

1 Introduction
In 1965 John Osborn published the first report 〚1〛 related to the use of well-defined rhodium-phosphine complex as catalyst for hydroformylation reaction: this pioneering work was done with Sir Geoffrey Wilkinson at the Imperial College of London. Since that time, a great deal of work has been done, mainly related to the use of a tremendous number of ligands with the aim to tune both the activities and selectivities.
Since the synthesis of the water soluble tris(sodium-m-sulfonatophenyl)phosphine (TPPTS) and its application in a large scale to propene hydroformylation in the so-called ‘Rhone-Poulenc-Ruhrchemie’ process (up to 500 000 tons per year), attention has been paid to the concept of phase separation, by which the catalyst is forced to remain in a different phase from that of the products of the reaction, allowing their easy separation. In that context, the fluorous biphase approach has recently attracted the attention of many academic and industrial researchers 〚2–5〛, due to the crucial advantages it offers over other purely two-phase systems, since, whereas at ambient temperature the fluorous phase containing the catalyst is separated from the organic phase, the reaction medium becomes homogeneous under the reaction conditions at higher temperatures 〚6–8〛. Obviously, this behaviour allows combining the activity and the selectivity of homogeneous catalysts with the simplicity of product isolation. This elegant approach has been successfully applied in hydroformylation of higher olefins by Horváth and Rábai in 1994 〚9〛, using a rhodium catalyst dissolved in a fluorous phase by the way of the trialkylphosphine P〚CH2CH2(CF2)5CF3〛3. This catalyst displays satisfactory activities in a perfluoromethylcyclohexane/toluene solvent system and the normal to branched aldehyde ratio (n/i) was comparable to that obtained in a conventional solvent with HRh(CO)(PPh3)3 (n/i = 2.9). Interestingly, these authors demonstrated in another detailed study that the long-term stability of the fluorous catalyst was greater than that of the Rh/PPh3 catalyst 〚10〛. Hydroformylation in fluorous biphasic conditions with fluorous triarylphosphites was briefly investigated by Hope and co-workers 〚11–13〛. The authors reported the hydroformylation of 1-hexene and 1-octene under 20 atm syngas at 60–80 °C using tris(4-perfluorohexylphenyl)phosphite as ligand. It was found that the n/i ratio was higher than that observed with the Rh/P(OPh)3 catalyst and that the catalytic system was also active for the hydroformylation of internal alkenes. Unfortunately, no information about the stability of the catalyst was reported in this work.
In previous studies, we described the synthesis of new fluorinated analogues of triphenylphosphite 〚14, 15〛. These fluorous soluble phosphites were obtained by a copper-mediated coupling reaction of 1-iodoperfluorooctane with various halophenol derivatives, followed by the reaction of the corresponding phenol with phosphorus trichloride. This procedure allowed introducing one or two perfluorooctyl groups on the aromatic ring. We report herein our efforts to develop a hydroformylation process of higher olefins in the presence of catalysts generated in situ from Rh(acac)(CO)2 and six fluorinated analogues of triphenylphosphite (Fig. 1). The stability of these catalysts and the possibility of recovering quantitatively the catalytic systems were carefully examined by performing several reaction cycles.
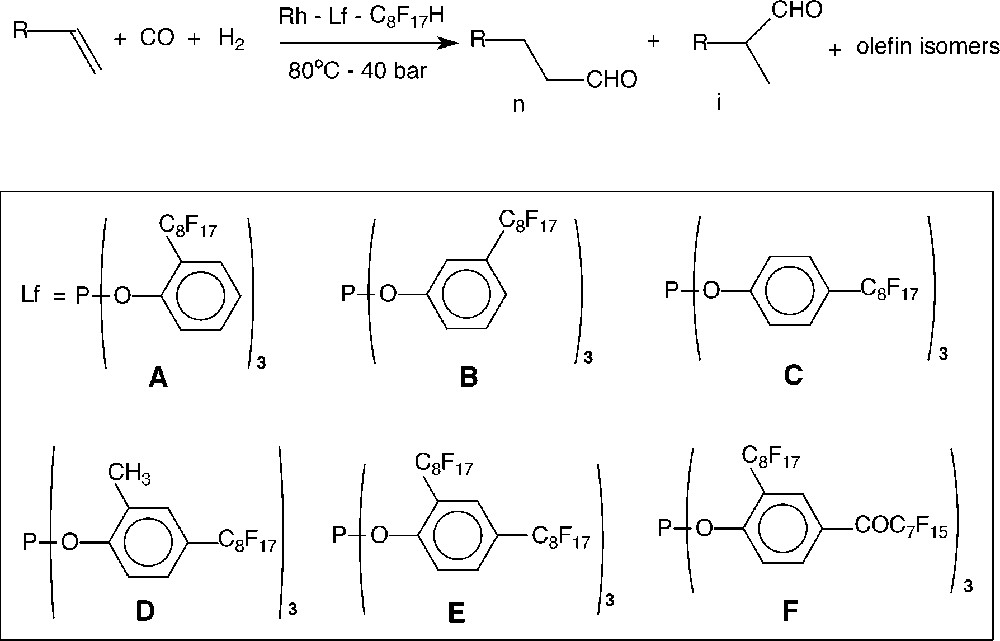
Hydroformylation of 1-decene in fluorous biphasic medium using phosphites A–F.
2 Experimental
All synthesis and catalytic reactions were performed under nitrogen using standard Schlenk techniques. All solvents and liquid reagents were degassed by bubbling nitrogen for 15 min before each use or by two freeze-pump-thaw cycles before use. Tris(4-(perfluorooctyl)phenyl) phosphite, tris(3-(perfluorooctyl)phenyl)phosphite, tris(2-(perfluorooctyl)phenyl)phosphite and tris(2,4-bis(perfluorooctyl)phenyl)phosphite were prepared following our previously described procedures. Dicarbonylacetylacetonato rhodium (I) was purchased from Aldrich Chemicals and used without further purification. Cyclohexene, 1-hexene, 1-octene, 2-octene, 4-octene, 1-decene, 1-dodecene were purchased from Acros Chemicals and used as received. 1H-perfluorooctane was generously supplied by Atofina and was distilled from CaH2. Carbon monoxide/hydrogen mixture (1:1) was used directly from cylinders (> 99.9% pure; Air Liquide). Gas chromatography was performed on Chrompack CP-9001 gas chromatograph equipped with a CPSil-5CB column (25 m × 0.32 mm) and FID detector. Nitrogen was the carrier gas and the temperature program was from 80 to 200 °C at a heating rate of 5 °C min–1.
2.1 Hydroformylation experiments
All the high-pressure hydroformylation experiments were carried out in a 50-ml stainless steel microclave supplied by Parr. The reactor was fitted with arrangements for liquid sampling, automatic temperature control, and variable stirring with precise speed measurement by a tachometer display and pressure gauge. A safety rupture disk was also fitted to the reactor. Rh(acac)(CO)2 (0.038 mmol) and the phosphite (0.193 mmol) were dissolved in 15 ml of 1H-perfluorooctane; the solution was stirred for 2 h. The resulting fluorous phase and an organic phase composed of the olefin (77.4 mmol) and undecane (7.74 mmol – GC internal standard) were charged under nitrogen into the reactor, which was heated at 80°C. Mechanical stirring equipped with a multipaddle unit was then started (1500 rpm) and the autoclave was pressurised with 40 bar of CO/H2 (1:1) from a gas reservoir connected to the reactor through a high-pressure regulator valve, allowing to keep a constant pressure in the reactor throughout the whole reaction. The reaction medium was sampled during the reaction for GC analyses of the organic phase after decantation. For kinetic measurements, the time corresponding to the addition of CO/H2 was considered as the beginning of the reaction.
2.2 Recycling experiments
The first batch was carried out as described above. After 10 min of reaction, the autoclave was cooled rapidly to room temperature and carefully depressurised. The reaction solution was transferred into a nitrogen-filled Schlenk tube. After 30 min, the fluorous and organic phases were separated and a new carefully deoxygenated solution of olefin (77.4 mmol) and undecane (7.74 mmol) was introduced into the Schlenk tube. The resulting mixture was charged under an atmosphere of N2 into the autoclave and used in another hydroformylation run as described above
3 Results and discussion
The different phosphites previously synthesised (see Fig. 1) were tested in fluorous biphasic hydroformylation of various olefins under standard conditions: 80 °C, 40 bar CO/H2 1:1, using Rh(acac)(CO)2 as catalyst precursor with a phosphite/Rh ratio of 5 and 1H-perfluorooctane as fluorous solvent.
The partition coefficient of phosphites C and F representative of phosphites with one or two perfluorous ponytails were determined by GC at room temperature. The distribution of C and F in 1-decene/C8F17H mixture were found to be 2.7/97.3 and 0.9/99.1, respectively.
3.1 Hydroformylation of 1-decene under fluorous biphasic conditions
In a first step, 1-decene was chosen as model of heavy olefin. The progress of the reaction was monitored by GC analysis of aliquot samples taken at regular time intervals. The main results observed are summarised in Table 1.
Hydroformylation of 1-decene under fluorous biphasic conditions.
| Entry | Phosphite | t (min) | TOFc (h–1) | n/id | Aldehyde selectivityd (mol%) | |
| 70%a | 100%b | |||||
| 1 | A | 5 | 10 | 10 000 | 2.3 | 71 |
| 2 | B | 10 | 60 | 6300 | 5.8 | 80 |
| 3 | C | 25 | 60 | 3500 | 5.3 | 85 |
| 4 | D | 10 | 30 | 7100 | 2.9 | 80 |
| 5 | E | 5 | 15 | 7900 | 2.4 | 46 |
| 6 | F | 10 | 30 | 6200 | 2.6 | 39 |
All of the catalytic systems obtained with the different fluorous phosphites exhibited high activity: initial TOF > 3000 h–1. Nevertheless, there was a great difference of reactivity between the different catalytic systems according to the position and the number of substituents on the aromatic ring of the phosphite used. For example, with the parasubstituted phosphite C, the activity is about three fold lower than the one resulting from the use of the bulky ortho-substituted phosphite A, whereas the meta-substituted ligand B led to an intermediate activity between A and C. Hydroformylation of 1-octene under biphasic conditions using a rhodium catalyst generated from P–(O–4-C6H4C6F13)3 has been recently reported 〚13〛. The TOF at 80 °C as well as the n/i ratio claimed in this paper are higher than in this work (TOF = 15 600 h–1, n/i = 6.3). However, the conditions are different and it is difficult to compare the results. In the same way, we have previously described the hydroformylation of 1-decene using ligands closely related to phosphites A and C, which are bearing ethylene spacers between the aryl ring and the perfluorous chain 〚16〛. Thus, under the same reaction conditions, tris(2-(1H,1H,2H,2H-perfluorodecyl)phenyl)phosphite and tris(2-(1H,1H,2H,2H-perfluorodecyl)phenyl)phosphite lead to TOF and n/i ratios close to those observed with A and C (TOF = 10 000 and 3900, n/i = 2 and 3.5, respectively).
It is also noteworthy that the only by-products observed in all experiments are the isomers of 1-decene with internal double bond and that at long reaction time the aldehyde selectivity increased to reach 100%, whereas at the same time the n/i ratio decreased. This behaviour probably arises from the fact that, when the reaction of 1-decene began to slow down (at low 1-decene concentration), the internal decenes (formed at the early stage of the reaction) were hydroformylated, resulting in the production of several branched aldehydes. Thus to compare the data, the n/i ratio and aldehyde selectivity presented in Table 1 are those obtained at the same conversion (70%). It appears that the aldehyde selectivity and the n/i ratio are also very different according to the nature of the phosphite. Indeed, ortho-substituted phosphite A and, to a greater extent, o,o’-disubstituted E and F induce lower aldehyde selectivities and lower n/i ratios than their meta- or para-substituted counterparts.
These differences of behaviour between the fluorous phosphites are probably related to the fact that, with the bulky o- or o,o’-disubstituted phosphites, only one phosphite coordinates the Rh centre 〚17〛 to give the active species HRh(CO)3L 1, 4, 5, 6 (L = A, D, E or F, respectively). On the other hand, with the meta or para substituted phosphite B or C, as in classical triphenyl phosphine modified catalyst, two phosphites are bonded to rhodium. Complexes 1, 4, 5, 6 similarly to HRh(CO)4 have a stronger aptitude to CO dissociation than complexes HRh(CO)2L2 2, 3 (L = B or C). Thus with only one phosphite bonded to the metal, the complex HRh(CO)3L after CO dissociation easily binds to the olefin, initiating a fast reaction cycle. More, due to the large space available in comparison with the HRh(CO)2L2 system, the reaction giving the branched aldehyde as well the β-H elimination proceeds with relative ease resulting in a modest linearity and aldehyde selectivity. However, as the use of triphenyl phosphite under similar conditions in homogeneous medium led to an aldehyde selectivity of 90% and to a n/i ratio of 2.3, largely different from those obtained with phosphites B and C; the steric hindrance is probably not the sole factor that has an influence on the behaviour of the fluorous phosphites.
Actually, the perfluorous ponytail of the fluorous phosphites has a strong electron-withdrawing effect, as proved for instance, in the case of phosphite B, by the large difference between the IR νCO value 〚16〛 of complexes HRh(CO)B3 and HRh(CO)(P(OPh)3)3, respectively 2066 and 2010 cm–1. Consequently, the electron density on complexes 2 and 3 decreases, which promotes the olefin insertion on its terminal carbon, giving a linear alkylrhodium and then, after CO insertion and hydrogenolysis, a linear aldehyde. On the other hand, a decrease of the electronic density on the metal has also the effect of favouring the formation of unsaturated rhodium species, which induces an easier β-H elimination and thus olefin isomerisation.
3.2 Hydroformylation of various terminal or internal olefins
As it was observed that internal decene could react under our reaction conditions, the hydroformylation of various heavy internal or terminal olefins was checked still using C8F17H as fluorous phase, in order to determine the scope and limitations of the process. For this study, phosphite B, which had allowed conjugating high activity and high n/i ratios, was chosen as ligand model.
Table 2 shows that terminal olefins behave similarly to 1-decene. Classically, the activity decreased with the size of the substrate, but more unexpectedly the n/i ratio and the aldehyde selectivity varied in the same way. For instance, an n/i ratio of 6.5 and an aldehyde selectivity of 94% were obtained with 1-hexene vs 5.8 and 80% with 1-decene, respectively.
Hydroformylation of various olefins under fluorous biphasic conditions using tris(3-perfluorooctylphenyl)phosphite B as ligand.
| Entry | Olefin | t (min) | TOFc | n/id | Aldehyde selectivityd | |
| 70%a | 100%b | |||||
| 1 | 1-hexene | 9 | 60 | 7800 | 6.5 | 94 |
| 2 | 1-octene | 10 | 60 | 6900 | 6.3 | 87 |
| 3 | 2-octene | 30 | 150 | 1400 | —e | 75e |
| 4 | 4-octene | 60 | 150 | 800 | —f | 76f |
| 5 | cyclohexene | — | — | 70 | — | 100 |
As expected, the reactivity markedly fell with internal alkenes. Indeed, the activity obtained with 2-octene was about four folds smaller than using 1-octene under the same conditions and slowed down again when going from 2-octene to 4-octene. Since the percentage of n-nonanal was about the same starting from 2- or 4-octene (10%), the difference of reactivity between these two olefins is not ascribable to an easier isomerisation of 2-octene into 1-octene, which is more reactive, but rather to a greater steric demand with 4-octene. On the other hand, the distribution of the aldehydes was markedly different with 2- and 4-octene. With 2-octene, the aldehydes resulting from the direct hydroformylation of the substrate (i.e. 2-methyloctanal: 60% and 2-ethylheptanal: 25%) were the main reaction products. Conversely, with 4-octene, isomerisation into 2-octene occurred to a large extend, leading to a large percentage of 2-methyloctanal (28%).
Finally, cyclohexene as model of cyclic olefin, was practically inactive with this class of phosphites and under these reaction conditions, since only 20% of aldehyde were obtained after 4 h.
3.3 Catalyst recovery and re-use
Phosphites A, B and F have been chosen to investigate the recovery and reuse of the catalytic system. The reaction was conducted as described above, except that the duration of each test was fixed at 5 min for A and 10 min for B and F, in order to stop the reaction at about 70% conversion and avoid the levelling of the n/i ratio and aldehyde selectivity that occurs when total conversion is reached (see above). At the end of each test, the phases were separated by decantation on cooling (30 min); after that, pure 1-decene was added to the mother solution of C8F17H containing the catalytic system and again allowed to react. Figs 2, 3 and 4 respectively show the evolution of the conversion, the n/i ratio and the aldehyde selectivity for four reaction cycles. With the three phosphites tested, the activity decreased after each reuse (see Fig. 2). Nevertheless, whereas this decrease was moderate in the case of phosphite A and to a lesser extent F, the activity dramatically dropped after the second cycle with the meta-substituted phosphite B, and practically no activity was observed during the fourth run. In this last case, the orange colour of the organic phase at the end of the three first runs and the fact that the fluorous phase was practically colourless after the last one indicate that the drop in activity with phosphite B is due to a rhodium leaching from the fluorous to the organic phase. In contrast, with phosphites A and F the organic phases are only slightly coloured, which is consistent with a much more limited leaching and with the preservation of some catalytic activity. From Figs 3 and 4, it appears also that with A and F the n/i ratio and the aldehyde selectivity remained practically constant during the different recycles. In contrast with phosphite B, the n/i ratio (initially twice as high as the one of A and F) dramatically fell after the first cycle and yet decreased during the following cycles. In the same way, the aldehyde selectivity markedly decreased after the first use with phosphite B. These observations with phosphite B are ascribable to a modification of the catalytic species that occurs during the first cycles.
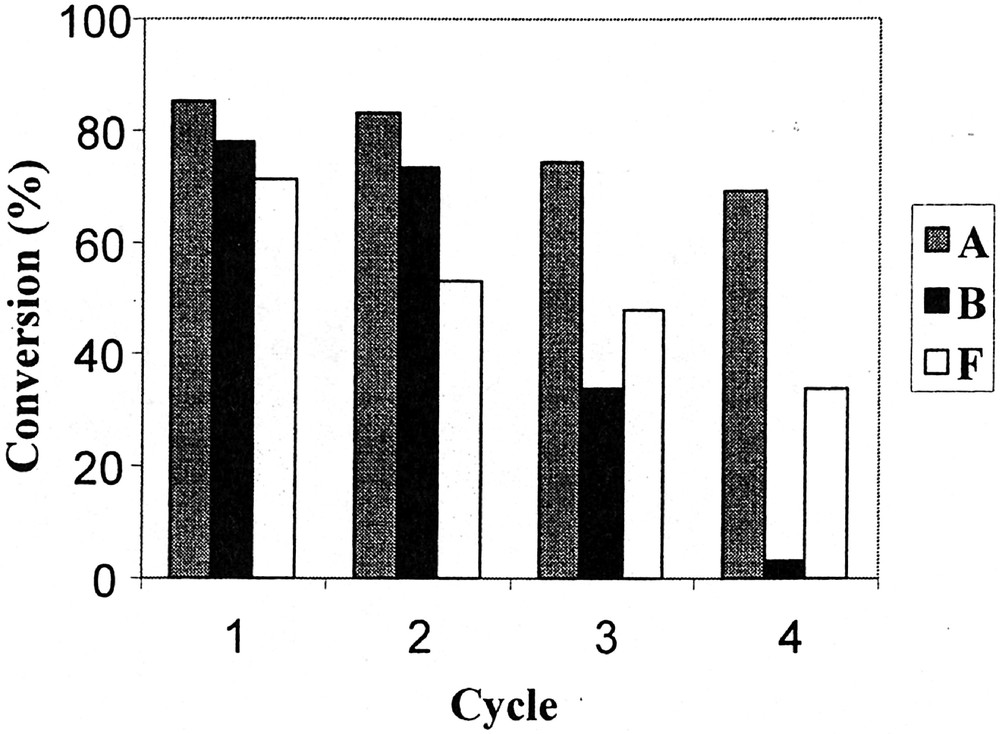
Conversion of 1-decene as a function of cycle under the conditions of Table 1, using rhodium catalysts generated from phosphites A, B and F.
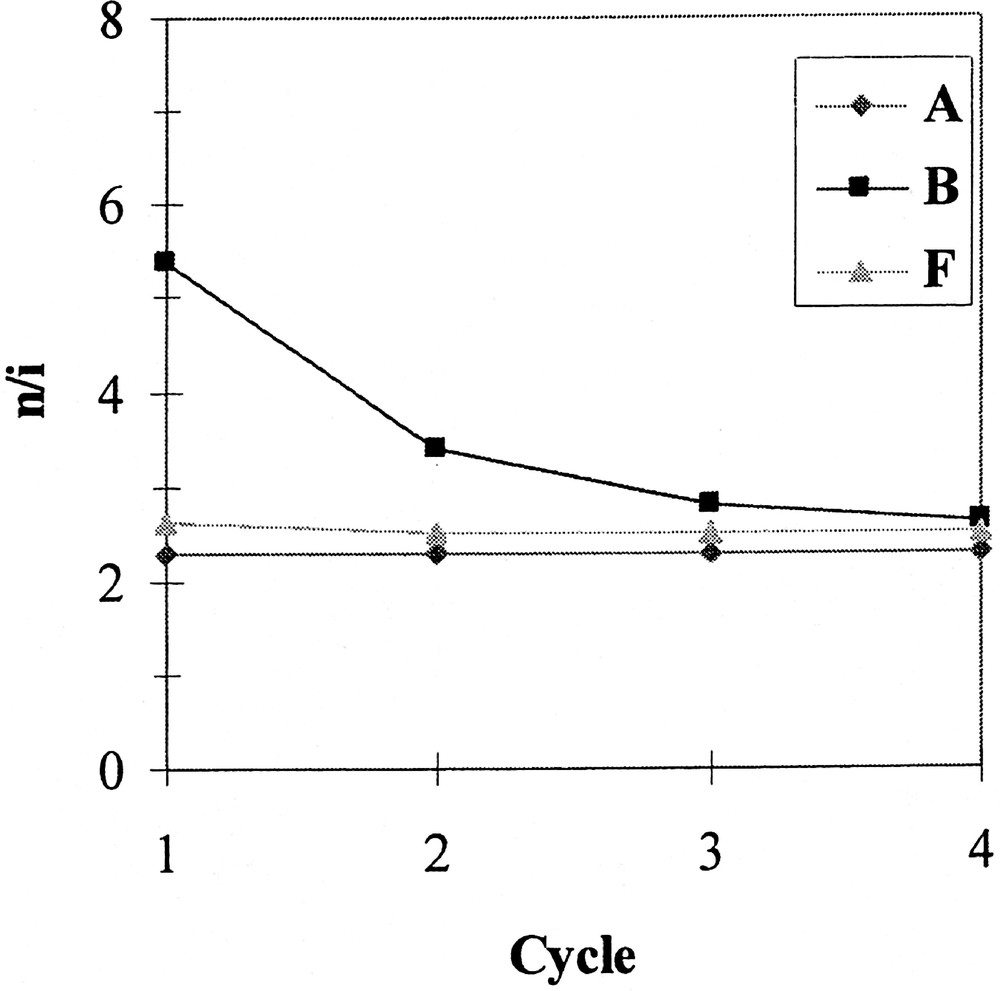
Hydroformylation of 1-decene – variation of the n/i ratio during recycle experiments, using Rh catalysts generated from phosphites A, B and F.
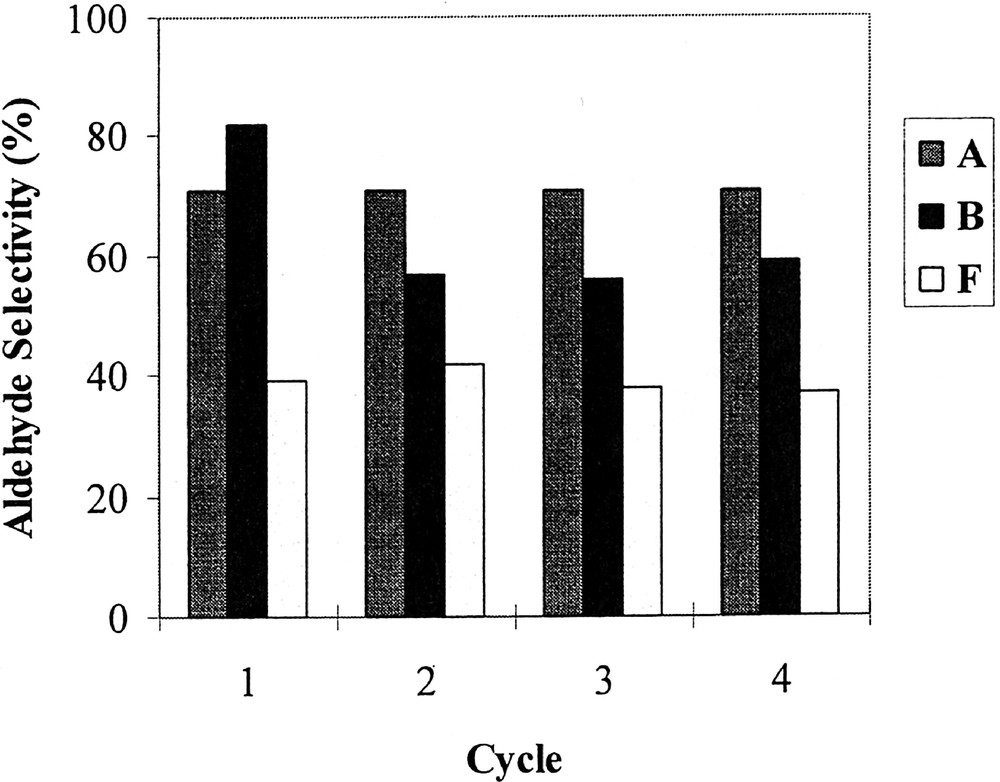
Aldehyde selectivity as a function of cycle under the conditions of Table 1, using rhodium catalysts generated from phosphites A, B, and F.
In order to verify this assertion, we have studied the stability of phosphites A and B under the reaction conditions and compared with the one of triphenylphosphite. A sample of each phosphite was heated at 80 °C during 1 h in mixtures of C8F17H and 1-decene on the one hand and C8F17H, 1-decene and undecanal (1:1:1, v/v/v) on the other hand. At the end of the test, a 31P NMR analysis of the fluorous phase was carried out. In the absence of undecanal, all phosphites remained unchanged (see Table 3). On the other hand, in the presence of aldehyde, the behaviour of A and B was very different. Actually about 30% of phosphite B were converted into oxidation products, whereas ortho substituted phosphite A appeared much more stable, since only about 3% decomposition occurred.
Stability of phosphites A and B in the presence of aldehyde.
| Ligand | % Decomposition | ||
| Decene/C8F17H | Decene/undecanal/C8F17H | Decene/undecanal/C8F17H/Rh | |
| P(OPh)3a | < 1 | 40 | 100 |
| A | < 1 | < 3 | < 5 |
| B | < 1 | 30 | 100 |
The attack of triphenylphosphite by aldehydes is a known process that occurs via the formation of dioxophospholanes (see Fig. 5), which evolves to give a phosphite oxide 〚18–21〛. The fact that the decomposition of B was of the same order of magnitude as the one of P(OPh)3 (see Table 3) suggests that the decay of B is not due to its fluorous group. As a matter of fact, phosphite A possesses also a perfluorous pony tail and is much more stable than B; moreover, the electron withdrawing effect of the fluorous group should reduce the nucleophilicity of the phosphorus atom and thus the attack on the carbonyl of the aldehyde. Thus, the difference of behaviour between A and B is probably connected with the steric hindrance of the perfluorous chains in ortho of the P–O–C bonds in the case of A, which has the effect of limiting the approach of the phosphorus atom to the aldehyde, preventing its oxidation. Nevertheless, the decomposition of B in the presence of aldehyde does not seem sufficient to explain the total decay of the catalyst after three cycles (with less than 30 min of contact between the phosphite B and aldehyde, the decomposition should not exceed about 15%). We have therefore examined the behaviour of the different phosphites in the presence of all the components involved in the reaction (i.e. in the presence of the rhodium catalytic precursor). Surprisingly, under these conditions, B and P(OPh)3 were completely decomposed after 1 h of heating, whereas A remained practically unchanged (< 5% decomposition).
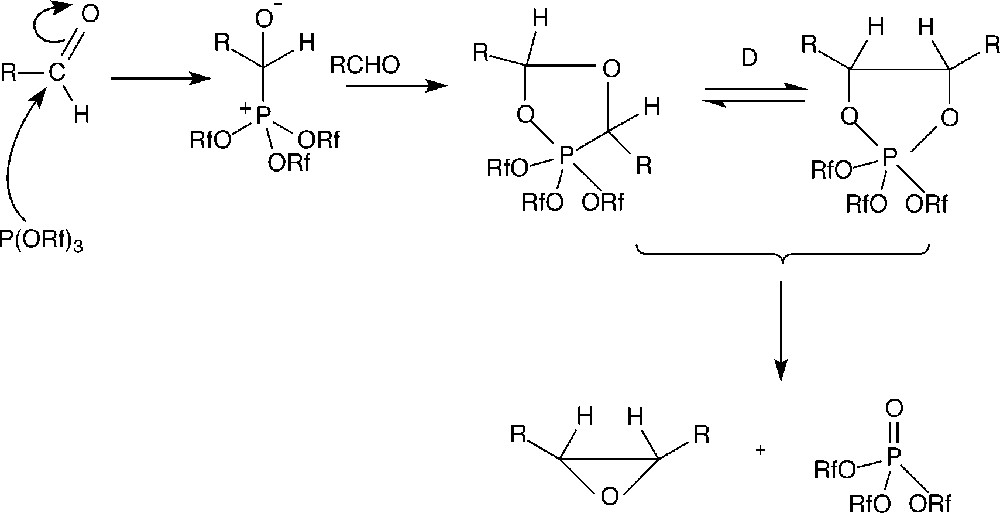
Proposed mechanism for the reaction of the phosphite with the aldehyde.
This last experiment allows us to suggest that the decreases of the n/i ratio and of the aldehyde selectivity in the case of B are probably ascribable to the decomposition of the phosphite in the presence of aldehyde during the first cycle. As phosphite oxides are poor ligands, the decay of the phosphite induces the decrease of the ligand/Rh ratio and equilibrium I in Fig. 6 between the different catalytic species is shifted toward the formation of low phosphane coordinated species. This last species, as mentioned above, leads to low n/i ratio and low aldehyde selectivity. Moreover, as at very low phosphite concentration, the complex HRh(CO)4 becomes the main rhodium species (equilibrium II, Fig. 6) and, as this one has no more a specific affinity for the fluorous phase, it can migrate into the organic phase, inducing rhodium leaching.
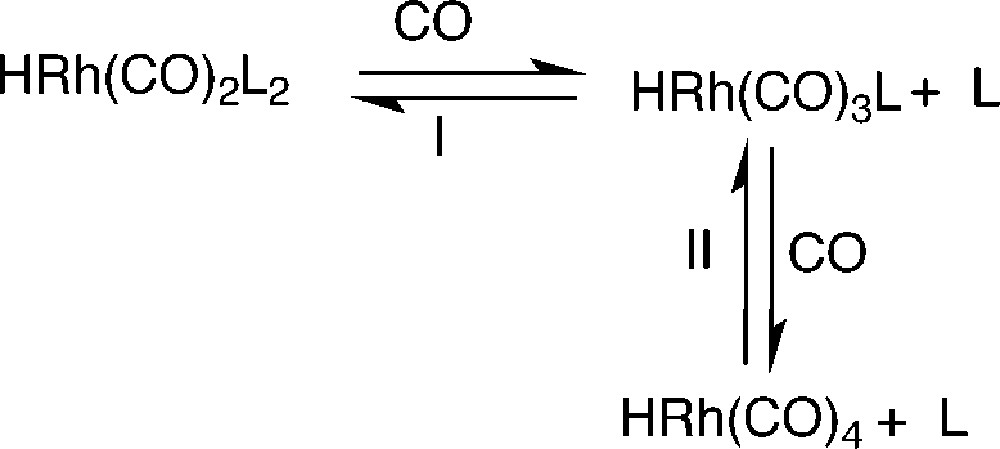
Equilibrium between the different rhodium species under CO.
In the case of phosphites A and F, the same catalytic species are probably involved during the different recycles, since the n/i ratio and aldehyde selectivity do not vary. That is in agreement with the stability of these ortho substituted phosphites that practically do not react with aldehydes. Nevertheless, the decrease of the conversion as well as the slightly yellow colour of the organic phase at the end of each cycle prove that leaching of the rhodium is also occurring. To explain this apparent contradiction, it should be noticed that, as previously mentioned, with the bulky orthosubstituted phosphite the main rhodium species are 1 or 6. With only three perfluorous groups per rhodium atom, the partition coefficient of 1 between the fluorous and the organic phases is probably too low to keep all of the complex in the former one. On the other hand, whereas 6, which contains six perfluorous chains per rhodium, should be more retained in the fluorous phase than its counterpart 1, the decrease of the conversion using F as ligand (see Fig. 2) suggests a greater leaching than with A. The presence of two electron-withdrawing substituents per phenyl ring in the case of F instead of one with A has the effect of diminishing the electronic density of the phosphorus atom of F vs the one of A thus shifting the equilibrium II (Fig. 6) toward HRh(CO)4. As a result, the HRh(CO)4 species formed moves from the fluorous to the organic phase.
4 Conclusion
All of the perfluorooctyl-substituted triphenylphosphites previously synthesised lead to modified rhodium hydroformylation catalysts that exhibit high efficiency under fluorous biphasic conditions, particularly in the case of terminal alkenes. Nevertheless, the position of the perfluoroalkyl substituents on the aromatic ring and their number have a marked influence on the activity and selectivity induced by the different phosphites.
For a catalyst recovery and re-use point of view, it appears that the presence of a perfluorosubstituent in the ortho position is essential in order to avoid the attack of the phosphite by the aldehyde formed. Unfortunately, with bulky orthosubstituted phosphites, the catalytic species contains only one phosphite per rhodium and the partition coefficient of this species between the fluorous and the hydrocarbon phase is too low to prevent rhodium leaching during the separation step, which limits the recycling character of this procedure.
Acknowledgements
This research was supported by funds from the French ‘Centre national de la recherche scientifique’ (CNRS) and Atofina. We would like also to thank Atofina for kind gifts of fluorinated chemicals.