

1 Introduction
Phosphorus ylides are reactive compounds, which take part in many reactions of value in the synthesis of organic products [1–4]. Resonance-stabilized phosphorous ylides, particularly the keto ylides, are used as important ligands in organometallic and coordination chemistry [5–9]. This delocalization also makes these ligands weak nucleophiles, but this does not reduce their interest as ligands. And it was their weak donor ability that allowed us to prepare new types of ylide complexes [9–11]. The reactivity and coordination chemistry of carbonyl stabilized [12–14] and bifunctionalized [2,15–18] ylides is an important research field of our group. In recent years, this area was developed while studying the organometallic chemistry of ketenylidenetriphenylphosphorane Ph3PCCO towards Pt substrates, obtaining complexes in which the ylide was σ-bonded to the metal through the ylidic carbon atom [19,20]. Phosphorus ylide complexes are versatile compounds in catalytic reactions, such as the hydrogenation of olefins [21], the cyclotrimerization [22] and polymerization of acetylenes [23]. Phosphorus ylides of the type Ph2P(CH2)nPPh2(CHC(O)R) (n = 1, or 2; R = Me, Ph or –OMe), containing phosphine and keto-stabilized phosphorus ylide moieties are also known, and their transition metal complexes are mostly confined to palladium and, to a lesser extent, to platinum [24–26]. We have tried to convert both phosphine moieties in 1,2–bisdiphenylphosphinoethane into keto-stabilized ylides to facilitate the coordination ability of the ylides. They coordinate to palladium(II) to form chelate ylide complexes of the type PdCl2(Y) (Y = chelate ylide) with a seven-membered chelate ring [17]. However, the synthesis of cyclic bis-ylides, and the study of their complexation to transition metals, is a less documented field and very few examples have been reported in the literature [27–29], in spite of their practical importance [30,31]. Palladium catalysis is ubiquitous in modern organic chemistry and a great number of catalytic systems have been designed and fine-tuned through careful ligand design and choice of experimental conditions (solvent, temperature, catalyst loading, etc.). The palladium-catalyzed Suzuki cross-coupling of aryl halides with arylboronic acid is among the most powerful C–C-bond-forming reactions available to synthetic chemists [32]. Current technologies demand new catalysts, which are inexpensive, readily accessible, moisture- and air-stable and, most importantly, highly effective under low catalyst loading [33]. Recently, palladacycle complexes have attracted much attention as exciting catalyst precursors to cross-coupling reactions [32b–h,33c]. In conjunction with our previous work on the synthesis and catalytic activity of palladacycle complexes [34], herein, we report the synthesis, characterization and application of a new Pd(II) complex as a catalyst in the Suzuki cross-coupling reaction of various aryl halides under aerobic and conventional heating conditions.
2 Experimental
2.1 Physical measurements and materials
All the reactions were carried out under an atmosphere of dry nitrogen. Methanol was distilled over magnesium powder and diethyl ether (Et2O) over a mixture of sodium and benzophenone just before use. All the other solvents were reagent-grade and used without further purification. Melting points were measured on a SMP3 apparatus. IR spectra were recorded on a Shimadzu 435-U-04 spectrophotometer from KBr pellets. 1H, 13C and 31P NMR spectra were recorded on 400 MHz Bruker and 90 MHz Jeol spectrometer in DMSO-d6 or CDCl3 as the solvent at 25 °C. Chemical shifts (ppm) are reported according to internal TMS and external 85% phosphoric acid. Coupling constants are given in Hz. Elemental analyses for C, H and N atoms were performed using a PerkinElmer 2400 series analyzer.
2.2 X-ray crystallography
The single-crystal X-ray diffraction data of suitable crystals of 5 were collected with an Oxford Diffraction single-crystal X-ray diffractometer using the mirror monochromated Mo Kα radiation (0.71073 Å) at 130 K (Table 3). Gaussian absorption corrections were carried out using a multifaceted crystal model using CrysAlisPro [35]. Both structures were solved by direct methods and refined by the full-matrix least-squares method on F2 using the SHELXTL-97 crystallographic package [36,37]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions using a riding model, with isotropic displacement parameters.
Crystal data and refinement details for 5.
| Compound 5 | |
| Empirical formula | C44H34Cl10O2P2 |
| Formula weight | 1011.15 |
| Temperature (K) | 130(10) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Triclinic |
| Space group | P |
| a (Å) | 11.4908(3) |
| b (Å) | 15.0297(5) |
| c (Å) | 16.0555(5) |
| α (̊). | 106.327(3) |
| β (̊) | 110.245(3) |
| γ (̊) | 103.841(3) |
| Z | 2 |
| Absorption coefficient (mm−1) | 0.708 |
| θ range for data collection (°) | 2.92 to 30.11 |
| Index ranges | –15 ≤ h ≤ 15 |
| –19 ≤ k ≤ 21 | |
| –22 ≤ l ≤ 20 | |
| Reflections collected | 24918 |
| Independent reflections | 12040 [R(int) = 0.0317] |
| Absorption correction | Gaussian |
| Max. and min. transmission | 0.965 and 0.980 |
| Refinement method | Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 | 1.046 |
| Final R indices [I > 2 σ(I)] | R1 = 0.0446, wR2 = 0.1034 |
| R indices (all data) | R1 = 0.0701, wR2 = 0.1230 |
| Largest diff. peak and hole (e·Å−3) | 0.59 and −0.52 |
2.3 Sample preparation
2.3.1 Synthesis of diphosphonium salts
2.3.1.1 General procedure
A solution consisting of bis(diphenylphosphino)ethane (dppe) (0.32 g, 0.8 mmol) and the related carbonyl compound (1.7 mmol) in acetone was stirred at room temperature for 24 h. The resulting solution was filtered off, and the precipitate obtained washed with diethyl ether and dried.
2.3.1.2 Data for [C10H7C(O)CH2PPh2(CH2)2PPh2CH2C(O)C10H7]Br2 (1)
Yield: 0.58 g, 81%. Mp 248–250 °C. Anal. calcd. for C50H42Br2O2P2: C, 66.98; H, 4.72. Found: C, 66.89; H, 4.70%. IR (KBr disk, cm−1) ν: 1658 (CO). 1H NMR (DMSO-d6) δH: 3.74 (br, 4H, CH2); 6.02 (br, 4H, PCH2CO); 7.80, 7.95, 8.02, 8.92 (m, 34H, Ph and 2-naphtyl). 31P NMR (DMSO-d6) δP: (ppm): 27.17 (s, PPh2). 13C NMR (DMSO-d6) δC: 23.43 (br, CH2); 36.19 (br, PCH2CO); 119.38, 123.68, 124.80, 129.32, 139.44, 130.50, 131.70, 132.85, 133.63, 133.83, 134.76, 139.43 (Ph and 2-naphtyl); 193.25 (s, CO).
2.3.1.3 Data for [Cl2C6H3C(O)CH2PPh2(CH2)2PPh2CH2C(O)C6H3Cl2]Cl2 (2)
Yield: 0.47 g, 69%. Mp 298–300 °C. Anal. calcd. for C42H34Cl6O2P2: C, 59.67; H, 4.05. Found: C, 59.72; H, 4.01%. IR (KBr disk, cm−1) ν: 1684 (CO).1H NMR (CDCl3) δH: 3.83 (br, 4H, CH2); 6.32 (br, 4H, PCH2CO); 7.66, 7.72, 8.02, 8.56, 8.75, 9.03 (m, 34 H, Ph). 31P NMR (CDCl3): δP (ppm): 23.97 (s, PPh2). 13C NMR (CDCl3) δC: 21.36 (dd, CH2, 1JPC = 64.58, 2JPC = 30.03); 38.09 (t, 2C, PCH2CO, 1JPC = 28.03 Hz); 116.29, 117.04, 117.9, 128.85, 130.72, 132.14, 133.33, 134.83,139.18, 139.29 (Ph); 191.79 (s, CO).
2.3.1.4 Data for [O2NC6H4C(O)CH2PPh2(CH2)2PPh2CH2C(O)C6H4NO2]Br2 (3)
Yield: 0.61 g, 85%. Mp 249–251 °C. Anal. calcd. for C42H46Br2N2O6P2: C, 56.90; H, 4.09. Found: C, 56.78; H, 4.12%. IR (KBr disk, cm−1) ν: 1682 (CO). 1H NMR (DMSO): δH = 3.8 (br, 4H, CH2); 6.58 (br, 4H, PCH2CO); 7.51, 7.59, 7.67, 7.71, 7.80, 7.91, 8.07 (d, 3JPC = 5.58), 8.42 (d, 2JPC = 18.0), 8.52 (d, 2JPC = 16.5), 8.71 (m, 28 H, Ph). 31P NMR (DMSO-d6): δP (ppm): 26.72 (s, PPh2). 13C NMR (DMSO-d6) δC: 15.30 (t, CH2, 1JPC = 25.66); 33.56 (br, PCH2CO); 122.72, 127.54, 128.67, 128.92, 129.21, 129.55, 132.65, 133.88, 134.63, 135.57, 146.66, 147.22; 190.1 (s, CO).
2.3.2 Synthesis of ligands
2.3.2.1 General procedure
Phosphonium salts 1–3 were further treated with sodium bis(trimethylsilyl)amide (1.0 mL), leading to the elimination of HX (X = Cl, Br), affording the free ligands 4–6 as the sole products.
2.3.2.2 Data for C10H7C(O)CHPPh2(CH2)2PPh2CHC(O)C10H7 (4)
Yield: 0.22 g, 60%. Mp 219–221 °C. Anal. calcd. for C50H40O2P2: C, 81.73; H, 5.48. Found: C, 81.60; H, 5.46%. IR (KBr disk, cm−1) ν: 1517 (CO). 1H NMR (CDCl3) δH: 3.40 (s, 4H, CH2); 4.36 (br, 2H, CH); 7.46, 8.02, 8.12, 7.80, 8.53 (m, 34 H, Ph and 2-naphtyl). 31P NMR (CDCl3) δP: 15.12 (s, PPh2). 13C NMR (DMSO-d6) δC:16.95 (br, CH2); 22.383 (t, CH, 1JPC = 57.15 Hz); 116.02, 117.6, 119.94, 123.52, 130.07, 133.37, 134.86, 147.87 (Ph); 191.24 (s, CO).
2.3.2.3 Data for Cl2C6H3C(O)CHPPh2(CH2)2PPh2CHC(O)C6H3Cl2 (5)
Yield: 0.22 g, 57%. Mp 258–260 °C. Anal. calcd. for C42H32Cl4O2P2: C, 65.30; H, 4.18. Found: C, 64.93; H, 4.24%. IR (KBr disk, cm−1) ν: 1582 (CO). 1H NMR (CDCl3) δH: 3.31 (br, 4H, CH2); 4.45 (br, 2H, CH); 7.49, 7.55, 7.63, 7.72,8.17, 8.25 (m, 26 H, Ph). 31P NMR (CDCl3) δP: 13.50 (s, PPh2). 13C NMR (CDCl3) δC: 18.14 (br, CH2); 25.01 (br, CH); 121.71, 125.88, 126.35, 127.53, 129.31, 131.63, 133.07, 134.28 (Ph); 185.11 (s, CO).
2.3.2.4 Data for O2NC6H4C(O)CHPPh2(CH2)2PPh2CHC(O)C6H4NO2 (6)
Yield: 0.25 g, 68%. Mp 203–205 °C. Anal. calcd. for C42H44N2O6P2: C, 68.65; H, 6.04. Found: C, 68.61; H, 6.06%. IR (KBr disk, cm−1) ν: 1585 (CO). 1H NMR (CDCl3): δH = 3.06 (bd, 4H, CH2, 2JPH = 3.20); 4.60 (t, 2H, CH, 2JPH = 11.21); 7.16, 7.24, 7.5, 7.62, 7.71, 8.24 (t, 3JPC = 7.83), 8.77 (m, 28 H, Ph). 31P NMR (CDCl3): δP (ppm): 15.49 (s, PPh2). 13C NMR (DMSO-d6) δC: 17.34 (br, CH2); 26.08 (br, CH); 120.80, 124.03, 125.47, 129.26, 129.32, 129.38, 129.50, 131.95, 132.01, 132.6, 133.25, 147.73 (Ph); 180.16 (s, CO).
2.3.3 Synthesis of the Pd(II) complexes
2.3.3.1 General procedure
To a [PdCl2(COD)] (0.085 g, 0.3 mmol) dichloromethane solution (5 mL), a solution of ylides 4–6 (0.3 mmol) (5 mL, CH2Cl2) was added dropwise. The resulting solution was stirred for 2 h at room temperature and then concentrated to ca. 2 mL in volume and treated with cool n-hexane (ca. 15 mL) to afford the products, which were collected and dried under vacuum.
2.3.3.2 Data for [C10H7C(O)CHPPh2(CH2)2PPh2CHC(O)C10H7]PdCl2 (7)
Yield: 0.17 g, 64%. Mp 212 °C dec. Anal. calcd. for C50H40Cl2O2P2Pd: C, 65.83; H, 4.42. Found: C, 65.71; H, 4.37%. IR (KBr disk, cm−1) ν: 1624 (CO). 1H NMR (CDCl3): δH = 3.42 (br, 4H, CH2); 5.25 (br, 2H, CH); 7.23, 7.27, 7.39, 7.57, 7.68, 7.79, 7.88, 8.30 (m, 34 H, Ph and 2-naphtyl). 31P NMR (CDCl3) δP (ppm): 28.03 (d, 3JPP = 5.00 Hz, P1), 30.34 (d, 3JPP = 3.23 Hz, P2).
2.3.3.3 Data for [Cl2C6H3C(O)CH = PPh2(CH2)2PPh2 = CHC(O)C6H3Cl2]PdCl2 (8)
Yield: 0.176 g, 62%. Mp 220 °C dec. Anal. calcd. for C42H32Cl6O2P2Pd: C, 53.11; H, 3.39. Found: C, 53.14; H, 3.38%. IR (KBr disk, cm−1) ν: 1625, 1655 (CO).1H NMR (DMSO-d6): δH = 4.20 (s, 4H, CH2); 5.38 (s, 2H, CH); 7.04, 7.039, 7.55, 8.29, 8.32, 8.92 (m, 26 H, Ph). 31P NMR (DMSO-d6) δP (ppm): 26.10 (d, 3JPP = 4.85 Hz, P1), 32.04 (d, 3JPP = 6.16 Hz, P2).
2.3.3.4 Data for [O2NC6H4C(O)CHPPh2(CH2)2PPh2CHC(O)C6H4NO2]PdCl2 (9)
Yield: 0.324 g, 72%. Mp 245–247 °C. Anal. Calc. for C42H34Cl2N2O6P2Pd: C, 55.92; H, 3.79. Found: C, 55.89; H, 3.76%. IR (KBr disk, cm−1) ν: 1611, 1643 (CO).1H NMR (DMSO-d6) δH (ppm): 2.38 (br, 4H, CH2), 6.15 (br, 2H, CH), 7.46, 7.63, 7.92, 8.05, 8.22 (m, 24H, Ph). 31P NMR (DMSO-d6) δP: 29.59 (br, P1), 32.76 (bd, PPh2, 3JPP = 4.24 Hz, P2).
2.4 General experimental procedure for Suzuki cross-coupling reactions
A mixture of an aryl halide (0.75 mmol), phenyl boronic acid (1 mmol), complex 9 (0.001 mol%), Cs2CO3 (1.5 mmol), and DMF (2 mL) was heated to 110 °C for specified time. The reactions were monitored by thin-layer chromatography (TLC). The reaction mixture was then cooled to room temperature. After completion of the reaction, the mixture was diluted with n-hexane (15 mL) and water (15 mL). The organic layer was washed with brine (15 mL), dried over CaCl2. The solvent was evaporated and a crude product was obtained, which was analyzed by 1H and 31C NMR. The liquid residues were purified by silica gel column chromatography (n-hexane:EtOAc, 80:20), whereas the solid ones were purified by re-crystallization from ethanol and water.
3 Results and discussion
3.1 Synthesis
Diphosphine Ph2P(CH2)2PPh2 reacts with 2 equiv of the appropriate ketones, forming the corresponding phosphonium salts 1–3. Further treatment with sodium bis(trimethylsilyl)amide leads to two-fold elimination of HX (X = Cl, Br), giving the free ligands 4–6. Reaction of these ligands with Pd(II) chloride in 1:1 molar ratio form C,C-chelated complexes [RC(O)CHPPh2(CH2)2PPh2CHC(O)R]PdCl2, where R = 2-naphtyl (7), 2,4-dichlorophenyl (8), 3-nitrophenyl (9) (Scheme 1). All complexes are moderately soluble in dichloromethane and insoluble in non-polar solvents, such as n-hexane.
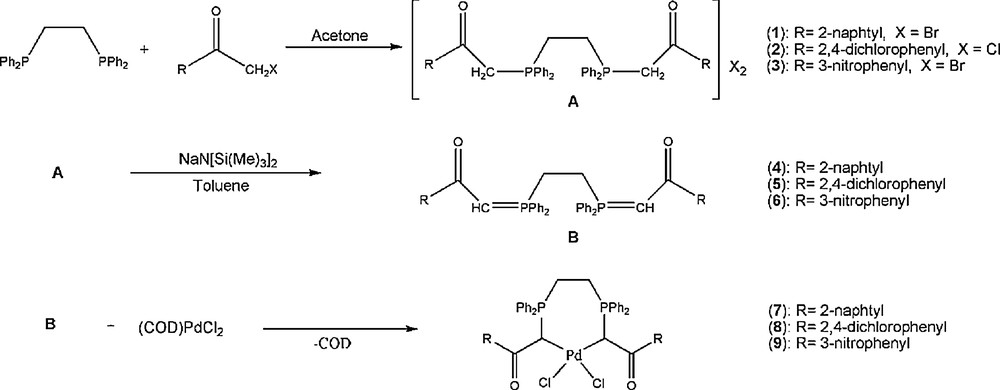
Synthetic route for the preparation of compounds 1–9.
3.2 Spectroscopy
In the IR spectra of diphosphonium salts, a strong stretching absorption due to carbonyl groups has been observed, which confirms the formation of salts 1–3 symmetrically. IR data confirm the complete formation of the phosphorus ylides with the vanishing of the phosphonium CO band between 1658 and 1684 cm−1 for diphosphonium salts 1–3 and the appearance of a new strong CO band relative to carbonyl stabilized ylides 4–6 at 1517 to 1585 cm−1 [17,20]. As we have noted earlier [38], the coordination of the phosphorus ylides through carbon or oxygen causes a significant increase or decrease, respectively, in the carbonyl stretching frequency. Thus, the infrared absorption bands observed for complexes 7–9 indicate that the coordination of the symmetric phosphorus ylides with the palladium center occurs through the ylidic carbon atoms. The two carbonyl groups of all complexes are non-equivalent and discriminable in IR spectra (Table 1).
IR data for compounds 1–9.
| Compound | ν(CO) cm−1 | Reference |
| 1 | 1658 | This work |
| 2 | 1684 | This work |
| 3 | 1682 | This work |
| PhC(O)CHPPh2(CH2)2PPh2CHC(O)Ph | 1509 | [17] |
| 4 | 1517 | This work |
| 5 | 1582 | This work |
| 6 | 1585 | This work |
| [PhC(O)CHPPh2(CH2)2PPh2CHC(O)Ph]PdCl2 | 1632, 1679 | [17] |
| 7 | 1624, 1659 | This work |
| 8 | 1625, 1655 | This work |
| 9 | 1611, 1643 | This work |
The 31P NMR spectra of all the compounds show all the expected resonances for the proposed structures. The 31P NMR spectra of phosphonium salts exhibit a singlet around 23–27 ppm due to PPh2 groups, which indicates that the two phosphorus atoms are equivalent. The 1H NMR spectra show, in addition to the expected aromatic resonances, two peaks centered around 4 and 6 ppm, which are attributable to the P(CH2)2P and PCH2CO protons, respectively. The 31P NMR spectrum of phosphorus ylides 4–6 shows a singlet around 15 ppm, which corresponds to the PPh2 groups. The phosphonium atoms of these compounds show upfield shifts compared to that of the parent phosphonium salt, suggesting some electron density increase in the P–C bonds [17,39]. The 1H NMR spectra show all the expected resonances of symmetric phosphorus ylides 4–6. Chemical shifts appear to be shifted upfield with respect to the parent phosphonium salts, indicating that synthesis of the ylides has occurred. The room temperature 31P NMR spectra of complexes 7–9 exhibit two doublets around 28 and 32 ppm, which are assigned to the P1 and P2 atoms, respectively. This chemical shift is at a lower field than that observed for the corresponding free ylide (around 15 ppm), indicating that the coordination of the ylides has occurred. As reported earlier [17,40], the downfield shift of the ylidic phosphorus signifies the coordination of the ylidic moiety. The coordination of the bis-ylides 4–6 to the palladium center creates two stereogenic centers of the same configuration, as indicated by the presence of two 31P NMR signals (doublets with a coupling constant 3JP–P of 2–7 Hz). In the 1H NMR spectra, the signals due to the methinic protons for all the complexes are broad and centered at 5–6 ppm. The 1H chemical shift values for the complexes appear to be shifted downfield with respect to the parent ylide, indicating also that the coordination of the ylides has occurred [40,41]. Selected NMR chemical shifts for compounds 1–9 are reported in Table 2.
Selected 1H and 31P NMR spectral data for compounds 1–9 [δ (ppm), J (Hz)].
| Compound | δ PCH (2JP–H) | δ PCH2CO (3JP–P) | δ PCH (3JP–P) |
| 1a | – | 27.17 (s) | – |
| 2b | – | 23.97 (s) | – |
| 3a | – | 26.72 (s) | – |
| 4b | 4.36 (br) | – | 15.12 (s) |
| 5b | 4.45 (br) | – | 13.50 (s) |
| 6b | 4.60 (t, 11.21) | – | 15.49 (s) |
| 7b | 5.25 (br) | – | 28.03 (5.00), 30.34 (3.23) |
| 8a | 5.38 (s) | – | 26.10 (4.85), 32.04 (6.16) |
| 9a | 6.15 (br) | – | 29.59 (br), 32.76 (4.24) |
a Record in DMSO-d6.
b Record in CDCl3. br, broad; s, singlet.
3.3 X-ray crystallography
Colorless crystals of 5 were obtained from a chloroform solution by slow evaporation of the solvent. Relevant parameters concerning data collection and refinement for 5 are given in Table 3. Selected bond distances and angles for the two independent but chemically identical molecules found in the unit cell of 5 are displayed in Table 4. The crystal structure (Fig. 1) shows that in this molecule the geometry around the phosphorus atoms is nearly tetrahedral, and that the O atoms are oriented cis with regard to the adjacent P atom. The P(1)–C(1) (1.7218(19)) and C(1)–C(2) (1.384(3)) bond lengths are shorter than the (P+–C (sp3) (1.800)) and (C–C (sp3) (1.511)) normal values [42], respectively. The crystals of this compound contain CHCl3 as the solvent.
Selected bond lengths (Å) and bond angles (°) for 5.
| Compound 5 | |
| C(1)–P(1) | 1.7218(19) |
| C(1)–C(2) | 1.384(3) |
| C(2)–O(1) | 1.267(2) |
| C(9)–P(1) | 1.815(2) |
| C(9)–C(9) | 1.522(4) |
| C(30)–P(2) | 1.814(2) |
| C(22)–C(23) | 1.384(3) |
| C(23)–O(2) | 1.266(2) |
| C(22)–P(2) | 1.7233(19) |
| C(30)–C(30) | 1.525(4) |
| C(9)–P(1)–C(1) | 114.08(9) |
| C(10)–P(1)–C(1) | 113.44(10) |
| C(16)–P(1)–C(1) | 108.55(9) |
| C(9)–P(1)–C(10) | 104.32(9) |
| C(16)–P(1)–C(9) | 110.02(9) |
| C(10)–P(1)–C(16) | 106.09(9) |
| C(22)–P(2)–C(30) | 113.58(9) |
| C(22)–P(2)–C(31) | 114.72(10) |
| C(22)–P(2)–C(37) | 108.22(10) |
| C(30)–P(2)–C(31) | 103.74(9) |
| C(30)–P(2)–C(37) | 110.56(10) |
| C(31)–P(2)–C(37) | 105.70(9) |
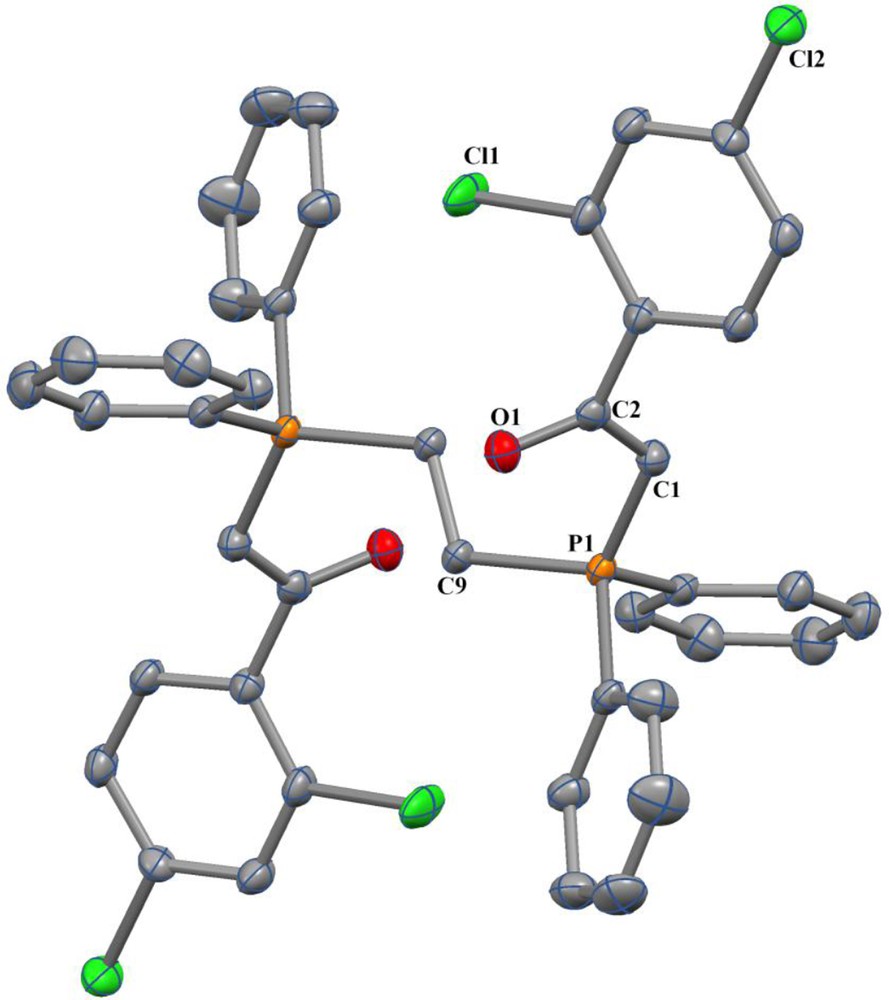
ORTEP view of the X-ray crystal structure of 5. H atoms are omitted for clarity. Color available online.
This bond shortening is due to the ylidic resonance and is halfway between the common values for single (P–C = 1.80) and double (P = C = 1.66) bonds. The CO bond lengths of 5 (1.267(2)) also are larger than the normal value (1.210). This bond distance suggests an electron delocalization in this molecule [17,42]. The P–C(H) (1.7218(19)) and C(1)–C(2) (1.384(3)) bond lengths are close to the corresponding distances P–C(H) (1.705(2)) and C–C (1.404(3) Å) found in similar phosphorus ylides, such as [Ph2PCH2PPh2C(H)C(O)Ph] [43]. The packing structure of 5 has short contacts between the adjacent molecules. In this case, the interaction between the O2 and H44 atoms of the chloroform as the solvent (1.988 Å) have a great importance in the formation of the packing structure (Supplementary data, Fig. 1S). The packing also shows two hydrogen bonds between oxygen and chlorine atoms (Fig. 2).
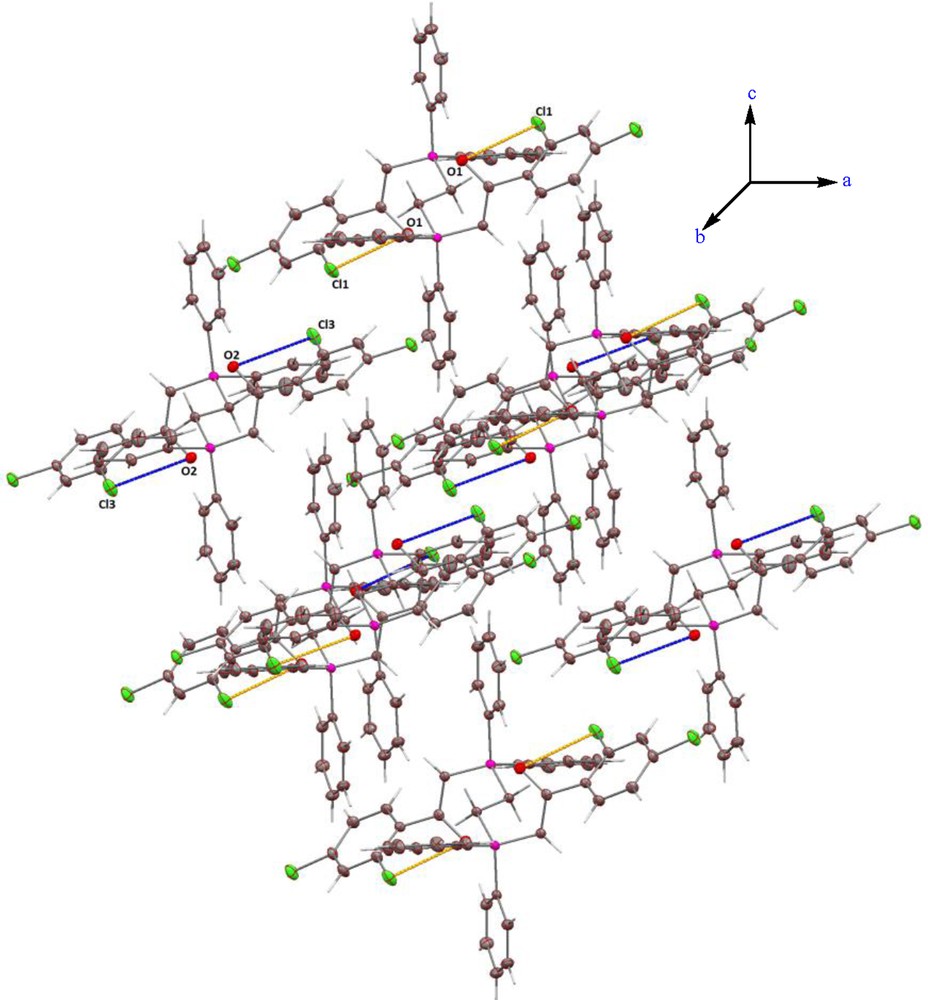
View, along b, of the packing in compound 5. H–bonds: O1···Cl1 (3.178 Å) and O2···Cl3 (3.212 Å). Color available online.
3.4 Suzuki cross-coupling reactions of aryl halides
The palladacycle complex 9 as a catalyst was assessed for its activity in the Suzuki cross-coupling reaction initially by studying the coupling of 4-bromoacetophenone with phenylboronic acid to form 4-phenylacetophenone as the sole product. Various parameters including solvent, base, and catalyst loading were screened to optimize the reaction conditions. The proper combination of base and solvent is extremely important; we have examined several different bases and solvents for the Suzuki reaction (Table 5). Initially, many commonly available bases were used with DMF as the solvent. Cs2CO3, K2CO3, and Na2CO3 as a base, which produced higher yields in short reaction times (Table 5, entries 1–3), respectively. The use of NaOAc, NaF, and NEt3 as bases gave lower yields (Table 5, entries 4–6), respectively. In the reaction, several commonly used solvents were also tested (Table 5). Solvents, such as THF and H2O were less effective (Table 5, entries 11–12) for this reaction. Dioxane, NMP, methanol, and toluene were moderately effective (Table 5, entries 7–10), respectively. After an optimization process, we found that our complex could perform the coupling of 4-bromoacetophenone with phenylboronic acid using a 0.001 mol% catalyst loading with Cs2CO3 at 110 °C in good yield in a 90-minute procedure when using technical-grade DMF as the solvent.
Optimization of base and solvent for the Suzuki cross-coupling reactiona.
| Entry | Base | Solvent | Temperature (°C) | Time (min) | Yield (%)b |
| 1 | Cs2CO3 | DMF | 110 | 90 | 91 |
| 2 | K2CO3 | DMF | 110 | 180 | 87 |
| 3 | Na2CO3 | DMF | 110 | 210 | 82 |
| 4 | NaOAc | DMF | 110 | 480 | 72 |
| 5 | NaF | DMF | 110 | 480 | 57 |
| 6 | Et3N | DMF | 110 | 480 | 42 |
| 7 | Cs2CO3 | Dioxane | 110 | 180 | 73 |
| 8 | Cs2CO3 | NMP | 110 | 210 | 72 |
| 9 | Cs2CO3 | Methanol | 65 | 480 | 68 |
| 10 | Cs2CO3 | Toluene | 110 | 480 | 65 |
| 11 | Cs2CO3 | THF | 60 | 480 | 51 |
| 12 | Cs2CO3 | H2O | 100 | 480 | 25 |
a Reaction conditions: 4-bromoacetophenone (0.75 mmol), phenyl boronic acid (1 mmol), base (1.5 mmol), solvent (2 mL), catalyst (0.001 mol%), in the air.
b Isolated yield.
In order to gauge the further potential of 9 towards the Suzuki coupling methodology, we chose various substituted aryl halides for the reaction with phenylboronic acid, as shown in Table 6. Aryl bromides with various functional groups efficiently reacted with phenyl boronic acid (Table 6, entries 1–11) using Cs2CO3 and 2 mL DMF at 110 °C in the presence of palladium(II) catalyst to give Suzuki products in high yields. In palladium-catalyzed carbon–carbon bond formation reactions, it is commonly believed that better yields are achieved for aryl halides with electron-withdrawing rather than electron-donating substituents [51]. The reaction of electron-deficient aryl bromides with phenylboronic acid afforded efficiently the coupling products in more than 90% yield in short reaction times (Table 6, entries 2–3, 5). The electron-poor, 4-bromobenzaldehyde and 4-bromoacetophenone, also afforded high yields (Table 6, entry 2, 90% and entry 5, 91%). The reaction of p-bromo nitrobenzene with phenylboronic acid showed also high yields (Table 6, entry 3, 90%). In the electron-rich or deactivated p-bromotoluene, the yields are good (Table 6, entry 4, 80%). The electronically neutral bromobenzene (Table 6, entry 1) produced good amounts of the desired product. As expected, very satisfactory yields were obtained with the 2-bromothiophene and the 1-bromonaphthalene of phenylboronic acid (Table 6, entry 6, 84% and entry 7, 86%). Next, we examined the reaction of aryl iodides with phenylboronic acid for the Suzuki reaction. And good yields of the corresponding products were obtained under the optimized conditions (Table 6, entries 8–9). Trying to apply aryl chloride as an efficient substrate was successful (Table 6, entries 10–11). Electron-deficient substrates, such as 4-chloroacetophenone (Table 6, entry 11) coupled with phenylboronic acid gave yields of the coupling product in the order of 82%. The main drawback of the Pd-mediated Suzuki cross-coupling reaction is that only aryl iodides and aryl bromides can be used efficiently. Recent progress with increasing the reaction rate of aryl chlorides permitted to overcome this problem [52–54]. The stronger C–Cl bond is, in fact, responsible for the slower reaction rate of aryl halides because the oxidative addition step was suggested to be the rate-determining step in cross-coupling catalytic cycles [55].
Suzuki cross-coupling reaction of aryl halides with phenylboronic acid catalyzed by Pd complex 9a.
| Entry | Aryl halides | Product | Reference | Time (min) | Yield (%)b |
| 1 | [44,45b] | 120 | 82 | ||
| 2 | [45] | 90 | 90 | ||
| 3 | [44b,46] | 90 | 90 | ||
| 4 | [44b,47] | 180 | 80 | ||
| 5 | [45b,48] | 90 | 91 | ||
| 6 | [47] | 150 | 84 | ||
| 7 | [46] | 150 | 86 | ||
| 8 | [49,50] | 90 | 86 | ||
| 9 | [50] | 90 | 80 | ||
| 10 | [44a,50] | 180 | 77 | ||
| 11 | [32c,49] | 180 | 82 |
a Reaction conditions: aryl halide (0.75 mmol), phenylboronic acid (1 mmol), Cs2CO3 (1.5 mmol), DMF (2 mL), catalyst (0.001 mol%), in the air, 110 °C.
b Isolated yield (yields were calculated against consumption of the aryl halides).
The homogeneous nature of the catalysis was checked by the classical mercury test [56]. Addition of a drop of mercury to the reaction mixture did not affect the conversion of the reaction, which suggests that the catalysis is homogeneous in nature, since heterogeneous catalysts would form an amalgam, thereby poisoning it. The fact that metallic Hg does not kill the catalyst is a good point in favour of the fact that no metallic Pd is being produced.
The catalytic activity of a seven-membered Pd complex (A, 9) was higher than that of the five-membered Pd complex (B) in all the reactions in our previous work [34]. This may be due to the fact that Pd(II) in palladacycles with five-membered rings is difficult to reduce to Pd(0). As it can be seen in Table 7, some of the aryl halides with an electron-withdrawing or an electron-donating substituent have been tested, among which the seven-membered Pd complex (A) has a higher efficiency as a catalyst in a shorter reaction time. Although several catalytic systems have been reported to support the Suzuki C–C coupling reaction, a catalyst of this type is novel for its seven-membered ring containing bisphosphine bis ylide as a ligand coordinated through the CH methinic bond. The high efficiency of this catalyst in low catalyst loading, under aerobic conditions and short reaction times, makes it valuable.
Comparison of the seven-membered (A)a and five-membered (B)b Pd complexes in the Suzuki cross-coupling reaction.
| Entry | Ar–X | Complexes | Time (h) | Yield (%)c |
| 1 | Ph–Cl | A | 3 | 77 |
| B | 12 | 76 | ||
| 2 | Ph–Br | A | 2 | 82 |
| B | 10 | 80 | ||
| 3 | Ph–I | A | 1.5 | 86 |
| B | 6 | 82 | ||
| 4 | p-Me–Ph–Br | A | 2 | 80 |
| B | 4 | 83 | ||
| 5 | p-CH3CO–Ph–Br | A | 1.5 | 91 |
| B | 4 | 87 | ||
| 6 | p-CH3CO–Ph–Cl | A | 3 | 82 |
| B | 6 | 80 |
a Reaction conditions: aryl halide (0.75 mmol), phenylboronic acid (1 mmol), Cs2CO3 (1.5 mmol), DMF (2 mL), catalyst (0.001 mol%), 110 °C.
b Reaction conditions: aryl halide (0.75 mmol), phenylboronic acid (1 mmol), K2CO3 (1.5 mmol), DMF (2 mL), catalyst (0.2 mol%), 130 °C [53].
c Isolated yield.
4 Conclusions
The present study describes the synthesis and characterization of a series of chelate palladium(II) complexes derived from palladium chloride and new symmetric phosphorus ylides. On the basis of the physicochemical and spectroscopic data, we propose that ligands herein exhibit a chelate C–C coordination behavior to the metal center, affording a seven-membered chelate ring. We used palladium(II) complexes as highly active and efficient catalysts for promoting the Suzuki cross-coupling reaction of various aryl halides to produce the corresponding products in high yields. The easiness of preparation of the complex, its high solubility in organic solvents, the low catalyst loading, and the short reaction time needed under aerobic conditions make it an ideal complex for the above transformations.
Acknowledgements
We are grateful to the Bu-Ali Sina University for a grant and Mr Zebarjadian for recording the NMR spectra. Our gratitude also goes to Ms Behranj for her cooperation.