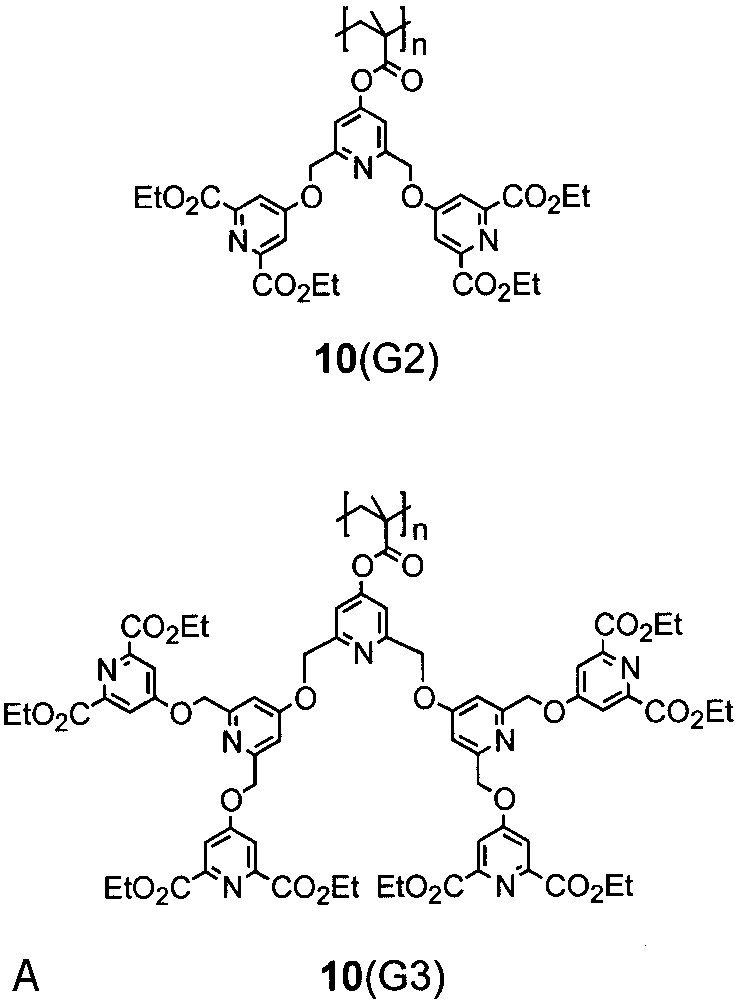
Dendronized polymers prepared according to chain-growth procedures (route b).
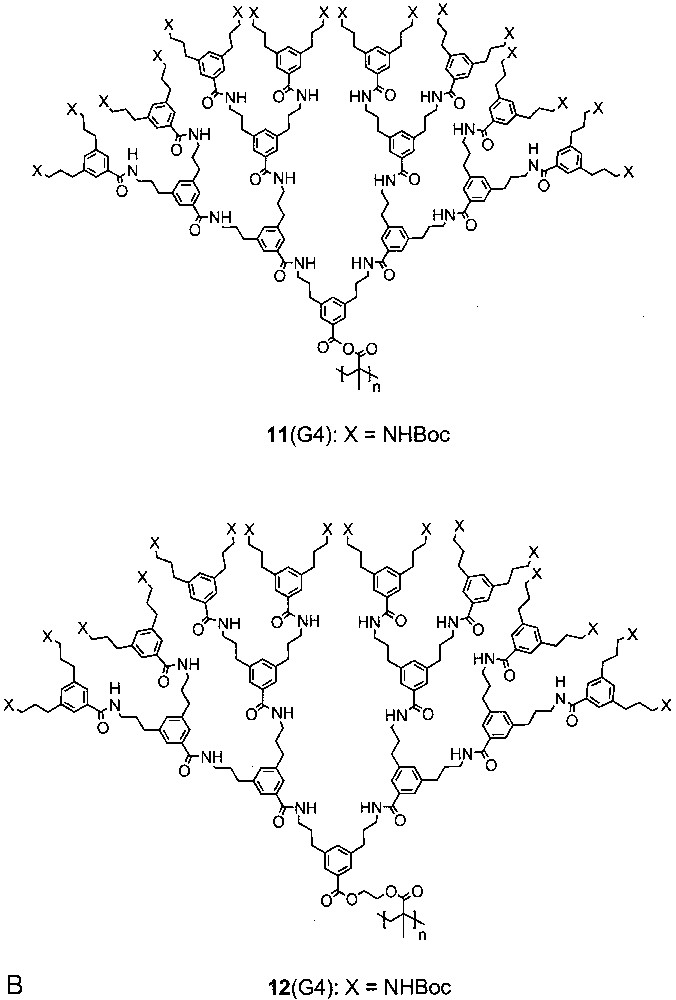
In an intense effort to see whether higher generation vinyl-type macromonomers can be polymerised to high molar mass product, the two homologous series of dendronized methacrylate-based macromonomers were prepared, obtained on the several-gram scale as analytically pure materials, and polymerised to 11(G1–G4) and 12(G1–G4) [8]. The only difference between these two series is the ethyleneoxy spacer of 12. This spacer was introduced because it was not clear from the literature whether or not a spacer facilitates polymerisation, as one intuitively may be inclined to think. Both series of macromonomers were polymerised under radical conditions. For series 11 either thermally induced radical polymerization (TRP) or atom transfer radical polymerization (ATRP [36]) was applied, and for series 12 only TRP. The molar masses were determined by GPC in THF (referenced to polystyrene standard) and in DMF (referenced to PMMA standard) [8]. (Under thermally induced radical polymerization, we understand the fully reproducible experimental observation that dendronized macromonomers upon removal of the solvent in vacuum at somewhat elevated temperatures start to polymerise to very high molar mass material at high conversion without any initiator added).
TRP was applied to all monomers of series 11 and gave polymers whose molar masses decrease from several millions for 11(G1) to several 100 000s for 11(G2) and 11(G3) and to approximately 100 000 for 11(G4). ATRP was only applied to 11(G1) and 11(G2). The highest monomer-to-initiator ratio which still gave monomodal molar mass distributions was 300:1 [for 11(G1)] and 100:1 [for 11(G2)] which corresponds to an achievable molar mass regime for both polymers of approximately Mn = 100 000 [DPcalc (11(G1)) = 300; DPcalc (11(G2)) = 90]. The polydispersities lay in the normal range (PDIs 1.1–1.2). The molar masses of series 12 (obtained by TRP only) were practically identical for all generations. In all TRP experiments it was of utmost importance to reach the highest synthetically achievable concentration. If fluidity was sufficient, they were carried out in bulk. Otherwise the molar masses dropped sharply, sometimes to a point where no polymer was obtained at all [37].
Which conclusions can be drawn from these surprising findings? (a) High molar mass dendronized polymers can be obtained at least from some vinyl-type macromonomers if high concentrations are applied. Even fourth generation macromonomers can be polymerised. (b) Qualitatively there is an inverse dependence of molar mass on generation number. (c) Controlled radical procedures are applicable for G1 and G2 monomers. As far as monomodal and narrowly distributed products are concerned, however, the maximum achievable molar mass is on the order of 100 000. Finally, (d) at least for the ethyleneoxy group there is no recognizable spacer effect. Monomer purity and concentration of the polymerisable unit seem to play the major role.
3 Comments on characterization
Characterization of dendronized polymers is a bit like taming a wild beast. Specifically for the high molar mass cases, which can easily be on the order of a few million, NMR spectroscopy, the synthetic chemist’s most favourite toy, cannot be reasonably used anymore. Lines can become so broad that they virtually disappear in the baseline not to mention showing any fine structure. Determinations of coverage as briefly discussed above have to therefore be done with quantitative UV absorbance or fluorescence measurements after appropriate labelling. NMR spectroscopy has been usefully applied in several cases where the molar mass did not exceed several 100 000s. A specific problem of high molar mass dendronized polymers is their work-up. Specifically if they have relatively low generation dendrons, their products should not be completely dried, because this sharply reduces their solubility and makes them completely insoluble, even when treated with high-boiling organic solvents for extended periods of time. This effect seems to be general with the level of gravity depending on the concrete structure. Only in freeze-dried form can dendronized polymers be re-dissolved. For polymers with higher generation dendrons, solubility is not such a critical problem. This may indicate that the lower generation representatives have a higher tendency to entangle and/or interdigitate. The presumed higher stiffness of higher generation ones keeps them from doing this. A broad investigation of the molar mass determination by light scattering and small angle neutron scattering techniques was started a while ago and is still being continued [38].
An interesting tool for molar mass determination is the scanning force microscope (SFM). Except when the aggregation is too strong, polymer samples can be spin-coated on a solid surface in a form so that basically all chains can be individually imaged. Under certain circumstances this allows measurement of contour lengths and lengths distributions and combination of these data into a histographic analysis. Such analyses have, for example, been done for dendronized polymers [7] and represent an interesting and relatively easy to do complementary tool for scattering techniques. The obtainment of correct data from elemental analysis is another problem. Because of the mere dimensions of these highly branched structures, solvents are frequently entrapped, even after prolonged treatment of freeze-dried material in high vacuum. Repetition of the same procedure at elevated temperatures can sometimes be helpful. Solvent-free NMR spectra can be obtained, if necessary, by repeated dissolution/drying cycles with deuterated solvents. This way non-deuterated solvent molecules are exchanged. This does not, of course, help achievement of correct data from elemental analysis.
4 Summary and outlook
Where does this lead us now that the main synthetic problems to high molar mass dendronized polymers with high generation dendrons at every repeat unit have been solved, at least for a few cases with different structures and polymerization methods and for the most part by following the macromonomer route? Two factors immediately come to mind under synthetic aspects: chain length control and surface decoration. Promising attempts have already been undertaken for both, but it will require a few more years, specifically regarding length control, until this matter can be considered as solved. Presently a non-trivial problem appears to be reaching narrowly distributed material, for example, by ATRP over a molar mass of 100 000. From a more speculative side, there is the question of whether there are other targets for the dendronization approach. Would it be feasible, for example, to merge this concept with the biological world of viruses, proteins, membranes, and alike, or the colloid sciences? One may also ask whether certain dendronized polymers have a biological function because their size resembles several biological functional units. Considerable potential is also seen in the use of dendronized polymers both with appropriate surface decoration and internal structural conditions as constituents for the bottom-up approach to functional units on the molecular level. They are amongst the largest man-made molecules ever, which enables application of physical tools like the SFM for manipulating, connecting, and oxidizing/reducing, whose resolution is just below the length scale of these cylinders’ diameter. Finally, it would be desirable to initiate more exchange between people doing dendronized polymers and others working with the so-called poly(macromonomers) [39–46]. The latter polymers have linear chains of considerable molar mass attached to their backbones and in a sense complement the ones with branched chains discussed here.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (Sfb 448, TP A1), which is gratefully acknowledged. I wish to thank my co-workers, mainly Afang Zhang, Lijin Shu, and Zhishan Bo for their invaluable input to the project ‘Synthesis of dendronized polymers’, which I greatly appreciated at all times.