

1 Introduction
Heterodifunctional P,O ligands, which associate a phosphine moiety and an oxygen functionality, are receiving much attention owing to the properties and reactivity of their transition-metal complexes [1–4]. Interest in this class of ligands has been further enhanced by the rich chemistry of phosphino-enolates, especially those derived from the β-carbonyl-phosphines A (Fig. 1). These anionic ligands usually behave as rigid 3-electron donor chelates as a result of the formation of an O–M covalent bond [5–9], and olefin oligomerisation catalysed by nickel complexes of the type B (SHOP process) is certainly their most remarkable application (Fig. 1) [10,11]. It has been shown that both catalytic activity and product selectivity (chain length distribution) could be tuned by substituent modifications on the phosphino-enolate ligands [12–15].
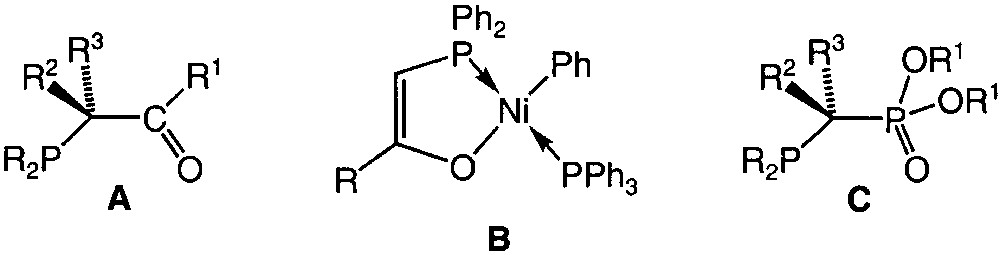
Formulae of compounds A, B, and C.
As part of our continuing interest in the design and chemistry of P,O analogues of β-carbonylphosphines A and the modified stereoelectronic influences thus brought about, we are currently studying the synthesis and coordination behaviour of new phosphonato-phosphines of the type C (Fig. 1). Since deprotonation of coordinated A is known to afford the anionic chelate present in B, we wondered whether a similar procedure with C could lead to the so far unknown corresponding anion. We wish to report here preliminary investigations on the transition-metal chemistry of the new P,O ligand rac-Ph2PCH(Ph)P(O)(OEt)2 1 and of its anion [Ph2PC(Ph)P(O)(OEt)2]–, which associate P(III) and P(V) centres.
2 Results and discussion
The new phosphonato-phosphine ligand 1 was synthesised from diethyl benzylphosphonate, upon deprotonation with n BuLi (or LDA), in the presence of LiCl [16, 17], and treatment of the resulting anion with Ph2PCl (Fig. 2 ). This reaction proceeded with a good selectivity and 1 was isolated, as a racemate, in more than 60% yield after work-up. This contrasts with the intractable mixtures of products that were obtained from diethyl methyl- or ethyl-phophonate under the same reaction conditions. This could be tentatively ascribed to the transient anion [PhCHP(O)(OEt)2]– having a less pronounced basic (nucleophilic) character than that of the anions derived from RCH2P(O)(OEt)2 (R = H, Me), and thus being less likely to be involved in side reactions.

(i) nBuLi (or LDA); (ii) Ph2PCl.
The 31P{1H} NMR spectrum of 1 consisted in two doublets at δ 25.3 and –6.0 ppm, with a 2JPP coupling of 51 Hz, ascribed to the Ph2P and phosphonate groups, respectively. In the 1H NMR spectrum (CDCl3) the CH resonance occurred as a doublet of doublets centred at δ 4.07 ppm (2JP(O)H = 9.9 and 2JPH = 1.3 Hz), whereas the CH2 protons, being diastereotopic, gave rise to three multiplets at δ 3.84, 3.67 and 3.56 ppm, with relative intensities of 2:1:1, respectively. In the IR spectrum (KBr), the ν(P=O) vibration was observed at 1243 cm–1. When 1 was reacted with 0.5 mol. equiv. of [Pd(NCMe)4](BF4)2 the bis-P,O-chelate complex 2 was formed in almost quantitative yield (equation (1)):
Coordination of the oxygen atom of the P=O function was evidenced in the IR spectrum, which showed the appearance of a new ν(P=O) stretch at 1168 cm–1. As expected, 2 was obtained as a mixture of diastereoisomers. Slow diffusion of hexane into a THF solution of 2 led to the formation of yellow crystals suitable for X-ray analysis, which contain exclusively the RR/SS pair of diastereoisomers. This allowed the assignment of the NMR spectra of the mixture. In the 31P{1H} NMR spectrum, each pair of diastereoisomers gave one set of two doublets. Those centred at δ 31.4 and 37.3 ppm were ascribed to the RR/SS pair, the signals for the RS/SR pair occurred at δ 33.5 and 38.2 ppm. Although the RS/SR diastereoisomers were preferentially formed (kinetic product) in the early stage of the reaction (31P{1H} NMR monitoring), as shown by a 65:35 RS/SR:RR/SS ratio observed after 10 min, an equilibrium was reached after a few days, in which the RR/SS pair was largely predominant (RS/SR:RR/SS ratio: 15:85). The 1H NMR spectrum showed two well-defined doublets of doublets at δ 5.46 (2JPH = 23.1 and 16.2 Hz) and 5.81 (2JPH = 27.2 and 16.2 Hz), which were ascribed to PCHP protons of the RS/SR and RR/SS isomers, respectively.
Selected crystal and data collection are given in Table 1 and selected bond lengths and angles are given in Table 2. The unit cell contains both RR and SS enantiomers. An ORTEP view of the cation of the RR enantiomer in 2 ·THF is represented in Fig. 3. It shows the chelation of the Pd centre by two phosphonato- phosphine ligands 1 through coordination of the P and O atoms of the phosphine moieties and the P=O groups, respectively. There is no symmetry element in the molecule. The coordinated P atoms are in cis positions around the Pd centre [P(3)–Pd–P(1) = 95.67(4)°]. The geometry at the Pd centre is square planar as shown in the O(3)–Pd–P(3) and O(4)–Pd–P(1) angles of 174.91(8) and 175.09(9)°, respectively. The bond distances within the P,O chelates are within the expected range. The P,O bite angles [O(3)–Pd–P(1) = 89.35(9)°; O(4)–Pd–P(3) = 89.14(9)°] compare with those found in other P CP(O) chelate complexes such as [PdCl(Me)(dppmO-κ2-P,O)] [89.2(1)°] [18] or [RhCl{PPh2CH2P(O)(OPri)2-κ2-P,O)] [88.1(2)°] [19]. In both five-membered ring chelates, there is a significant deviation of the C atoms from the best plane containing the other four atoms, 0.721(5) and 0.674(5) Å for C(7) and C(32), respectively. The dihedral angle between the mean planes of the P,O chelates is 10.9(2)°. Interestingly, the dihedral angle between the mean plane containing the Pd, O(3), P(1), C(7) and P(2) atoms and that of the C(8)–C(13) phenyl ring is larger [72.1(5)°] than that observed for the similar planes within the second P,O chelate [59.2(5)°].
Selected crystal and data collection parameters for 2·THF and 4
| 2·THF | 4 | |
| Formula | C46H52B2F8O6P4Pd·C4H8O | C32H37NO3P2Pd |
| Mr | 1176.88 | 651.97 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| a (Å) | 19.994(5) | 17.706(5) |
| b(Å) | 14.413(5) | 9.394(5) |
| c(Å) | 19.243(5) | 18.435(5) |
| β (°) | 93.559(5) | 96.697(5) |
| V(Å3) | 5535(3) | 3045(2) |
| Z | 4 | 4 |
| T(K) | 293(2) | 173(2) |
| ρ(g cm–3) | 1.412 | 1.422 |
| λ(Å) | 0.71069 | 0.71069 |
| R[I > 2 σ(I)] | 0.0719 | 0.0534 |
| Rw[I > 2 σ(I)] | 0.1760 | 0.1111 |
| gof | 1.039 | 0.838 |
Selected bond lengths (Å) and angles (°) for 2·THF, with esd’s in parenthesis
| Pd–O(3) | 2.109(3) | O(3)–Pd–O(4) | 85.8(2) |
| Pd–O(4) | 2.113(3) | O(3)–Pd–P(3) | 174.91(8) |
| Pd–P(1) | 2.240(2) | O(4)–Pd–P(3) | 89.14(9) |
| Pd–P(3) | 2.239(2) | O(4)–Pd–P(1) | 175.09(9) |
| P(1)–C(7) | 1.869(4) | P(1)–Pd–O(3) | 89.35(9) |
| P(2)–C(7) | 1.814(4) | P(3)–Pd–P(1) | 95.67(4) |
| P(3)–C(32) | 1.873(5) | Pd–P(1)–C(7) | 100.6(2) |
| P(4)–C(32) | 1.808(4) | C(7)–P(2)–O(3) | 108.0(2) |
| P(2)–O(1) | 1.548(3) | C(32)–P(3)–Pd | 101.0(2) |
| P(2)–O(2) | 1.560(3) | P(2)–O(3)–Pd | 118.4(2) |
| P(2)–O(3) | 1.494(3) | P(4)–O(4)–Pd | 117.2(2) |
| P(4)–O(4) | 1.490(3) | P(1)–C(7)–P(2) | 106.2(2) |
| P(4)–O(5) | 1.550(3) | P(4)–C(32)–P(3) | 104.6(2) |
| P(4)–O(6) | 1.550(3) |
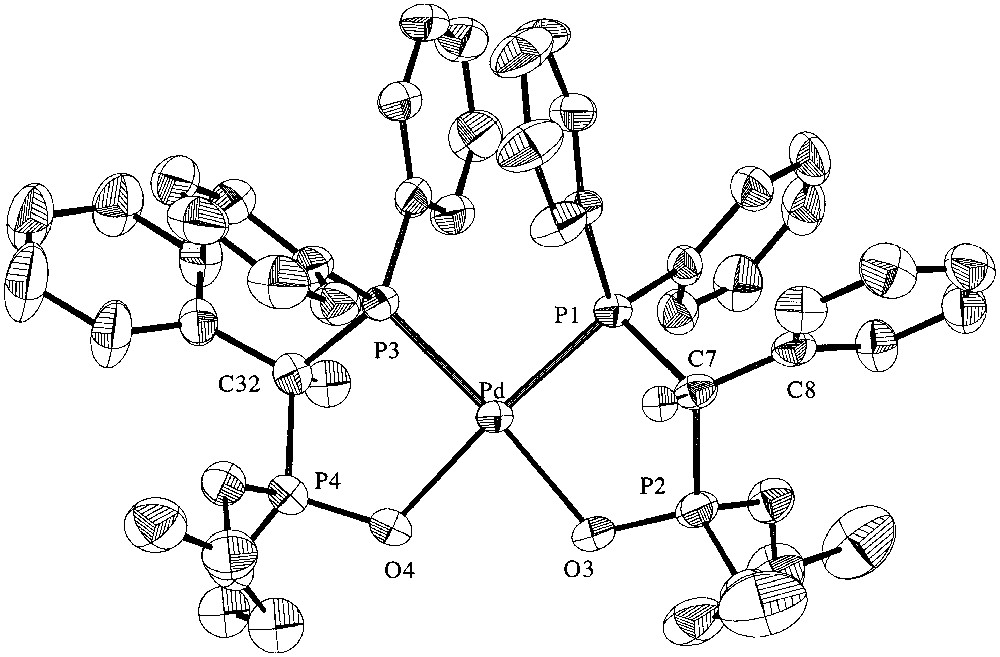
ORTEP representation of the cation of the RR enantiomer in 2·THF. Ellipsoids are shown at the 50% probability level; hydrogen atoms are not shown for clarity.
Reaction of [Pd(dmba-C,N)(μ-Cl)]2 with 2 mol equiv of 1 led the formation of complex 3 in which 1 behaves as a monodentate phosphine ligand (Fig. 4). As a result, there was no significant shift in the ν(P=O) vibration (1248 cm–1) by comparison with the free ligand. The 31P{1H} NMR spectrum contained two signals at δ 43.8 and 20.6 ppm, which were ascribed to the phosphine and phosphonate moities, respectively. The JPP coupling constant was only 5.5 Hz. In the 1H NMR spectrum, the PCHP proton gave a doublet of doublets at δ 6.02 ppm (2JPH = 14.6 and 10.3 Hz), while complicated patterns were observed for the OEt signals, owing to the OCH2 protons being diastereotopic. Note that the 13C resonance of the PCP carbon atom occurred as a doublet of doublets at δ 45.0 ppm, with 1 JPC coupling constants of 131 and 14 Hz. Addition of KH in excess (ca. 2 mol equiv) to a clear yellow THF solution of 3 rapidly afforded a dark green suspension (30 min), which turned black/red after 24 h. After work-up, the new complex 4 was obtained in 92% yield (Fig. 4).

Synthesis of the complex 4.
Compared to 3, the change in the nature of the P,O ligand in 4 was clearly reflected in the 31P{1H} NMR spectrum. Thus the resonance of the phosphine moiety underwent a low-field shift of ca. 11 ppm (δ 55.4 ppm), whereas that of the phosphonate function shifted to higher field (δ 14.5 vs 20.6 ppm). In addition, a large 2JPP coupling of 126 Hz was observed. The 1H NMR spectrum did not contain any signal characteristic of a PCHP proton. A striking feature was the occurrence of the PCP 13C resonance at δ 141.1 ppm (dd, 1JPC = 10.8 and 6.5 Hz). The proposed structure of 4 was confirmed by an X-ray diffraction study. Selected crystal and data collection are given in Table 1 and selected bond lengths and angles are given in Table 3. An ORTEP view of 4 is represented in Fig. 5. The coordination geometry around the Pd centre is approximately square planar, as reflected in the P(1)–Pd–N and O(3)–Pd–C(20) angles of 175.76(9) and 171.5(2)°, respectively. The bond distances and angles within the dmba ligand are unexceptional. By contrast with the situation encountered in 2, the Pd–P(1)–C(7)–P(2)–O(3) ring is close to planarity and the dihedral angle between the best plane passing through these atoms and that containing the phenyl ring attached to C(7) is only 8.4(2)°. The geometry around the C(7) atom being almost trigonal planar is indicative of a sp2 hybridisation [P(1)–C(7)–P(2) = 113.3(2)°; P(1)–C(7)–C(8) = 121.0(2)° and C(8)–C(7)–P(2) = 124.4(2)°]. A comparison of the bond distances within the P,O chelate between 2 and 4 reveals a slight lengthening of the P(1)–Pd, P(2)–O(3) and O(3)–Pd bonds of 0.01 to 0.017 Å, the most striking feature is the marked shortening of the P(1)–C(7) and P(2)–C(7) bond distances of 0.087 and 0.148 Å, respectively. Furthermore, the Pd–P(1)–C(7), P(1)–C(7)–P(2) and C(7)–P(2)–O(3) angles are more obtuse in 4 than in 2 by ca 6°. These structural features are consistent with the proposed charge delocalisation within the P,O chelate of 4 represented in Fig. 5. To the best of our knowledge, this complex is the first example of an anionic phosphino-phosphonate P,O chelate. Having now established the availability of these new ligands and their bonding mode to a metal centre, their reactivity is now being investigated.
Selected bond lengths (Å) and angles (°) for 4, with esd’s in parentheses.
| Pd–N | 2.133(3) | P(1)–Pd–O(3) | 87.02(8) |
| Pd–C(20) | 1.991(4) | Pd–P(1)–C(7) | 105.7(2) |
| Pd–O(3) | 2.119(2) | P(1)–C(7)–P(2) | 113.3(2) |
| Pd–P(1) | 2.257(2) | P(2)–O(3)–Pd | 118.5(2) |
| P(1)–C(7) | 1.782(4) | O(3)–P(2)–C(7) | 113.7(2) |
| P(2)–C(7) | 1.666(4) | O(3)–Pd–C(20) | 171.5(2) |
| P(2)–O(3) | 1.511(3) | P(1)–Pd–N | 175.76(9) |
| P(1)–C(7)–öC(8) | 121.0(2) | ||
| C(8)–C(7)–P(2) | 124.4(2) |

ORTEP representation of 4. Ellipsoids are shown at the 50% probability level; hydrogen atoms are not shown for clarity.
3 Experimental
3.1 General
All the reactions and manipulations were carried out under an inert atmosphere of purified nitrogen using standard Schlenk tube techniques. Solvents were dried and distilled under nitrogen before use: hexane, pentane and toluene over sodium, tetrahydrofuran and diethyl-ether over sodium-benzophenone, dichloromethane over calcium hydride. Nitrogen (Air liquide, R-grade) was passed through BASF R3-11 catalyst and molecular sieves columns to remove residual oxygen and water. Elemental C, H and N analyses were performed by the “Service de microanalyses” (Université Louis-Pasteur, Strasbourg, France). Infrared spectra were recorded on a IFS 66 Bruker FT–IR spectrometer. The 1H, 31P{1H} and 13C{1H} NMR spectra were recorded at 300.1, 121.5 and 75.5 MHz respectively, on a Bruker AC300 instrument. Phosphorus chemical shifts were externally referenced to 85% H3PO4in H2O, with downfield chemical shifts reported as positive.
3.2 Syntheses
Complexes [Pd(NCMe)4](BF4)2 [20] and [(dmba)Pd(μ-Cl)]2 [21] were prepared according to literature procedures. PhCH2P(O)(OEt)2 (Aldrich) was distilled before use.
Ph 2PCH(Ph)P(O)(OEt)2 1. To a THF/Et2O (1:1 ratio, 100 ml) solution of PhCH2P(O)(OEt)2 (5.00 g, 21.9 mmol) and LiCl (0.50 g, 11.8 mmol) cooled to –78 °C, 13.7 ml of a 1.6 M solution of nBuLi (21.9 mmol) in hexane were added dropwise over a period of 5 min. The temperature was then allowed to warm up to –20 °C and the mixture was stirred at this temperature for 30 min, before it was cooled again to –78 °C. Freshly degassed Ph2PCl (4.83 g, 21.9 mmol) in THF (10 ml) was then added dropwise and the reaction mixture was allowed to warm up to ambient over a period of 1 h. Degassed water (10 ml) was added and vigorous stirring maintained for 2 min. After decantation, the organic phase was collected and dried over degassed MgSO4. After filtration, the volatiles were removed under reduced pressure and the residue was washed with Et2O (2 × 25 ml) and pentane (1 × 25 ml), affording 1 as a white solid (5.530 g, 62%). 1H NMR [ppm, CDCl3]: 0.96 (m, 6H, CH3), 3.56 (m, 1H, CH2), 3.67 (m, 1H, CH2), 3.84 (m, 2H, CH2), 4.07 (dd, 1H, PCHP, 2JP(O)H = 9.9 Hz, 2JPH = 1.3 Hz), 7.05–8.10 (m, 15H, aromatics). 31P{1H} NMR [ppm, CDCl3]: –6.0 (d, P(O), 2JPP = 51 Hz), 25.3 (d, PPh2, 2JPP = 51 Hz). IR [KBr, cm–1]: 1310(m), 1243(vs), 1201(m), 1161(s). Anal: expt. (calcd) for C23H26O3P2: C, 67.12 (66.99); H, 6.51 (6.35).
cis-[Pd{Ph2PCH(Ph)P(O)(OEt)2-κ2-P,O}2](BF4)2 2. Solid [Pd(NCMe)4](BF4)2 (0.146 g, 0.33 mmol) and 1 (0.271 g, 0.66 mmol) were placed in a Schlenk flask and CH2Cl2 (15 ml) was added at ambient temperature. The yellow reaction mixture was stirred for 1 h and the volatiles were removed under reduced pressure. The residue was washed with Et2O (2 × 15 ml) and pentane (2 × 10 ml), which afforded 2 as a yellow solid (0.325 g, 89%). Complex 2 was obtained as a mixture of diastereoisomers, see text. IR [KBr, cm–1]: 1287(w), 1238(w), 1168(s). Anal: expt. (calcd) for C46H52B2F8O6P4Pd: C, 49.75 (50.01); H, 4.47 (4.74).
NMR data for RR/SS isomers: 1H NMR [ppm, CDCl3]: 1.21 (m, 12H, CH3), 4.32 (m, 2H, CH2), 4.43 (m, 2H, CH2), 4.65 (m, 4H, CH2), 5.81 (dd, 2H, PCHP, 2JPH = 27.2 and 16.2 Hz), 6.70-7.75 (m, 30H, aromatics). 31P{1H} NMR [ppm, CDCl3]: 31.4 (d, P(O), 2 JPP = 39 Hz), 37.3 (d, PPh2, 2 JPP = 39 Hz).
NMR data for RS/SR isomers: 1H NMR [ppm, CDCl3]: 1.18 (m, 12H, CH3), 4.18 (m, 2H, CH2), 4.35 (m, 6H, CH2), 5.46 (dd, 2H, PCHP, 2JPH= 23.1 and 16.2 Hz), 6.70-7.75 (m, 30H, aromatics). 31P{1H} NMR [ppm, CDCl3]: 33.5 (d, P(O), 2JPP = 40 Hz), 38.2 (d, PPh2, 2JPP = 40 Hz).
[(dmba)PdCl{Ph2PCH(Ph)P(O)(OEt)2}] 3. Solid [(dmba)Pd(μ-Cl)]2 (1.436 g, 2.6 mmol) and 1 (2.146 g, 5.2 mmol) were placed in a Schlenk flask and CH2Cl2 (25 ml) was added at ambient temperature. The yellow reaction mixture was stirred for 30 min and the volatiles were removed under reduced pressure. The residue was washed with Et2O (2 × 20 ml) and pentane (2 × 20 ml), which afforded 2 as a pale yellow solid (3.330 g, 93%). 1H NMR [ppm, CDCl3]: 1.16 (m, 6H, OCH2CH3), 2.82 (d, 6H, NCH3, 4JPH = 2.6 Hz), 3.72 (m, 2H, OC H2CH3), 3.93 (m, 1H, OC H2CH3), 3.96 (s, 2H, NCH2), 4.05 (m, 1H, OC H2CH3), 6.02 (dd, 2H, PCHP, 2 JPH = 14.6 and 10.3 Hz), 6.09–8.23 (m, 19H, aromatics). 31P{1H} NMR [ppm, CDCl3]: 20.6 (d, P(O), 2 JPP = 5.5 Hz), 43.8 (d, PPh2, 2JPP = 5.5 Hz). 13C{1H} NMR [ppm, CDCl3]: 15.9 (d, OCH2CH3, 3JPC = 6.4 Hz), 16.2 (d, OCH2CH3, 3JPC = 6 Hz), 45.0 (dd, PCP, 1JPC = 131 and 14 Hz), 50.3 (d, NCH3, 3JPC = 27 Hz), 62.3 (d, OCH2CH3, 2JPC = 6.7 Hz), 62.6 (d, O CH2CH3, 2JPC = 7.2 Hz), 73.0 (s, NCH2), 122.0-151.9 (aromatics). IR [KBr, cm–1]: 1293(w), 1248(vs), 1177(s), 1096(s). Anal: expt. (calcd) for C32H38ClNO3P2Pd: C, 55.64 (55.83); H, 5.33 (5.56); N, 2.05 (2.03).
[(dmba)Pd{Ph2PC(Ph)PO(OEt)2-κ2-P,O}] 4. To a THF (30 ml) suspension of KH (0.150 g, 3.75 mmol) was added solid 3 (1.500 g, 2.18 mmol) in one portion at ambient. This resulted in a gas (H2) evolution. The solution darkened and turned deep black–green after ca. 1 h (H2 evolution could still be observed). The reaction mixture was stirred overnight. The resulting black–red solution was filtered over a pad a dry Celite (1.5 cm) and the volatiles were removed under reduced pressure. The brown residue was washed with pentane (2 × 25 ml), which afforded 4 as pale brown–kaki solid (1.300 g, 92%). 1H NMR [ppm, CDCl3]: 1.36 (t, 6H, OCH2CH3, 3JHH = 7 Hz), 2.72 (d, 6H, NCH3, 4JPH = 2.3 Hz), 3.84 (s, 2H, NCH2), 4.15 (m, 4H, OC H2CH3), 6.60–7.88 (m, 19H, aromatics). 31P{1H} NMR [ppm, CDCl3]: 14.6 (d, P(O), 2JPP = 126 Hz), 55.4 (d, PPh2, 2JPP = 126 Hz). 13C{1H} NMR [ppm, CDCl3]: 16.7 (d, OCH2CH3, 3JPC = 6.8 Hz), 49.6 (s, NCH3), 60.9 (d, OCH2CH3, 2JPC = 3.6 Hz), 70.9 (s, NCH2), 118.3–148.9 (aromatics), 141.1 (dd, PCP, 1JPC = 10.8 and 6.5 Hz). IR [nujol, cm–1]: 1291(m), 1258(vs), 1156(s), 1117(s), 1108(s). Anal: expt. (calcd) for C32H37NO3P2Pd: C, 58.91 (58.95); H, 5.81 (5.72); N, 2.12 (2.15).
3.3 X-ray data collection
A yellow crystal of 2·THF, grown from slow diffusion at RT of hexane into a THF solution of 2, or a yellow crystal of 4, grown from slow diffusion at RT of pentane into a CDCl3 solution of 4, was selected and mounted on a Kappa CCD diffractometer. Data were collected using phi-scans and the structure was solved using direct methods and refined against F2 using SHELX 97 software [22,23]. No absorption correction was used. For 2·THF, a total of 16 110 reflections was collected with 1.02 < θ < 30.03°, of which 9327 unique reflections had intensities I > 2 σ(I). For 4, a total of 8869 reflections was collected with 1.02 < θ < 30.03°, of which 4929 unique reflections had intensities I > 2 σ(I). All non-hydrogen atoms were refined anisotropically with H atoms introduced as fixed contributors (dC–H = 0.95 Å, U11 = 0.04). Full data collection parameters, and structural data are available as supplementary material.
4 Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif files No. CCDC 199571 (2. THF) and 199572 (4) ..., and can be obtained by contacting the CCDC.
Acknowledgements
We gratefully acknowledge the support provided by the French ‘Centre national de la recherche scientifique’ and ‘Ministère de la Recherche’.