

1 Introduction
Immobilisation of biomolecules within mineral hosts has become a popular, yet challenging, approach to design new materials with biotechnological applications [1]. The sol–gel-based bioencapsulation route, proposed more than ten years ago by Avnir and co-workers [2], has made considerable progress [3–5]. Whole-cell entrapment has now been reported by various authors [6–10], opening future opportunities for biosensing [11], medical diagnostics [12] and even artificial organs [13].
The porous structure of the matrix is a major parameter for the efficiency of the immobilisation process. An ideal host would consist of large cavities accommodating the species to be trapped, connected to tailored channels allowing substrates and products diffusion. However, the conditions of formation of mesostructured materials [14,15] are, at this time, incompatible with biospecies, so that only the immobilisation of enzymes within a pre-formed silica host, rather than in-situ formation of the organised matrix, is accessible [16]. As a consequence, in order to avoid biomolecules leaching, functionalisation of the silica pores is often needed, which may result in a loss of enzyme catalytic activity [17]. As a matter of fact, the interactions that may arise between the entrapped species and the matrix surface are also of primary importance. Using hybrid materials that incorporate organic molecules may result in enhanced stability and reactivity [18,19].
In this context, we report here the synthesis and characterisation of new biocomposites associating alginic acid and mesoporous silica. The biopolymer should provide a suitable environment for bioencapsulation, as previously demonstrated [20,21]. The organised inorganic porous network may control the diffusion properties of the matrix together with conferring further stability [22,23].
2 Experimental
2.1 Biocomposite synthesis
MCM-41 silica matrix was synthesised following the procedure of Grün et al. [24]. 0.1 g of the thus-obtained silica powder was dipped overnight in a 2-ml aqueous solution containing 0.5 to 5 w% of alginate (alginic acid, sodium salt from brown algae, MW = 150 000 g mol–1, 70% guluronic acid, Fluka). Only limited amounts of polymer could be incorporated in the silica matrix because of the increasing viscosity of the starting alginate solution with concentration. After this delay, the remaining solution was withdrawn and 1 ml of an aqueous solution of CaCl2 0.1 M was added to induce alginate gelation [25].
2.2 Biocomposite characterisation
The obtained solids were recovered and dried 12 h under vacuum. Thermogravimetric analyses were performed on a Netzch apparatus under O2 flow with a heating rate of 10 °C min–1. Scanning electron micrographs (SEM) were obtained after gold coating on a Cambridge Stereoscan 120 instrument at an accelerating voltage of 20 kV. X-ray powder diffraction (XRD) data were recorded using a Philips diffractometer with the Cu Kα wavelength. Nitrogen-sorption experiments were performed at 77 K on a Micromeritics 2010 sorptometer. Specific surface areas SBET were determined by the BET method in the 0.05–0.35 relative pressure range, single-point total pore volume Vp was determined at p/p0 = 0.99 and average pore diameter Dp was calculated on the basis of 4 Vp/SBET [26,27].
2.3 Enzymatic activity
Immobilization of β-galactosidase was performed by preparation of a 5 w% alginate solution containing 0.02 mg ml–1 of enzyme (β-galactosidase from Kluyveromyces fragilis, Fluka) which was used for biocomposite preparation as described above, except that the samples were kept at 5 °C during all the reaction steps. A reference compound was prepared by gelation of 2 ml of the above-mentioned solution with 1 ml of CaCl2 0.1 M. Enzymatic activity was monitored using the abilityof β-galactosidase to catalyse the hydrolysis of 4-nitrophenyl-β-D-galactopyranoside (p-NPG) into β-D-galactose and p-nitrophenol, a yellow compound that can be titrated by UV-visible spectroscopy.
Typically, a fraction of 1/2 in mass of gel was washed two times with 2 ml Tris.HCl buffer (0.05 M, pH = 7.2) in order to remove non-trapped enzymes. It was then added to 4.25 ml of Tris.HCl and kept under stirring at constant temperature (20 °C). A chronometer was triggered when the p-NPG solution (50 mM, 0.75 ml) was added. At regular intervals, a suitable volume of the supernatant (Vs = 10–100 μl) was sampled and diluted in (1–Vs) ml of a potassium carbonate solution (1.4 M) in order to quench the enzymatic reaction. The amount of p-nitrophenol was calculated from optical density measurements at λ = 400 nm, applying the Beer-Lambert law (ϵ = 18 900 mol–1 l cm–1 in aqueous K2CO3 (1.4 M)). Enzymatic activity, expressed in U or μmol min–1, was then deduced from the initial reaction velocity of p-nitrophenol formation. Experiments were performed in triplicate.
3 Results
3.1 Characterisations of the biocomposites
Composition of the obtained solids was determined using thermogravimetric analysis (not shown). Alginate contents varied between 8 wt% to 50 wt%, indicating that 80–90% of the initial polymer was incorporated into the composites.
X-ray powder diffraction data of the pure mesoporous silica and of a biocomposite containing 50 wt% alginate (composite 1) are shown in Fig. 1. The initial calcinated MCM-41 phase (Fig. 1a) shows three well-defined peaks as well as a weaker one in the 2°–10° 2 θ range that could be indexed as the (100), (110), (200) and (210) reflections corresponding to a hexagonal network [16]. From a d100 value of 36.5 Å, an average pore size of 32 Å can be calculated that fits well with literature data [24]. Upon incorporation of alginate (Fig. 1b), the three main peaks are still present. However, they are clearly broadened and the (210) reflection is no longer observed, indicating a poorer structural order. Moreover, a slight shift in the three remaining peaks is observed leading to a d100 value of 34.5 Å, corresponding to an average pore diameter of 30 Å.

X-ray diffraction patterns of (a) the initial MCM-41 silica and (b) of the biocomposite 1.
More information on the composite porous structure was obtained from nitrogen-sorption experiments. Comparison of adsorption and desorption curves for the starting MCM-41 and composite 1 is shown in Fig. 2. An important loss of adsorbed volume is observed as well as a shift towards low p/p0 values for the mesoporous uptake curve (0.3 < p/p0 < 0.35 for MCM-41, 0.25 < p/p0 < 0.3 for Ca-alginate/SiO2), indicating a decrease in the mesoporosity of the material. Specific surface area SBET, porous volume Vp and average pore size Dp are reported in Table 1. In the case of the biocomposite, SBET and Vp could also be calculated per gram of SiO2, clearly underlining the loss in accessible porosity. However, the average pore size is not strongly modified, in agreement with the XRD data.
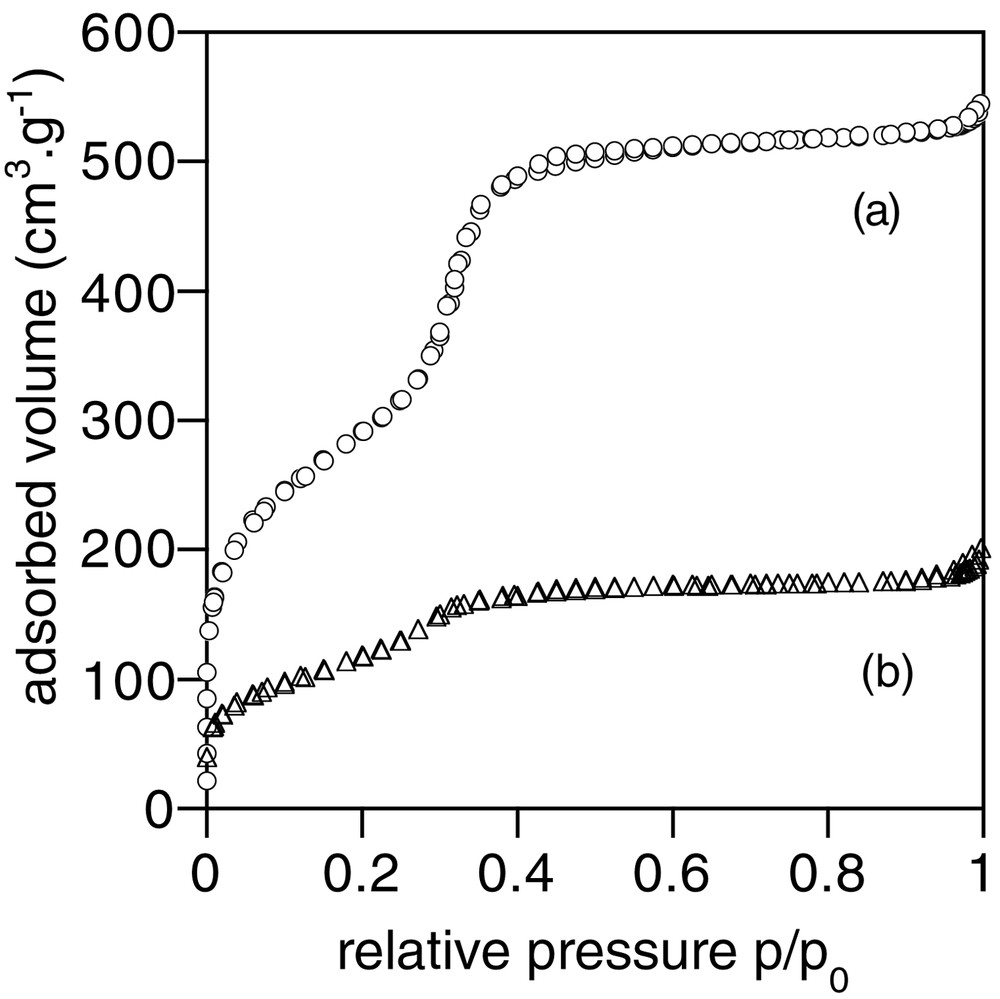
Nitrogen sorption curves of (a) the initial MCM-41 silica and (b) of the biocomposite 1.
BET specific area (SBET), total porous volume (Vp) and pore size (Dp) for the initial MCM-41 silica and the biocomposite 1, as determined by nitrogen-sorption measurements.
| SBET (m2 g–1) | VP (cm3 g–1) | DP (Å) | |
| MCM-41 | 1050 | 0.8 | 32 |
| 1 | 435 (870)a | 0.3 (0.6)a | 29 |
Scanning electron micrograph of the biocomposite 1 is shown in Fig. 3, together with the starting silica phase. As previously reported [24], the initial mesoporous system is seen as aggregated spheres of less than 1 μm. These particles seem to remain unchanged after alginate incorporation. However, they now appear rather homogeneously mixed with the biopolymer, which fills up the interparticle voids.

Scanning electron micrographs of (top) the initial MCM-41 silica and (bottom) of the biocomposite 1.
3.2 β-Galactosidase enzymatic activity
The kinetics curves of the hydrolysis of p-NPG catalysed by β-galactosidase in Ca-alginate gel and within the composite 1 after 24-h ageing at 5 °C are shown in Fig. 4a. Both systems present similar enzymatic activity of 0.1 ± 0.01 U. Limited enzyme leaching was ascertained by monitoring the activity of washing solutions, indicating ~ 10% β-galactosidase loss.

Kinetics of p-NPG hydrolysis by entrapped β-galactosidase in Ca-alginate gels (circle) and biocomposite 1 (triangle) after 24-h (a) and one-week (b) ageing at 5 °C.
After a one week ageing at 5 °C, the enzymatic activity of the biocomposite appears unperturbed, whereas an important decay (0.05 ± 0.01 U) is observed for the biopolymer gel; moreover, the p-NPG conversion to p-nitrophenol is not complete (Fig. 4b). In the meantime, enhanced enzyme leaching (~ 40%) was observed for the Ca-alginate gel after washing, whereas only a ≈ 15% value was measured for the composite.
4 Discussion
Alginic acid is a copolymer of l-guluronic acid and d-mannuronic acid [25] (Fig. 5), whose gelation properties have been widely used for cell immobilisation [20]. Adding divalent cationic metal ions (Ca2+, Sr2+) to sodium alginate solution induces cross-linking of the polymer chains. When kept in contact with aqueous solutions containing a complexing agent (citrate) or monovalent cations (Na+, K+), the gel starts to swell until all divalent cations have been withdrawn or exchanged, leading to alginate re-dispersion.

Representation of the two constituents of alginic acid.
This swelling process is the major drawback of biopolymers used for bioencapsulation. In contrast, mineral hosts are more stable towards solvents or ionic strength effects. However, they may lack the biocompatibility of natural polymers. Trying to design new biocomposites associating both components may therefore be a fruitful approach.
In this work, impregnation of mesoporous silica powders with alginate solutions results in biocomposites where the starting porous structure of the silica phase is maintained. However, the important loss in specific surface area and porous volume suggests that part of the initial mesoporosity is no longer accessible. This could be attributed to the presence of alginate within the silica porous network. However, this hypothesis does not appear compatible with the limited decrease in average pore size observed after impregnation. Another possibility lies in the filling-up of pore channels openings by the biopolymer. This is consistent with SEM studies indicating that alginate is located in the interparticle macroporosity of the silica powder, thus partially coating particle surface.
On a short term, the presence of silica particles does not appear to modify the reactivity of entrapped β-galactosidase. Measured catalytic activities are similar for the biopolymer gel and the biocomposite. Enzyme leaching is limited because of the strong cross-linking of the alginate network. Moreover, it suggests that p-NPG and p-nitrophenol transport is not hindered by the silica particles, indicating that their diffusion is possible within the mesopores. However, on a longer time scale, the two host matrices behave differently. Whereas the biocomposite reveals to be stable, the Ca-alginate gel shows an important loss of enzymes upon washing, leading to a decrease in catalytic activity. It suggests a loosening of the polymer network upon ageing, allowing an easier release of the entrapped species. This swelling may be limited within the composite, because the alginate chains are trapped in the inter-particle porosity of the silica.
5 Conclusion
This work shows that the association of an alginate gel to silica mesoporous particles at the microscale allows us to benefit from both the biocompatibility of the organic component and the stability of the mineral phase. Enzyme leaching appears limited, especially on the long term, whereas substrates and products diffusion are not hindered. Such an approach should permit the encapsulation of large species with dimensions above the accessible range of pore size for mesoporous materials. However, the main drawback of the materials here presented is that the biopolymer is confined in an open porosity of the silica network. Incorporating alginate within the cavities of a pre-formed mineral network exhibiting both controlled meso- and macroporosity would certainly lead to optimised properties.