

1 Introduction
One of the most active subjects in the area of molecular magnetism is the preparation and the study of the properties of cyanide-bridged systems. During the last ten years, chemists have succeeded by using metallocyanides as constructing units to prepare 3D, 2D, 1D and 0D systems that can behave as bulk or as single-molecule magnets [1–4]. We have focused on the preparation of mixed valence FeII–FeIII polynuclear complexes that may possess the same electronic structure as Prussian blue. Prussian blue is a cyanide-bridged 3D mixed valence mixed spin compound that behaves as a ferromagnet below TC = 5.5 K due to the ferromagnetic interaction between the high spin FeIII ions through the low spin (S = 0) FeII.4 The origin of the ferromagnetic interaction (instead of the expected antiferromagnetic interaction) has been ascribed to the mixed valence nature of the compound, that is to say the presence of FeII to FeIII charge-transfer excited state [5]. We have reasoned that if the charge-transfer excited state is responsible of such ferromagnetic interaction, such interaction may be tuned if one is able to tune the energy of the charge transfer state [6]. The only possibility to tune the energy of the charge transfer state, keeping the same structure of Prussian blue, is to prepare molecules that mimic Prussian blue where it may be possible to control the ability of the FeIII ions to accept the FeII electron density. The first step consists in preparing mononuclear FeIII complexes with different organic ligands chosen to tune the electrochemical response for the FeIII-to-FeII reduction potential. The second step is the synthesis of polynuclear Prussian blue complexes using the mononuclear ones. We propose that the optical transfer from FeII to FeIII within the polynuclear complexes will depend on the reduction potential of the FeIII ion in given surroundings. In this paper, we describe the preparation, the characterization and the magnetic properties of two heptanuclear complexes obtained from the reaction of K4[FeII(CN)6] and two mononuclear FeIII complexes chelated by a pentadentate ligand.
2 Results and discussion
2.1 Mononuclear FeIII complexes
2.1.1 Preparation and characterization
Two mononuclear FeIII complexes were prepared by reacting FeCl3 with the organic ligands H2L1 (bis–(3salicylideneaminopropyl)–methylamine) and HL2 (N,N-bis-(2-méthylpyridine)-N’-salicylidene-1,2-diaminoéthane) (Fig. 1) in the presence of triethylamine. Elemental analysis corresponds to the formula Fe(L1)Cl for 1 and [Fe(L2)Cl](B(C6H5)4) for 2, which was obtained by adding an excess of NaB(C6H5)4 to the mother solution (see experimental section).

Chemical formula of the pentadentate ligands H2L1 and HL2.
The structure of complex 1 (Fig. 2) shows the presence of the deprotonated pentadentate ligand and of a chloride atom occupying the sixth position [7].
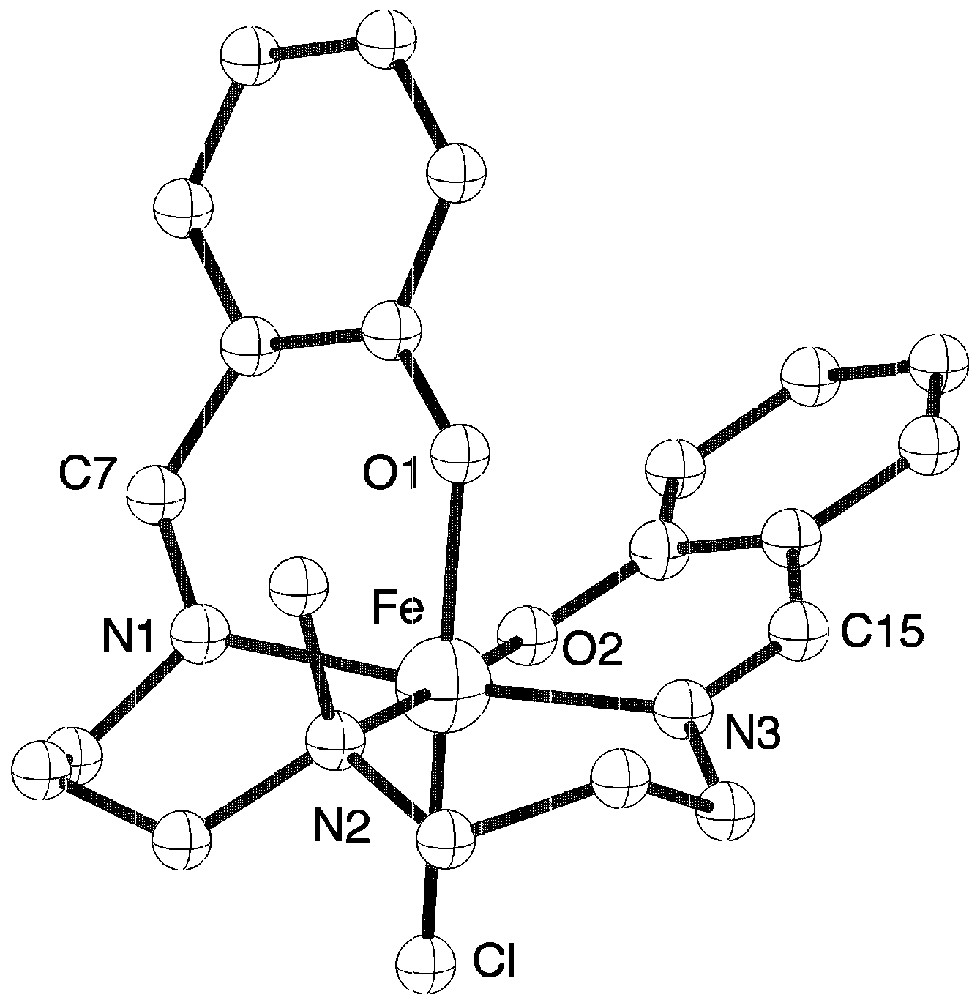
View of the molecular structure of complex 1.
The pentadentate ligand adopts a conformation around iron with one phenolate oxygen atom in trans position to chloride. Because of this particular coordination of the ligand, each molecule is chiral, but within the crystal the two enantiomers are present. The presence of the imine groups are confirmed since the C15–N3 and C7–N1 distances are equal to 1.286 Å against 1.49 Å expected for a single C–N bond. Complex 2 has the expected structure with a chloride atom in the sixth position. Tetraphenylborate anions are present in the structure since the mononuclear complex has a positive charge (Fig. 3).
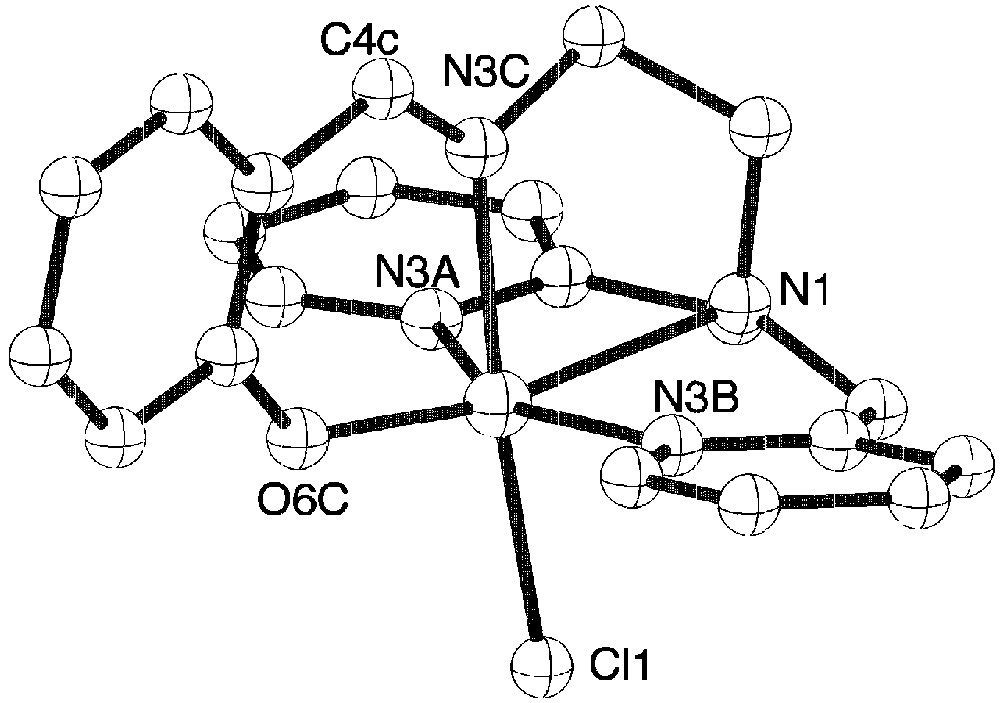
View of the molecular structure of the monocation of complex 2.
The Mössbauer spectrum of complex 1 was measured at T = 300 K. It presents a signal with an isomeric shift δ = 0.347 mm s–1 and a quadruple splitting ΔQ = 1.435 mm s–1.
2.1.2 Electrochemical properties
The electrochemical studies of complexes 1 and 2 were performed in the same conditions (see experimental section). The cyclic voltammogram of 1 in reduction mode shows an irreversible signal with two waves (Fig. 4).

Cyclic voltammograms in reduction for 1 after progressive addition of water (100 μl = 1110 equiv, CH3CN, c = 10–3 mol l–1, (NEt4)ClO4: 0.1 mol l–1, T = 20 °C, 100 mV s–1).
The origin of the two waves may be the presence of an equilibrium between two species: Fe(L1)Cl and Fe(L1)S+, where S is a solvent molecule. The neutral species is expected to be reduced at a lower potential than the charged one. In order to obtain a well reversible signal with only one wave, a progressive addition of water was carried out since it was noticed that the shape of the waves depend on the presence of water traces. The progressive addition water leads quickly (1200 equiv) to one reversible wave that is shifted towards positive potential upon water addition. A stationary state is reached when 3500 equiv of water is added. During the water addition, we have a very fast equilibrium between the two species Fe(L1)S+ and Fe(L1)H2O+, so that only an average wave is observed at the sweeping rate of 100 mV s–1. We may suggest the following scheme for the equilibrium of the different species when water is added to the solution and before reaching saturation (Fig. 5) [8].

Scheme of the equilibrium between the different species of complex 1 during the electrochemical process.
After addition of 4600 equiv of water, a single reversible wave is obtained with E1/2 = –0.145V (Fig. 6). This potential corresponds to the reduction of the Fe(L1)(H2O)+ 1′ species. The potentials of the Fe(L1)S+ species cannot be determined precisely but can be estimated to be around –0.25 and –0.15 V. For complex 2, the results are similar to that of 1. The addition of 3300 equiv of water leads to a signal with a quasi-reversible wave at E1/2 = +0.053V for the species Fe(L1)(H2O)2+ 2′ (Fig. 6).
Cyclic voltammograms for 1′ (—) and 2′ (----) in reduction (CH3CN, c = 10–3 mol l–1, N(Et)4ClO4 0.1 mol l–1, T = 20 °C, 100 mV s–1).
It is important to note here that the addition of water allowed the formation of species where the sixth position is occupied by the same ligand, i.e. water, which present reversible waves at the sweeping rate that we used. It is thus possible to compare the effect of the organic ligand on the reduction potential of FeIII. L2 gives a species that is more easily reducible than L1 [7].
This evolution of the reduction potential of FeIII in these complexes can be rationalized, following qualitatively the ideas developed by A.B.P. Lever [9]. First, since the phenolate group is a strong π-donor ligand, it should stabilize FeIII. We can thus justify that, in 1′ FeIII is more difficult to reduce than in 2′, because the metal is linked to two phenolate groups in the former and to only one in the latter compound. Secondly, since imine and pyridine groups (which we consider as equivalent in this rough approach) are π-acceptor ligands, the presence of two pyridine and one imine groups in 2′ and only two imines in 1′ is in line, as well, for the stabilization of the reduced FeII state in 2′.
2.2 Heptanuclear FeII FeIII6 complexes
2.2.1 Preparation and characterization
The reaction of K4[Fe(CN)6] with the mononuclear complexes 1 and 2 leads to two heptanuclear complexes of formula [FeII(CNFeIII(L1))6]Cl2 3 and [FeII(CNFeIII(L2))6](B(C6H5)4)8 4 respectively (see experimental section). The crystal structure of 3 shows the presence of discrete heptanuclear molecules with comparable distances and angles for the Fe(CNFe)6 unit as for Prussian blue (Fig. 7) [10]. No suitable single crystals for structural determination of complex 4 could be obtained, mainly because of the decomposition of the complex during the crystallization process that leads to the formation of the binuclear μ-oxo complex (L2)FeIIIOFeIII(L2). However, elemental analysis together with infrared and Mössbauer studies are in line with the formation of the expected heptanuclear complex.
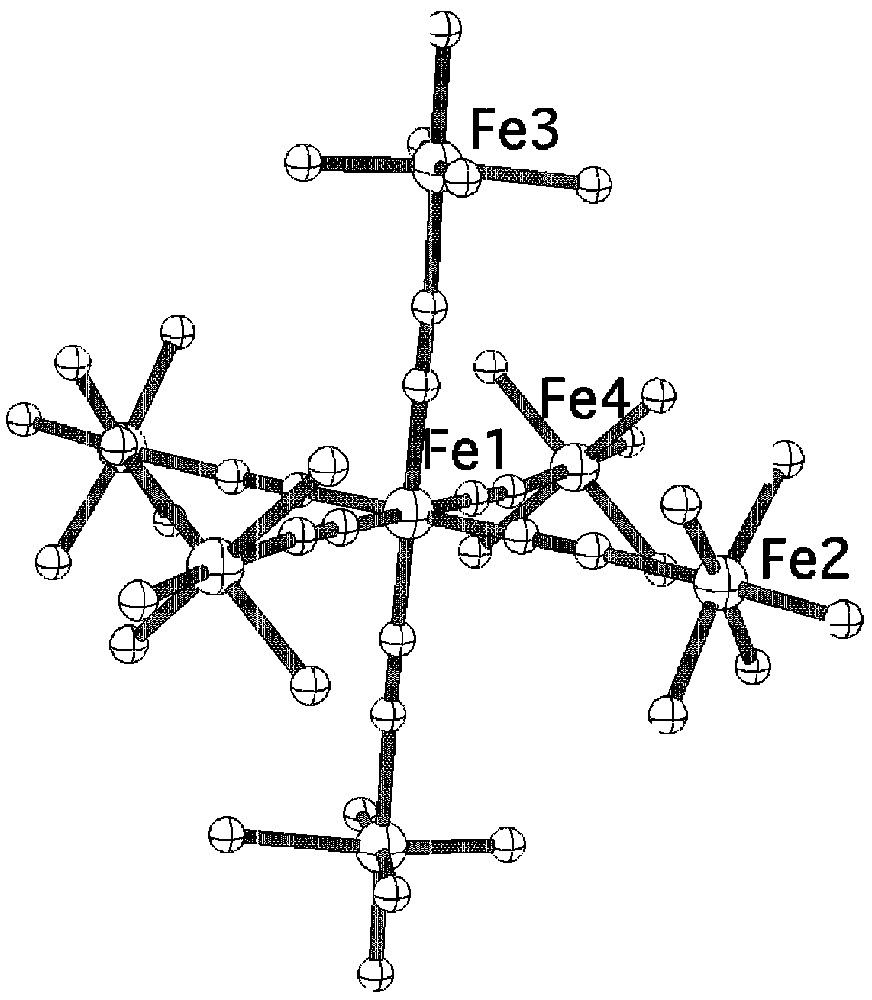
View of the molecular structure of the heptanuclear dication for complex 3.
2.2.2 Electronic properties
The UV–visible spectra of 3 and 4 (Fig. 8a and b) are almost identical to those of the mononuclear complexes. The only difference appears in the range between 10000 and 22000 cm–1, where an additional band seems to occur. Fig. 8c shows the spectra corresponding to the difference between those of the polynuclear and mononuclear complexes. These bands were assigned to the mixed valence nature of the complexes as due to the charge transfer from the central low-spin FeII to the peripheral high-spin FeIII ions (Fig. 8c) [7]. A tetranuclear complex [CoIII(CNFeIII(L1))3] (CoFe3, see experimental section) where FeII was replaced by the isoelectronic low spin CoIII was isolated; it has an identical electronic spectrum as that of complex 1 and thus does not show the presence of an additional band (Fig. 8a). This is an indirect manner to confirm that the additional bands observed in the polynuclear complexes are indeed due to FeII → FeIII charge transfer. Unfortunately, the similar complex with ligand L2 was not isolated.
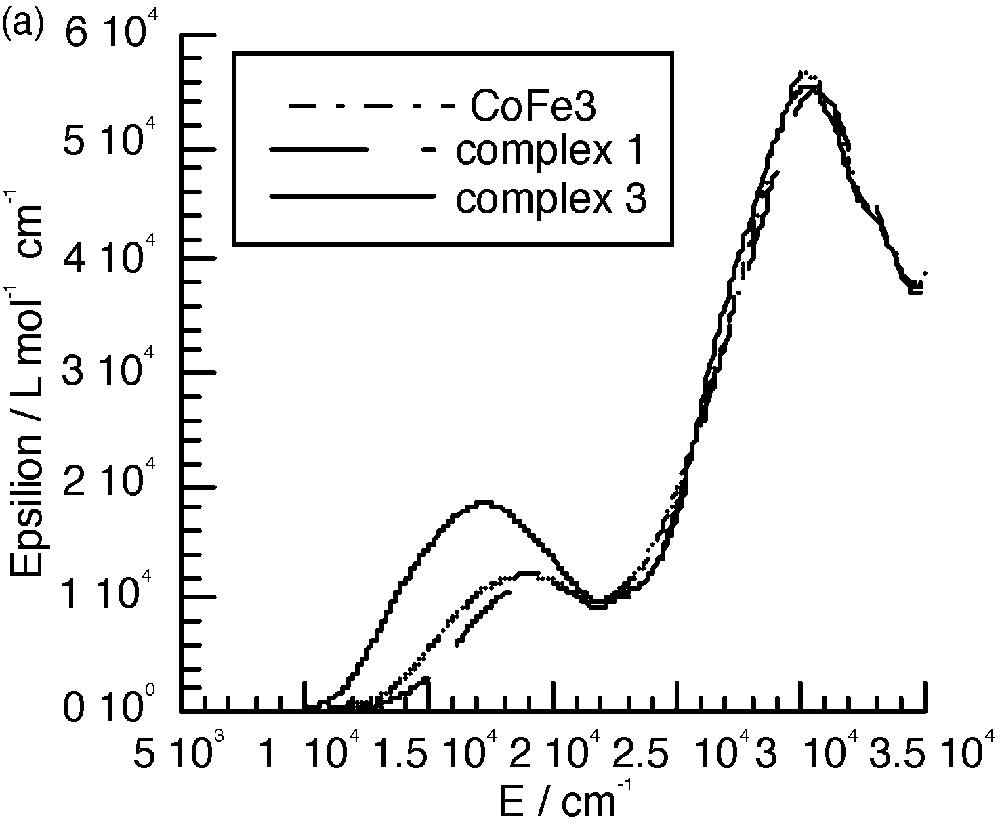
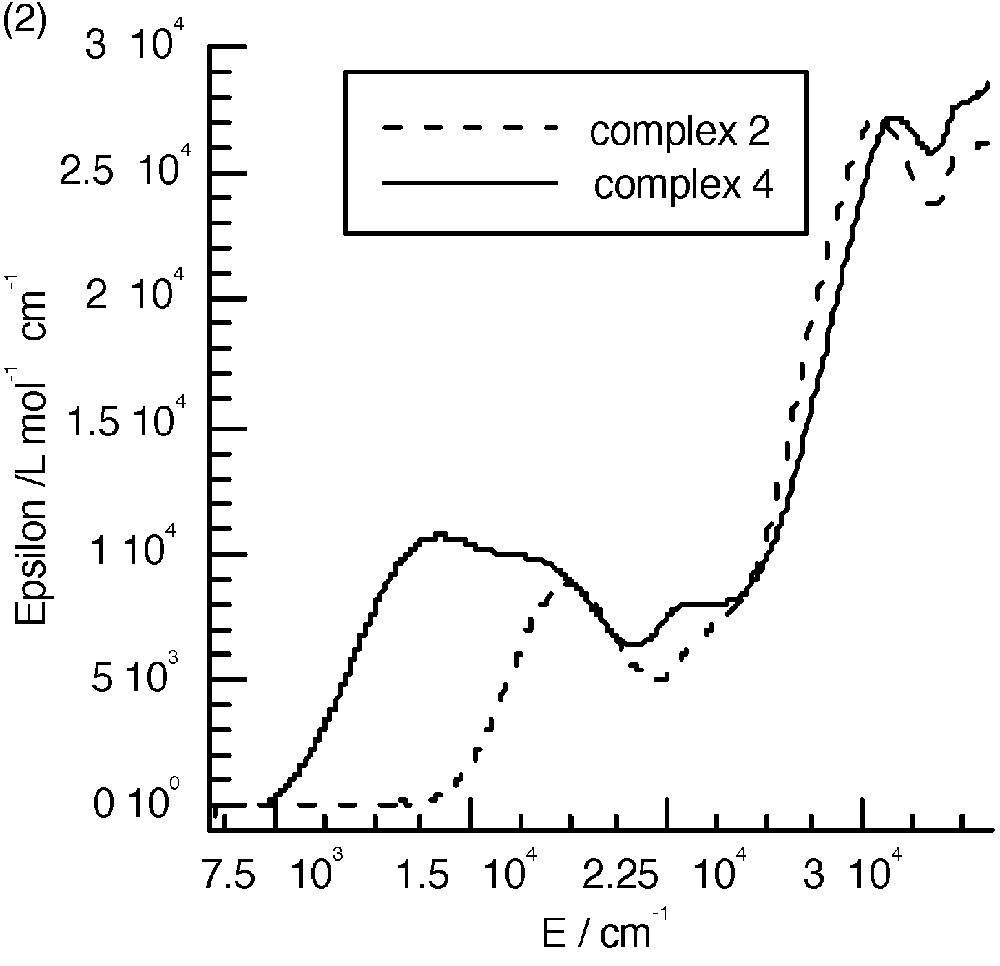
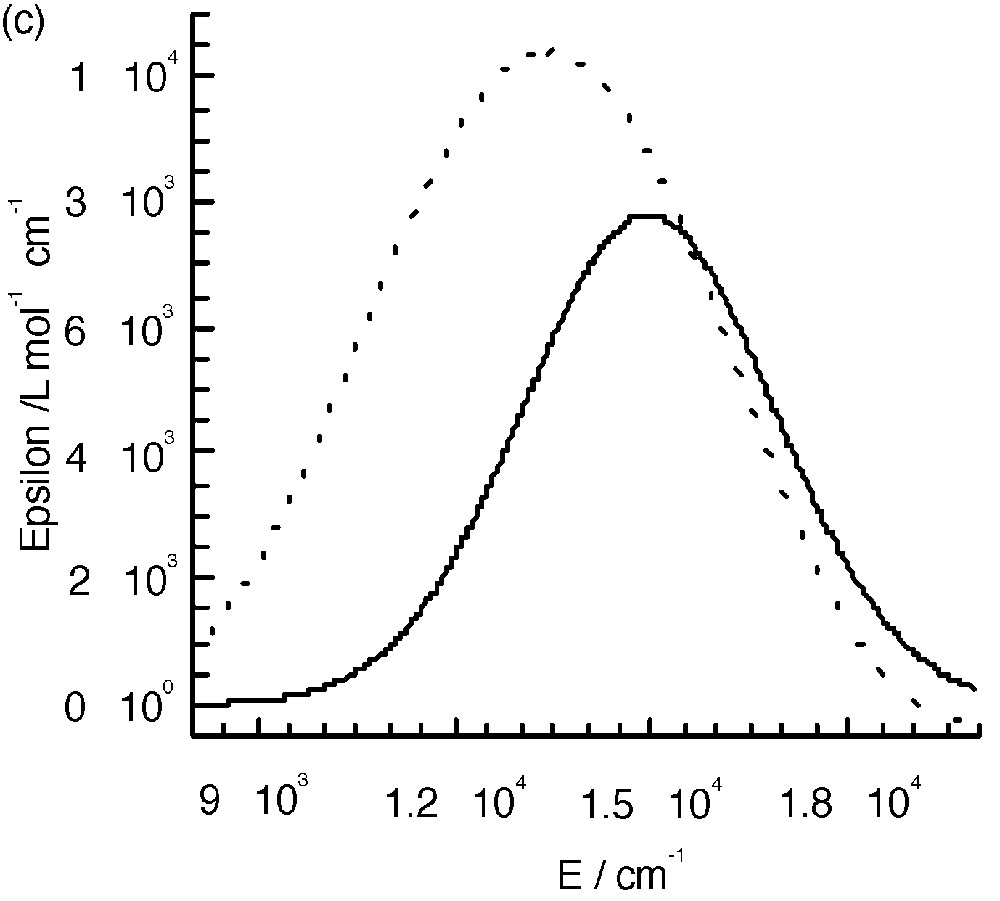
(a) Electronic spectra of complexes 1, 3 and CoFe 3 in CH2Cl2. (b) Electronic spectra of complexes 2 and 4. (c) Intervalence spectra for 3 (—) in CH2Cl2 and 4 (----) in CH3CN.
The energy of the intervalence band is at lower energy when the FeIII chelating ligand is L2. This is due as expected to the more easily reducible ability of this complex that shifts down the excited charge transfer state.
2.2.3 Magnetic properties
The χM T values at room temperature for 3 and 4 correspond to the expected values for isolated S = 5/2 FeIII ions (26.2 cm3 mol–1 K). Upon cooling from room temperature, χM T remains almost constant down to T = 70 K for complexes 3 and 4. For 4, χM T starts to increase slowly below 70 K and then abruptly and reaches a value of 71.8 cm3 mol–1 K at T = 3K. For compound 3, χM T starts to increase only below T = 35 K and reaches a much lower value (32.2 cm3 mol–1 K) than for compound 4 at T = 3 K (Fig. 9) [11].
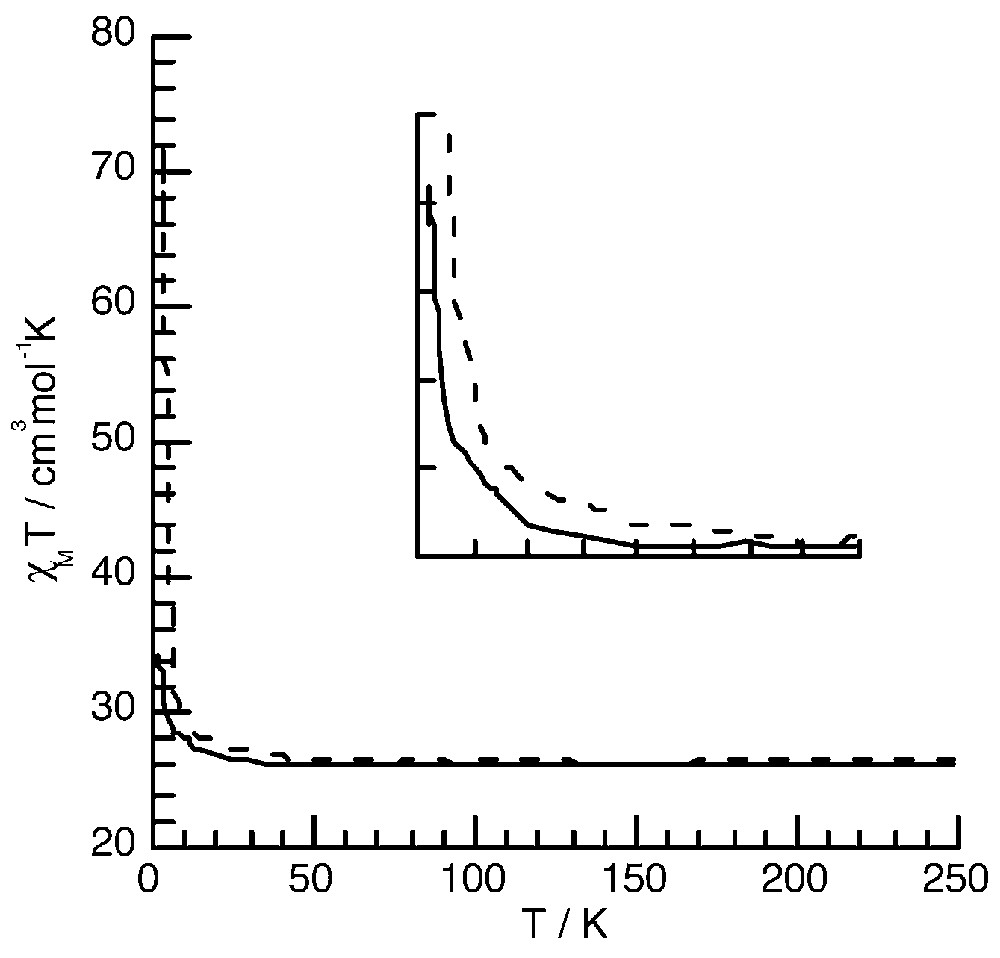
Thermal dependence of the χM T product for 3 (—) and 4 (----). The inset shows the faster increase of χM T for complex 4.
This behaviour is due to the presence of a ferromagnetic exchange coupling interaction between the high-spin S =5/2 FeIII through the diamagnetic FeII ions, as in Prussian blue. The fact that χM T increases more abruptly between 70 and 20 K in 4 (Fig. 9, inset) suggests that the magnitude of the exchange interaction is larger in this latter compound than in 3. The Curie–Weiss constant θ, obtained by fitting the 1/χ = f(T) data (not shown here), was found to be equal to 0.48 and 1.27 K for 3 and 4 respectively, as expected for a larger ferromagnetic interaction, for compound 4. The magnetic studies at low temperature (below 2 K) show the occurrence of an antiferromagnetic order for 3 at T = 0.2 K due to intermolecular interactions [7]. This is probably why the χM T value at T = 2 K does not reach the expected value for S = 15. No low temperature measurements were carried out for complex 4. In order to preclude the occurrence of intermolecular interactions, new bulky peripheral ligands are being developed.
The origin of the ferromagnetic interaction was assigned in these compounds to the presence of the charge-transfer excited state. An indirect proof is given from the study of the magnetic behaviour of the tetranuclear complex CoFe3 mentioned above, where the interaction between the peripheral FeIII ions was not found to be ferromagnetic (Fig. 10). Since no additional band is observed in the electronic spectrum of the CoFe3 tetranuclear complex, the charge-transfer excited state CoIVFeII is at a much higher energy than for the FeIIFeIII6 complexes and thus cannot contribute to stabilize the ferromagnetic state.

Thermal dependence of the χM T product for CoFe3.
3 Conclusion
We have shown that organic ligands that usually play an ‘innocent’ role in polynuclear magnetic complexes may be used to tune their magnetic properties. The Prussian blue complexes may not be the best examples, because of the rather weak magnitude of the exchange coupling interaction. However, the same ideas may be applied for other systems and particularly for complexes that possess magnetic properties sensitive to light.
4 Experimental section
4.1 Synthesis
4.1.1 Complex 1 [FeL1Cl]
7.76 g (22 mmol) of H2L1are dissolved in 30 ml of absolute ethanol. A 50-ml ethanol solution containing 5.42 g (20 mmol) of [Fe(H2O)6]Cl3 is added to the previously prepared one while stirring. Then 7.0 ml (44 mmol) of triethylamine are added dropwise. The mixture is heated at 60° C for 20 minutes. Upon cooling, a microcrystalline powder is obtained. It is filtered, thoroughly washed with ethanol and dried under vacuum. Recrystallisation from an acetonitrile solution gives crystals suitable for structural studies. Elemental analysis: % exp. (calc.) : C : 56.79 (56.96), H : 5.68 (5.65), N: 9.41 (9.49), Cl : 8.01 (8.06), Fe : 12.60 (12.51).
4.1.2 Complex 2 [FeL2Cl](B(C6H5)4)
0.687 g (1.98 mmol) of HL2 are dissolved in 15 ml of methanol. A 20-ml methanolic solution containing 0.536 g (1.98 mmol) of [Fe(H2O)6]Cl3 is added to the previously prepared one while stirring. Then 0.275 ml (1.98 mmol) of triethylamine in 10 ml of methanol is added dropwise. The mixture is stirred for 30 min at room temperature and 2.58 g (7.56 mmol) of NaB(C6H5)4 in 10 ml of methanol are added. The solution is left to cool in an ice bath until a microcrystalline powder separates. The powder is filtered washed with methanol dried under vacuum and recrystallized from an acetonitrile solution. Crystals suitable for structural studies are thus obtained. Elemental analysis: % exp. (calc.): C, 71.35 (71.50); H, 5.52 (5.47); N, 7.29 (7.42); Cl, 4.70 (4.63); B, 1.46 (1.46); Fe, 7.28 (7.41).
4.1.3 Complex 3 [FeII(CNFeIII(L1))6]Cl2·6 H2O
1.33 g (3 mmol) of 1 are dissolved in 50 ml of methanol. A 40 ml 4:1 methanol/water solution containing 0.211 g (0.5 mmol) of K4[Fe(CN)6]·3 H2O is added dropwise while stirring. The microcrystalline powder obtained is filtered, washed with methanol and water and then dried under vacuum. Elemental analysis: % exp. (calc.): C, 55.89 (55.92); H, 5.79 (5.76); N, 11.93 (11.86); Cl, 2.50 (2.50); Fe, 13.65 (13.79). Crystals suitable for structural studies are obtained by slow diffusion of the two solutions in an H tube during two months.
4.1.4 Complex 4 [FeII(CNFeIII(L2))6](B(C6H5)4)8
0.8 g of complex 1 are dissolved in 50 ml of acetonitrile. An aqueous solution containing 0.074 g (0.176 mmol) of K4[Fe(CN)6]·3H2O is added dropwise while stirring. The mixture is stirred for 10 min at room temperature and then 0.240 g (0.704 mmol) of NaB(C6H5)4 dissolved in 10 ml of acetonitrile are added. A deep blue microcrystalline powder is obtained upon cooling the solution. It is filtered, washed with cold acetonitrile and dried under vacuum. Elemental analysis: % exp (calc.) C, 74.77 (75.18); H, 5.45 (5.57); N: 8.33 (8.12), B, 1.90 (1.70); Fe, 8.01 (7.57). No crystals suitable for structural studies were obtained for this compound.
4.1.5 Complex CoFe3[CoIII(CNFeIII(L1))3]6H2O
1.33 g (3 mmol) of complex 1 are dissolved in 50 ml of methanol. A 40 ml of 4:1 methanol/water solution containing 0.166 g (0.5 mmol) of K3[Co(CN)6] is added dropwise. A precipitate is obtained, it is filtered and thoroughly washed with cold methanol and water and dried under vacuum. Elemental analysis: % exp (calc.) C, 53.65 (53.62); H, 5.59 (5.68); N, 13.26 (13.60); Fe, 8.01 (7.57); Co: 3.69 (3.82).
4.2 Electrochemical studies
A three-electrode cell was used for the electrochemical studies; working electrode: a 3 mm diameter disc of vitreous carbon which is polished before each scan; counter electrode: a gold wire; reference electrode: Saturated Calomel Electrode (E0SCE – E0NHE = 0.246 V). All experiments were carried out under argon atmosphere at 20 °C, freshly distilled acetonitrile as solvent and (NEt4)ClO4 (0.1 mol l–1) as supporting electrolyte were used. Sweeping rate: 100 mV s–1, c = 10–3 mol l–1.
4.3 Magnetic studies
The magnetic studies were performed on powder samples between 2 and 300 K within an applied field of 1000 Oe using a Quantum Design SQUID magnetometer. For all samples, no saturation effect was detected at the working field and in the temperature range of the experiments.
Acknowledgements
The authors thank CNRS (France), ‘Université Paris-Sud’ (France), the European community (Contract HPRN–CT–1999–00012/TMR) and the European Science Foundation (Molecular Magnets Programme) for financial support.