

1 Introduction
It has been well documented that chemical properties of organotransition metal complexes may be finely tuned by supporting ligands both from electronic and steric points of view [1]. In this context, diversity of ligand choice is a highly desirable situation for designing the most appropriate catalysts for individual reactions. From the electronic point of view, ligands are roughly classified into two categories, σ-donors and π-acceptors. The former type has a wide variety of selections, including tertiary phosphines, nitrogen bases (e.g., 2,2′-bipyridine, diimines), cyclopentadienyl and its derivatives, and Arduengo-type carbenes (i.e., 1,3-diarylimidazol-2-ylidenes) [2]. On the other hand, the supporting ligands that possess strong π-accepting ability are still limited. Thus, although carbon monoxide and isocyanides are well-known π-acceptors for transition metals, they are frequently reactive toward organometallic species and therefore unfit as efficient ligands.
We recently demonstrated that 1,2-diaryl-3,4-diphosphinidenecyclobutenes bearing sp2-hybridized phosphorus as coordinating atoms (DPCB–Y, Chart 1) can be used as highly effective π-acceptors for palladium and platinum [3–10]. They are fairly stable in catalytic systems, and form novel organometallic complexes with unique catalytic properties. This account summarizes those results. The related chemistry for other low-coordinated phosphorus compounds has been reviewed [11].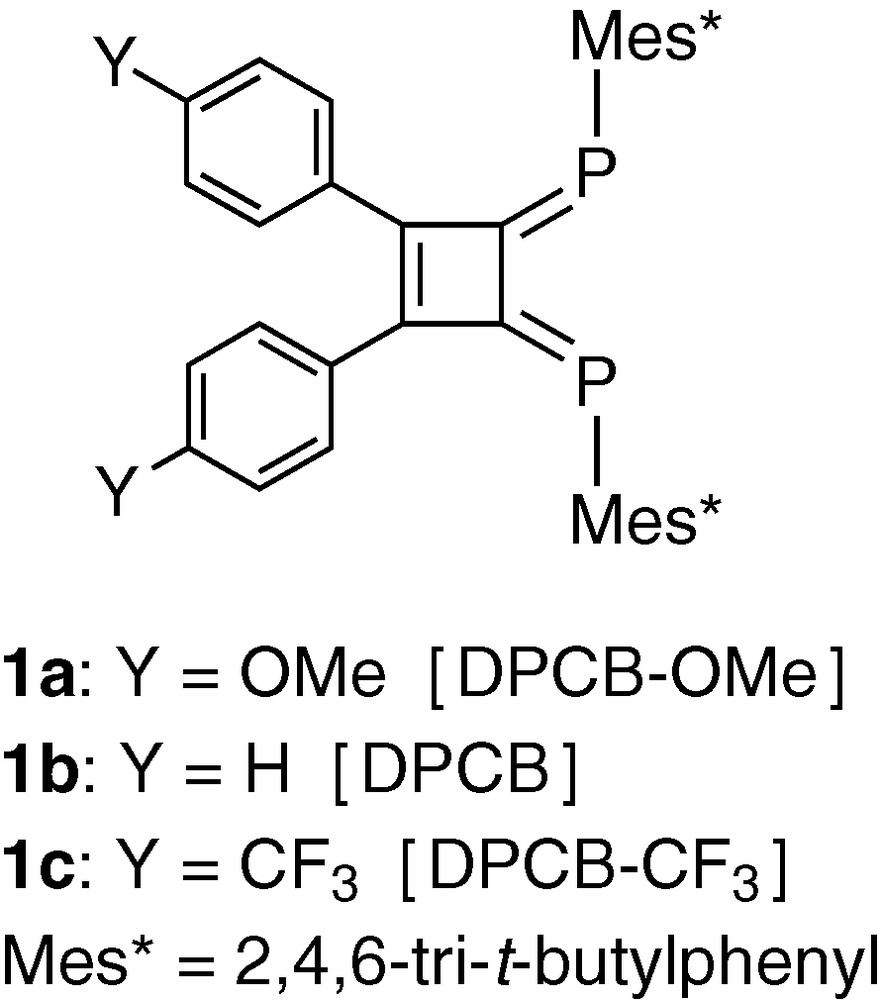
2 Preparation and structures of DPCB–Y complexes
2.1 Preparation of DPCB–Y ligands
The synthetic routes to diphosphinidenecyclobutenes have been examined by three groups [12–14]. In the present study, we prepared DPCB–Y ligands by a slightly modified method of Appel's route (Scheme 1) [8,15]. Thus, arylacetylenes were lithiated and allowed to react with ClP(NMe2)2 [16] in Et2O at –78 °C (steps (i) and (ii)). The (arylethynyl)bis(dimethylamino) phosphines thus formed were converted without isolation to dichlorophosphines by the treatment with dry HCl at room temperature (step (iii)). Dimethylammonium chloride generated in the systems was removed by filtration after precipitation with pentane, and 2,4,6-tri-t-butylphenyl group (Mes*) was introduced in THF using Mes*Li [17] (step (iv)). Treatment of the resulting chlorophosphines bearing arylethynyl and Mes* groups with zinc powder at room temperature led to P–P coupling, followed by Cope rearrangement to give bis(phosphaallenyl) intermediates, which are spontaneously converted into 1 in the reaction systems (step (v)). The products were isolated by silica-gel column chromatography with hexane elution as bright yellow crystalline solids in 30–50% yields based on arylacetylenes employed. Step (v) must be operated in the dark, otherwise (E,Z)-isomers of 1 are obtained as major products [14]. This step takes half a day for completion. The reaction progress may be followed by 31P NMR spectroscopy.
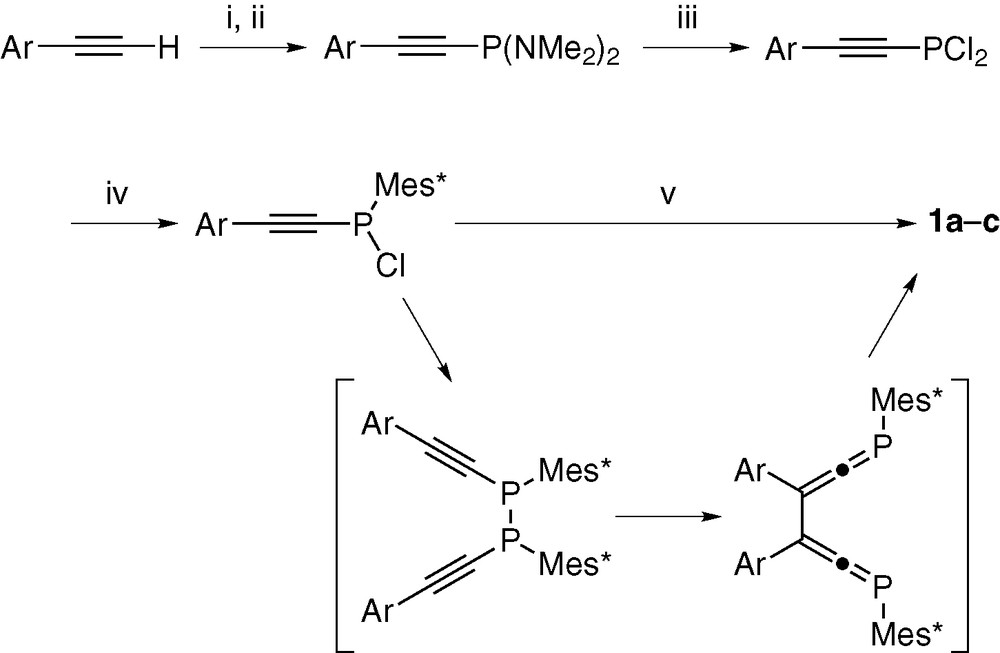
(i) n-BuLi/Et2O/–78 °C, (ii) ClP(NMe2)2/Et2O/–78 °C, (iii) dry-HCl/Et2O/room temp., (iv) Mes*Li/THF/–78 °C, (v) Zn/THF/room temp.
2.2 Preparation and structures of DPCB–Y complexes
Dichloro complexes MCl2(DPCB–Y) (M = Pd, Pt) were prepared by the reactions of MCl2(MeCN)2 with the ligands in benzene or THF, and the X-ray structure of PdCl2(DPCB) was reported [3]. On the other hand, dimethyl and monomethyl complexes were synthesized as summarized in Scheme 2 [8]. Ligand displacement of Pt2Me4(μ-SMe2)2 and PdMe2(tmeda) (tmeda = N,N,N′,N′-tetramethylethylenediamine) with DPCB–Y ligands proceeded in Et2O at room temperature to give reddish orange or orange crystalline solids of dimethyl complexes 2 and 3 in 80–99% yields. The use of Et2O as solvent was of particular importance for synthesizing the complexes in high yields. Thus, the synthetic reactions proceeded heterogeneously in Et2O, and this condition was profitable to minimize undesirable side reactions leading to degradation of DPCB–Y ligands. The dimethyl complexes thus prepared reacted smoothly with trifluoromethanesulfonic acid (HOTf) in Et2O at room temperature to give monomethyl complexes 4 and 5 in over 82% yields after isolation [8,9].
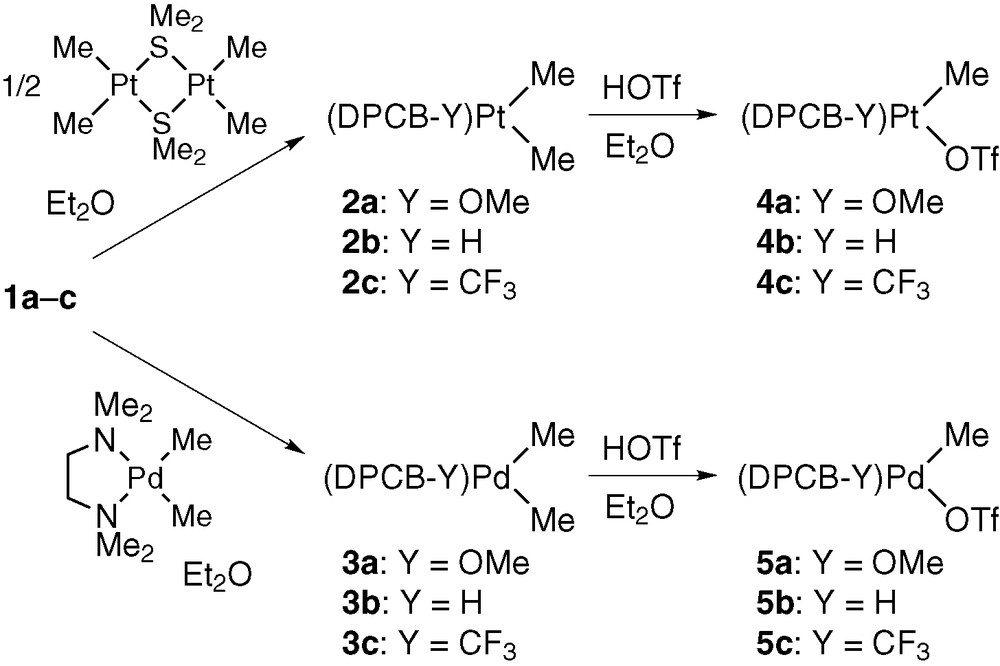
Fig. 1 shows X-ray structures of 2a and 2b [4,8]. Both complexes adopt square planar geometry around the platinum. The coordination plane (A) is coplanar to the cyclobutene ring (B), but almost perpendicular to the aryl rings (C and D) on the phosphorus atoms. The most interesting feature is significantly the parallel orientation of the two benzene rings (E and F), which are nearly coplanar with the cyclobutene ring (B). Thus, the dihedral angles between B and E (or F) are 25.2(2) and 25.7(2)° for 2a and 23.2(1)° for 2b, respectively, and these values are significantly smaller than that of free DPCB (40.0 and 42.6°) [12]. This fact could be rationalized by assuming the presence of a wide π-conjugated system spread over the molecule including platinum, diphosphinidenecyclobutene ring, and two benzene rings [8]. The effective π-conjugation between platinum and DPCB–Y is caused by back-donation of dπ electrons on platinum to the π* orbitals of the ligand.
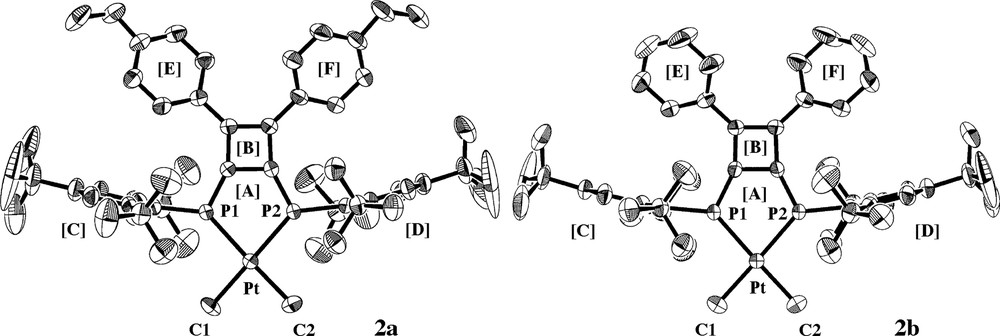
Molecular structures of 2a and 2b. Hydrogen atoms are omitted for simplicity. Selected bond distances (Å) and angles (deg): (2a) Pt–P1 = 2.2948(7), Pt–P2 = 2.2878(8), Pt–C1 = 2.089(4), Pt–C2 = 2.074(3), P1–Pt–P2 = 83.33(3), P1-Pt–C1 = 96.2(1), P2–Pt–C2 = 94.5(1), C1–Pt–C2 = 85.9(2); (2b) Pt–P1 = Pt–P2 = 2.2909(8), Pt–C1 = Pt–C2 = 2.093(3), P1–Pt–P2 = 82.85(4), P1-Pt–C1 = P2–Pt–C2 = 96.1(1), C1–Pt–C2 = 85.1(2). Dihedral angles between least-square planes (deg): (2a) [A]–[B] = 0.8(1), [A]–[C] = 95.71(8), [A]–[D] = 90.93(8), [B]–[E] = 25.2(2), [B]–[F] = 25.7(2); (2b) [A]–[B] = 1.4(1), [A]–[C] = [A]–[D] = 92.5(1), [B]–[E] = [B]–[F] = 23.2(1).
The occurrence of strong π-back-donation interaction between platinum and DPCB–Y ligands was also indicated by unusually downfield chemical shifts of the ethylene protons in [PtMe(η2-ethylene)(DPCB–Y)]OTf complexes (δ 5.02–5.22). On the other hand, DPCB–Y ligands were found to be intermediate σ-donors between diphosphines and diimines [8].
The (π-Allyl)palladium complexes bearing DPCB–Y ligands (6) were synthesized in 85–96% yields by the reactions of [Pd(η3-C3H5)(μ-Cl)]2 with DPCB–Y and AgOTf in CH2Cl2 at room temperature (Scheme 3) [9]. Fig. 2 shows the X-ray structure of 6b [5]. The C1–C2 and C2–C3 distances for the allyl ligand (1.376(8) and 1.383(8) Å, respectively) were very similar to those of [Pd(η3-C3H5)(dppf)]OTf (1.372(8) and 1.305(8) Å; dppf = 1,1′-bis(diphenylphosphino)ferrocene). Furthermore, the three Pd–C distances (2.168(5)–2.178(4) Å) were also comparable to those of the dppf complex (2.171(6)–2.187(4) Å).
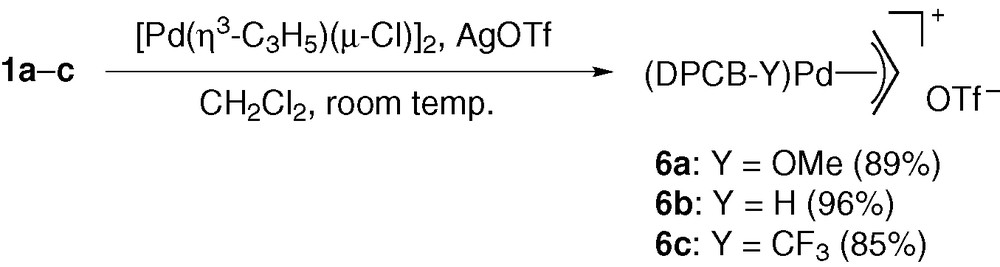
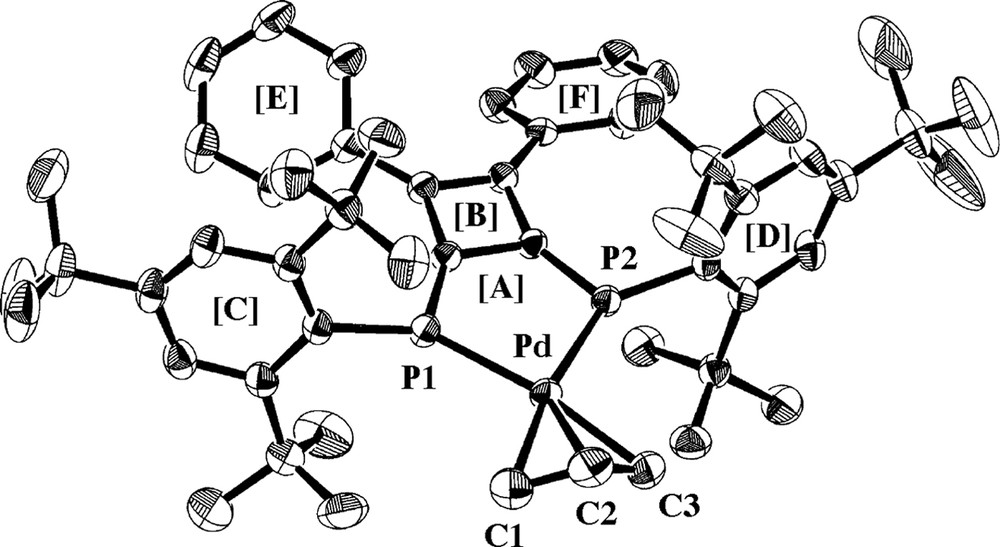
Molecular structure of 6b. Triflate anion and hydrogen atoms are omitted for simplicity. Selected bond distances (Å) and angles (deg): Pd–P1 = 2.322(1), Pd–P2 = 2.326(1), Pd–C1 = 2.178(4), Pd–C2 = 2.168(5), Pd–C3 = 2.168(5), C1–C2 = 1.376(8), C2–C3 = 1.383(8), P1–Pd–P2 = 85.50(4), C1–C2–C3 = 119.9(5). Dihedral angles between least-square planes (deg): [A]–[B] = 2.3(1), [A]–[C] = 100.3(1), [A]–[D] = 93.2(1), [B]–[E] = 32.2(2), [B]–[F] = 28.2(2).
Table 1 lists the 1H NMR data for the allyl ligands of 6a–c, together with the data for dppf and diimine analogues. Although the signals tend to shift to downfield in the order [diimine < dppf < DPCB–OMe (1a) < DPCB (1b) < DPCB–CF3 (1c)], the variation of the chemical shifts with ligands is rather small. Thus, the (π-allyl)palladium moiety of 6a–c has a similar structural feature to that of diimine and diphosphine analogues. However, it was found that the DPCB–Y complexes exhibit extremely high reactivity in stoichiometric and catalytic systems (vide infra).
1H NMR chemical shifts (δ ppm) for (π-allyl)palladium triflates in CDCl3 at 20 °C
| Ligand | anti-H | syn-H | central-H |
| DPCB–OMe (1a) | 3.64 | 4.92 | 5.86 |
| DPCB (1b) | 3.73 | 4.99 | 5.94 |
| DPCB–CF3 (1c) | 3.91 | 5.11 | 6.06 |
| dppf | 3.47 | 4.00 | 5.87 |
| diiminea | 3.53 | 3.65 | 5.83 |
a diimine = bis(phenylimino)acenaphthene.
3 Catalytic reactions using DPCB–Y complexes
Reflecting the strong π-accepting ability of DPCB–Y ligands, their palladium and platinum complexes show rather unique chemical properties leading to highly efficient metal-catalysed reactions including Sonogashira coupling of p-bromonitrobenzene with trimethylsilylacetylene [3], ethylene-polymerisation [4,18], hydroamination of 1,3-dienes with aniline [5], dehydrogenative silylation of ketones [6], and direct conversion of allylic alcohols into N- and C-allylation products [7]. The following sections deal with the last two reactions, mainly focusing on their mechanisms.
3.1 Dehydrogenative silylation of ketones
Dialkyl and alkyl-aryl ketones readily react with HSiMe2Ph at 50 °C in the presence of catalytic amounts of methylplatinum triflate 4b (3 mol%) and pyridine (3 mol%), affording exclusively silyl enol ethers as dehydrogenative silylation products (Scheme 4) [6]. This reaction may be conducted in toluene or without solvent. The lack of hydrosilylation products (i.e., R1CH(OSiMe2Ph)CH2R2) is of particular interest, because platinum complexes are generally highly efficient for hydrosilylation.
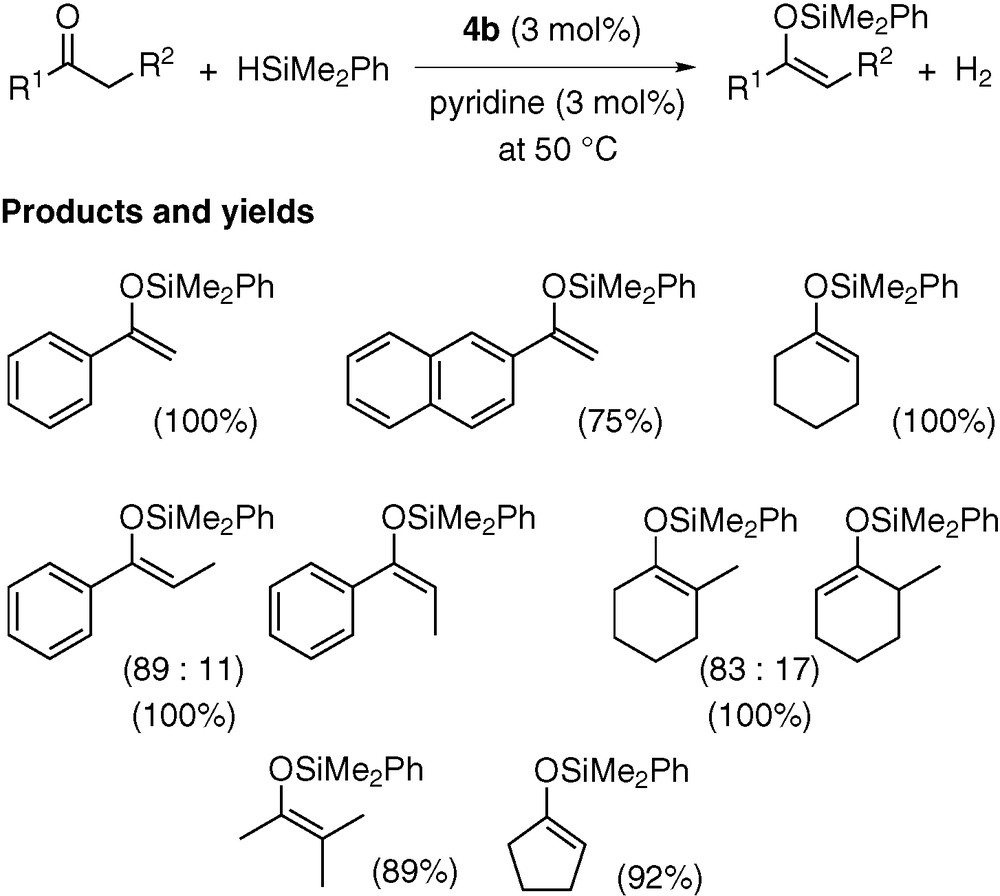
Scheme 5 illustrates our proposed mechanism for the reaction of acetophenone. Initially, 4b reacts with HSiMe2Ph, pyridine, and residual water in the system to give methane, PhMe2SiOSiMe2Ph, pyridinium triflate, and hydrido(silyl)platinum 7b (step (i)). This complex is interconverted with a platinum(0) species A (step (ii)). Oxidative addition of HOTf to A forms hydridoplatinum B, which is in equilibrium with its dimer 8b (steps (iii) and (iv)). Coordination of ketone to B, followed by proton-abstraction from C by pyridine, gives a platinum enolate D and pyridinium triflate (steps (v) and (vi)). The Pt–O bond in D is then cleaved by hydrosilane to afford silyl enol ether as the catalytic reaction product and dihydridoplatinum E (step (vii)). Finally, reductive elimination of H2 from E regenerates A (step (viii)).
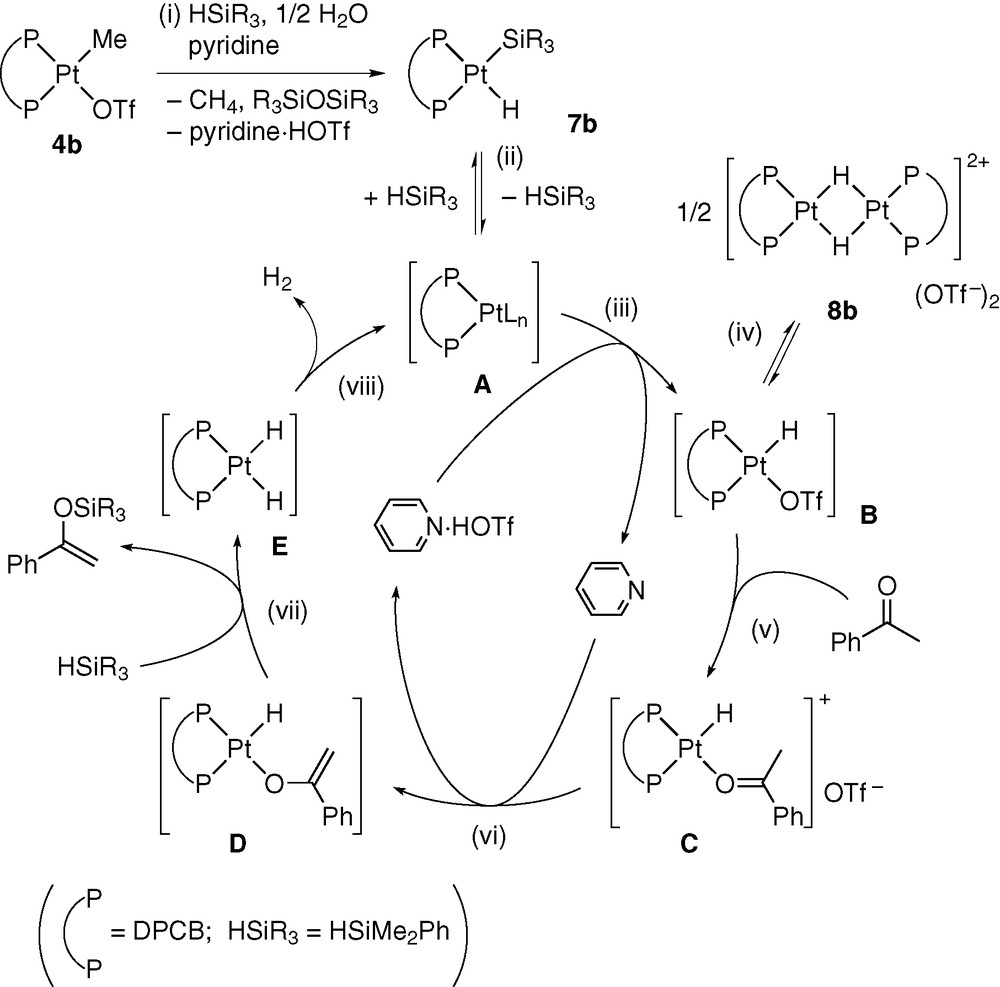
Several steps in this mechanism could be confirmed by stoichiometric reactions [9]. Thus, 4b actually formed 7b in quantitative yield by the treatment with pyridine (1 equiv) and an excess amount of HSiMe2Ph (10 equiv) in wet CD2Cl2 at room temperature. In this reaction, the formation of methane and PhMe2SiOSiMe2Ph was confirmed by 1H NMR spectroscopy and GLC. On the other hand, in the absence of pyridine and with 1 equiv of HSiMe2Ph, 4b was converted to 8b in quantitative yield. Hence, the sequence of the reactions in steps (i) to (iv) was reproduced. Complexes 7b and 8b, independently prepared from Pt(cod)2 (cod = 1,5-cyclooctadiene), efficiently catalysed the dehydrogenative silylation. The reaction of acetophenone with HSiMe2Ph (1 equiv) in the presence of 7b (3 mol%) and pyridinium triflate (6 mol%) at 50 °C was complete in 3.5 h to give a quantitative yield of CH2=C(OSiMe2Ph)Ph. Furthermore, the reaction in the presence of 8b (1.5 mol%) and pyridine (3 mol%) formed the dehydrogenative silylation product, quantitatively, in 3.5 h at 50 °C. These catalytic activities were comparable to that observed for 4b. Thus, the reaction with 4b and pyridine (each 3 mol%) took 4.5 h at 50 °C for completion.
3.2 Catalytic conversion of allylic alcohols into N- and C-allylation products
The hydridoplatinum complex 8b exhibited interesting reactivities toward other organic substrates (Scheme 6) [9]. For example, 8b reacted with dienes in ClCH2CH2Cl at 50 °C to give the corresponding π-allyl complexes 9a and 9b. Furthermore, the reactions of 8b with allylic alcohols formed π-allyl complexes 9c–e. All reactions proceeded cleanly without side-reactions as confirmed by 31P NMR spectroscopy.
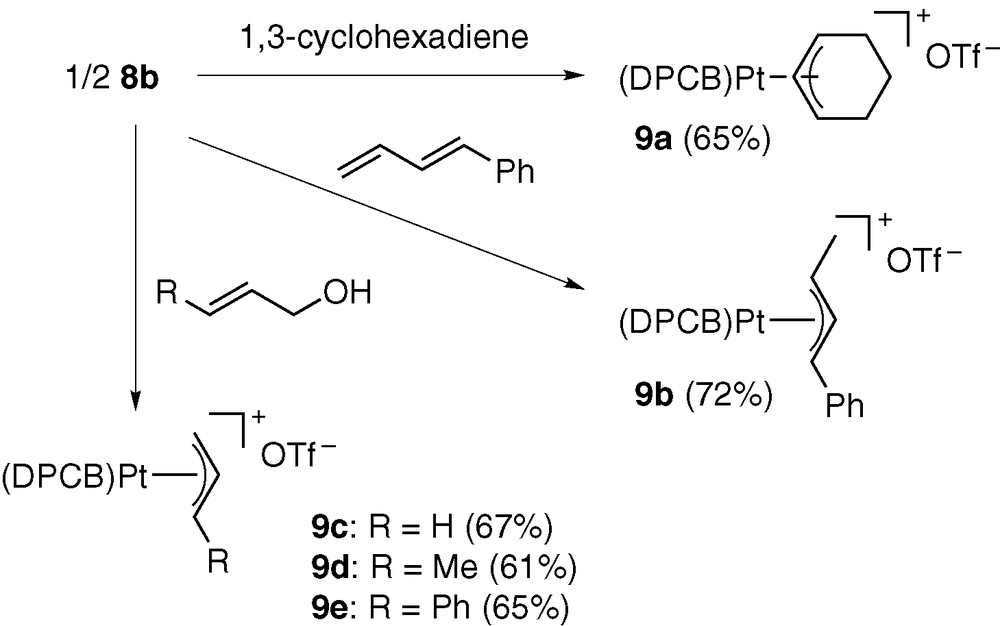
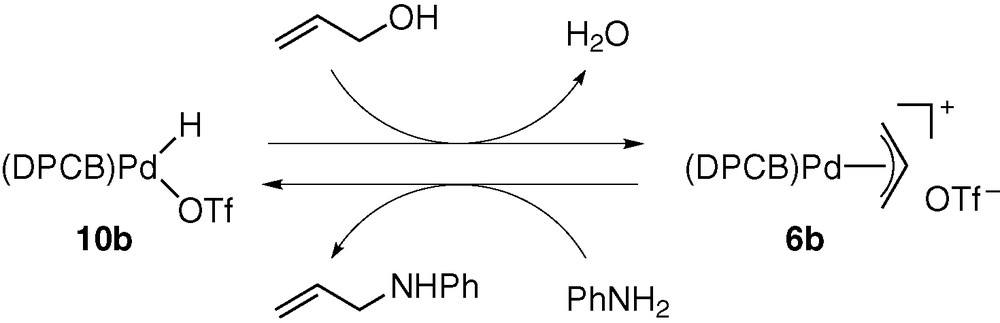
The hydridopalladium triflate 10b, in situ generated from 5b, HSiMe2Ph (1 equiv), and residual water in CH2Cl2, showed much higher reactivity than the platinum analogue [9]. The reactions with phenylbutadiene and cyclohexadiene spontaneously formed the corresponding π-allyl complexes. Furthermore, 10b rapidly caused C–O bond cleavage of allylic alcohols to give π-allyl complexes 6b, 6d, and 6e (Scheme 7).
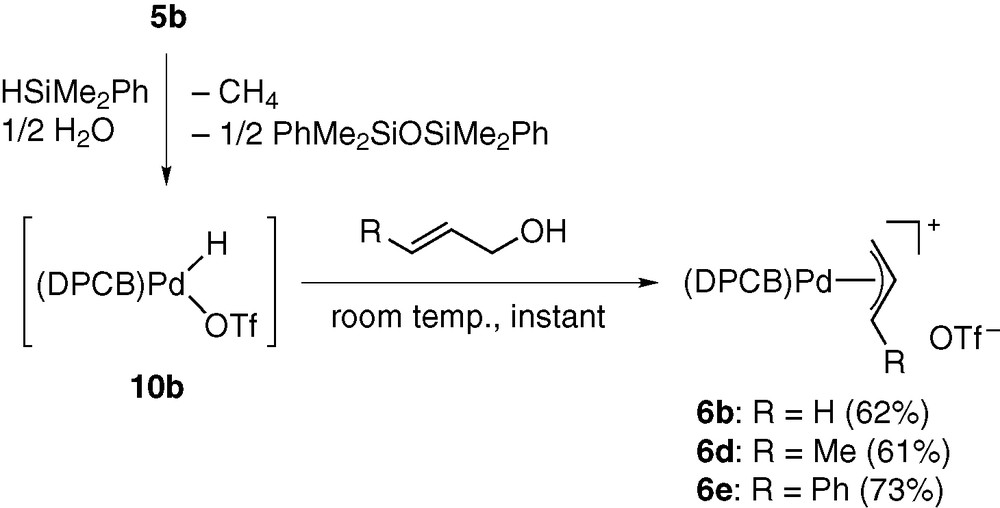
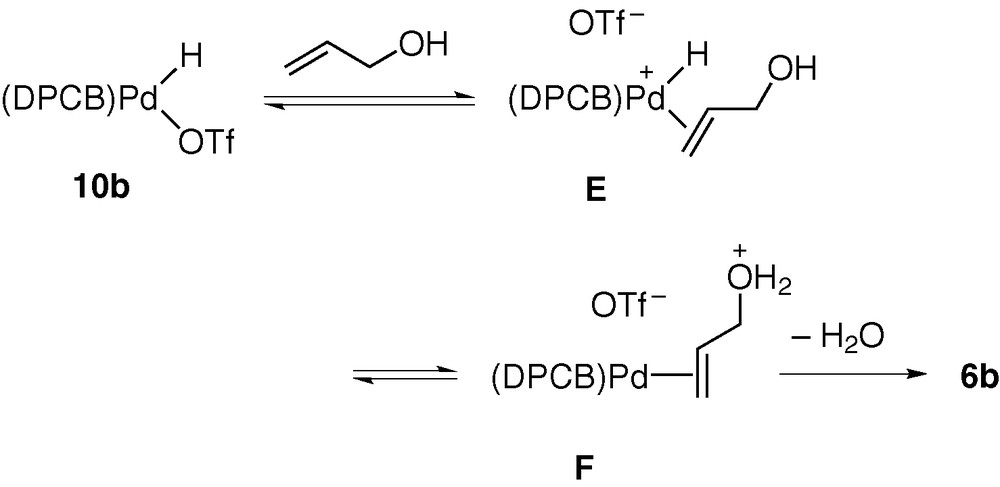
The π-allyl complex formation from 1,3-dienes should be a key elementary process for catalytic hydroamination of these substrates [5]. On the other hand, the direct conversion of allylic alcohols into π-allyl complexes is of great significance, in connection with catalytic allylation of active methylene compounds and amines [19]. Thus, the N- and C-allylation catalysed by palladium complexes (i.e., the so-called Tsuji–Trost reaction) is a versatile synthetic means of constructing C–N and C–C bonds in organic chemistry. This reaction generally employs allylic esters derived from allylic alcohols as substrates, because (π-allyl)palladium intermediates are usually generated in catalytic systems by oxidative addition of allylic substrates (allyl–OY), and this process involves nucleophilic substitution of the OY groups by Pd(0) species. Owing to the poor leaving ability of the OH group, it is necessary to convert allylic alcohols to esters having OY groups with higher leaving ability. The extremely facile cleavage of the non-activated allyl–OH bonds induced by 10b is therefore of particular interest and possibly serves as a key elementary process for direct conversion of allylic alcohols into N- and C-allylation products [7]. We proposed the mechanism given in Scheme 8 for the allyl–OH bond cleavage. It was considered that the strong π-accepting ability of DPCB ligand makes 10b highly acidic to allow the proton transfer from Pd to OH group in intermediate E. The resulting F undergoes elimination of water to give 6b [9].
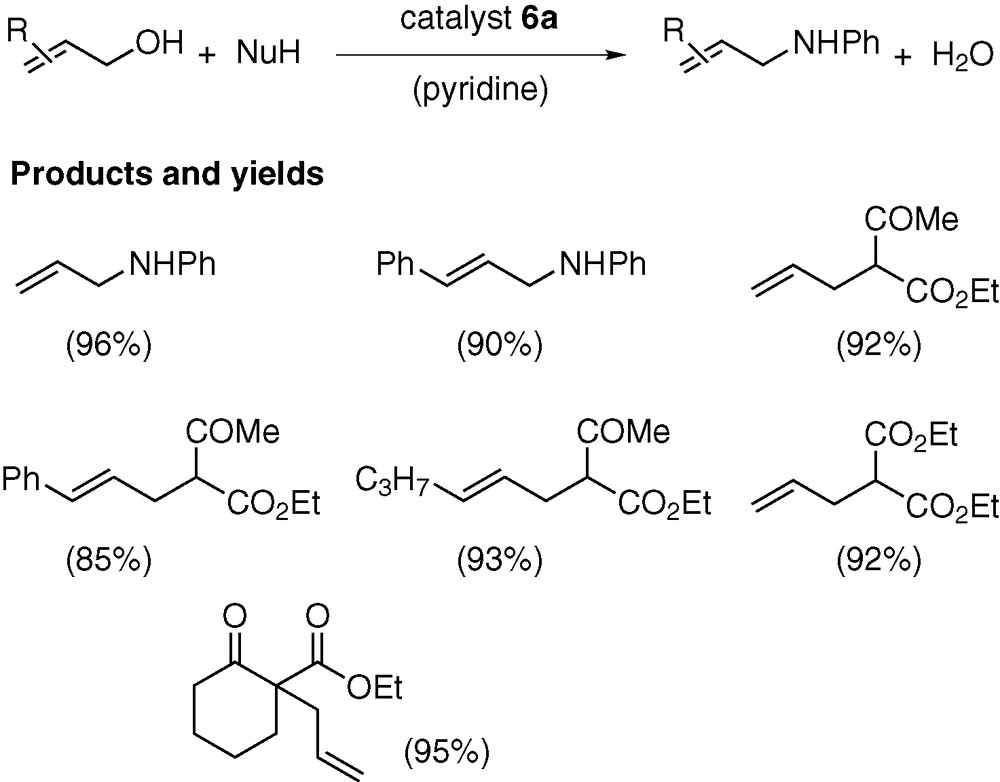
Representative examples of the catalytic allylation are given in Scheme 9. N-Allylation of aniline proceeded with 0.1 mol% of 6a at room temperature, whereas C-allylation of active methylene compounds was operative with catalytic amounts of 6a (2 mol%) and pyridine (10 mol%) at 50 °C for several hours. In all runs, monoallylation products were obtained in over 85% yields, along with the formation of water as the only co-product. The catalytic activity was significantly affected by the nature of Y on the DPCB–Y ligands (6a > 6b > 6c).
Scheme 10 shows the catalytic mechanism proposed for the reaction of allyl alcohol with aniline in the presence of 6b. The C–O bond cleavage promoted by palladium hydride 10b forms π-allyl complex 6b, as already confirmed by stoichiometric examinations (Scheme 7). Complex 6b then reacts with aniline to afford N-allylaniline and 10b. The latter process was confirmed by a stoichiometric experiment using 6b and aniline. Thus, the reaction proceeded rapidly at room temperature (Scheme 11). In contrast, (π-allyl)palladium triflates bearing dppf and diimine ligands were unreactive under the same reaction conditions [5].
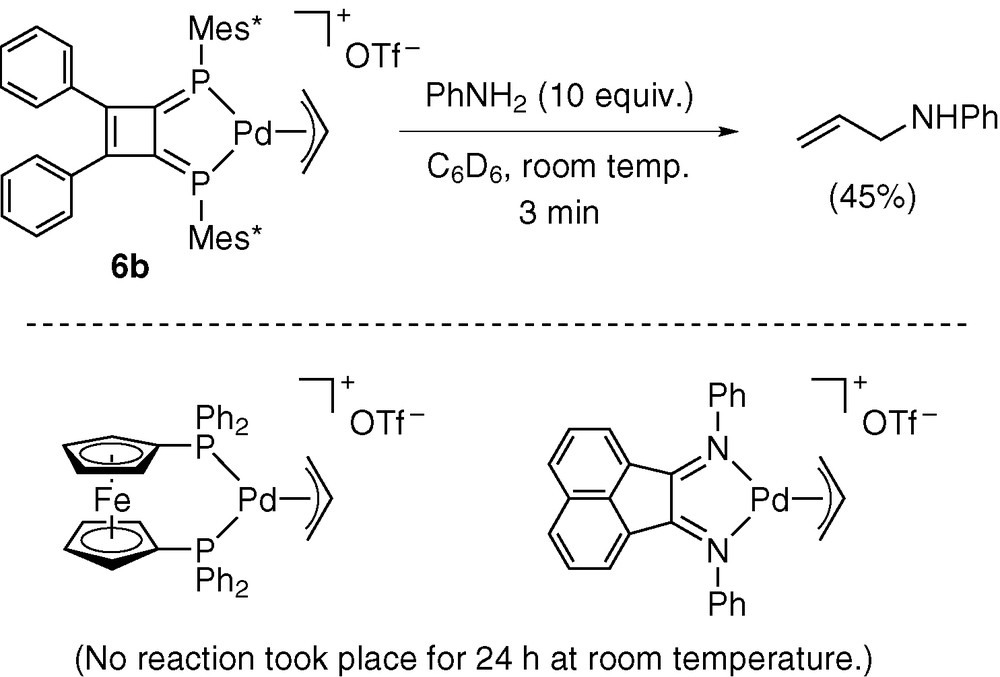
As already discussed with X-ray structural parameters and NMR data, the π-allyl complexes listed in Scheme 11 have very similar structures to each other. Nevertheless, DPCB complex 6b showed much higher reactivity toward aniline than the dppf and diimine complexes. Accordingly, the highly reactive nature of 6b must be attributed to the stability of transition state. The reaction of 6b with aniline very probably proceeds via nucleophilic attack of aniline to the π-allyl ligand from the opposite side of palladium. This process causes increase in the electron density of palladium. The strong π-accepting ability of DPCB will effectively reduce the increasing electron density to stabilize the transition state.
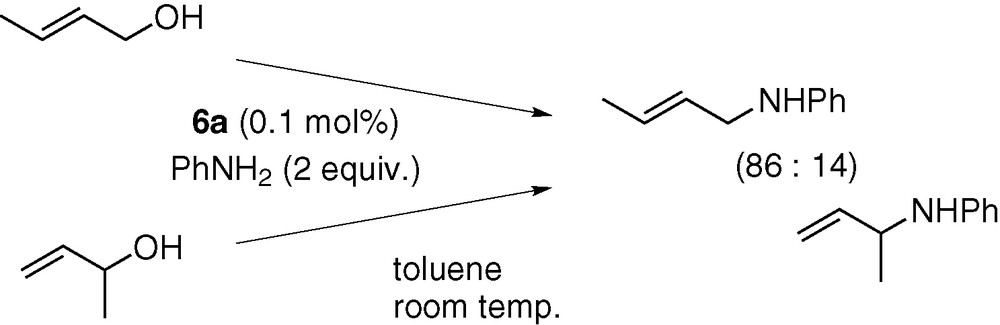
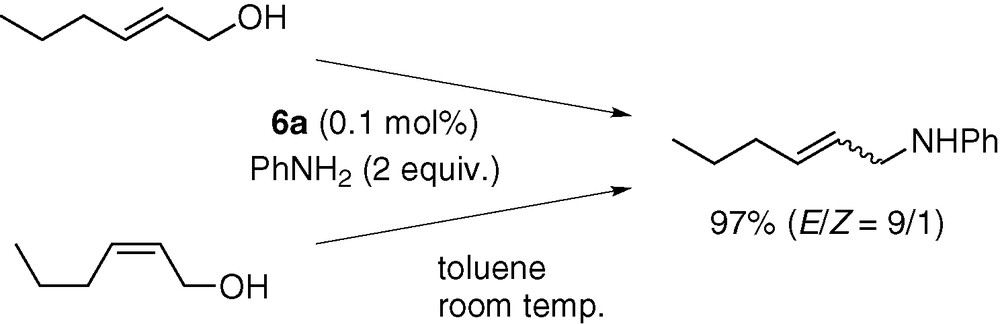
Although the novel C–O bond cleavage process promoted by a hydridopalladium complex is involved, the present catalysis preserved the regio- and stereochemical courses generally observed for palladium-catalysed allylation reactions [7]. Thus, the two regioisomers of butenyl alcohol were converted to N-(2-butenyl)aniline and N-(1-methyl-2-propenyl)aniline in almost the same regioselectivity (Scheme 12). Similarly, the reactions of the (E)- and (Z)-isomers of 2-hexenyl alcohol gave a nearly identical distribution of stereoisomers (Scheme 13). These results are consistent with a catalytic mechanism involving rapid interconversion between the syn- and anti-isomers of π-allyl intermediates.
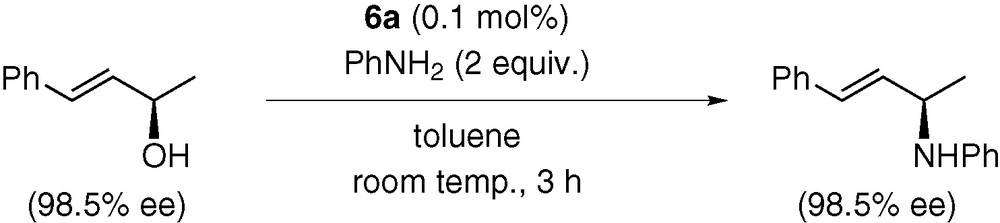
Furthermore, an optically active alcohol shown in Scheme 14 was converted to the corresponding allylated aniline with retention of the configuration of allylic carbon [7]. This type of stereochemistry has been observed for common allylation systems and attributed to the occurrence of two inversion processes at the C–O bond cleavage and the external attack of aniline to the π-allyl intermediate [19].
4 Conclusion
The palladium and platinum complexes bearing DPCB–Y ligands exhibited rather unique structures and reactivities, significantly different from common organometallic complexes having tertiary phosphine and nitrogen-based ligands. The highly reactive nature observed in several catalytic reactions was reasonably rationalized by considering the strong π-accepting property of sp2-hybridized phosphorus compounds toward transition metals.
Acknowledgments
We thank the co-workers whose names are described in the references for their dedicated research work on this study. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan.