

1 Introduction and background
Switchable molecular systems featuring electronic, magnetic, or optical bistability are attracting a strong research interest because of their potential use as molecular memories, switches, actuators, or sensors [1–3]. Although their integration into actual devices is challenging, various encouraging results show promise in emerging fields such as molecular electronics [4–7]. The cyanide coordination chemistry has proven successful in providing access to a variety of responsive systems, whose optical and magnetic properties can be reversibly switched [8–10]. One of the emblematic examples is the FeCo Prussian blue analogue, which shows an increase in magnetization under light irradiation [11]. In these materials, the so-called photomagnetic effect was ascribed to a metal–metal electron transfer coupled with a spin-state change induced by light irradiation [12,13]. The system can then relax from the photoinduced metastable state to the ground state upon heating. Since few years various groups have been investigating polymetallic complexes showing photomagnetism [14,15]. To reduce the dimensionality of the FeCo Prussian blue analogues (PBA), a common strategy is based on the use of [FeIII(L)(CN)x]− metalloligand toward partially blocked Co precursors [16]. More specifically, many of these systems were obtained with fac-[FeIII(Tp)(CN)3]− building blocks, where Tp is a derivative of the hydrotris(pyrazol-1-yl)borate scorpionate ligand. These low-dimensional systems can be used as model to better understand the photomagnetic properties [17,18] or they can also be integrated using soft chemistry approaches into molecule-based devices [19]. In the past years we have been interested in the study of {Fe2Co2} squares obtained by self-assembling the [FeIII(Tp)(CN)3]− with partially blocked [Co(bik)2(S)2] complexes (S = solvent, bik = bis(1-methylimidazol-2-yl)ketone) [16,20,21]. In addition, we also explored the use of [FeIII(Tp)(CN)3]− complexes as ligand toward other metal ions. The use of partially blocked FeII complexes is of particular interest as the resulting material exhibits FeIIN6 coordination sites, which provide a suitable surrounding for observing spin-crossover (SCO) phenomenon. As for the FeCo switchable materials, the spin-state change from the FeII low spin (LS) to the FeII high spin (HS) can be thermally induced and photoinduced at low temperature. In that case the photomagnetic properties arise from the so-called “light-induced excited spin-state trapping” (LIESST) effect. We first reported in 2012 the tetrametallic mixed–valence complex, {[FeIII(Tp)(CN)3]2[FeII(bik)2]2}·[FeIII(Tp)(CN)3]2·18H2O·4CH3OH (1), which exhibits a square core structure {FeIII2FeII2}, whereas two [FeIII(Tp)CN)3]− act as counterions [22]. The spin-state changes occurring in this complex are very dependent on the solvation state of the material. The solvated phase shows a transition temperature located slightly above room temperature, T1/2 = 330 K, whereas in the desolvated phase the transition is shifted toward low temperatures, with T1/2 = 240 K. Moreover, the fresh solvated compound is not photomagnetic whereas the desolvated one shows an efficient LIESST effect at 750 nm, with Trelax approx. 45 K (the relaxation temperature was measured by heating the sample at 0.3 K/min).
In the past years, we have decided to extend this work to study the influence of the N– donor on the switchable properties of the [FeII(bik)2(N–)2] SCO subunit, where N– stands for N-donor organic/inorganic ligand or metalloligands [23–25]. We observed that the spin transition temperature changed with the nature of the N-donor (metallo)ligand. Different N–donors can lead to different ligand fields on the FeII SCO units and thus a change in the LS/HS energy gap. However, it is difficult to draw correlations in solid-state studies as other factors, such as the intermolecular interactions [26,27] or the interligand interactions, also affect the T1/2 [28,29]. For those cases where the compared SCO complexes exhibit a similar structure such as in the case of the neutral [FeII(bik)2(NCX)2] SCO complexes (X= S, Se) the rationalization of the properties is easier. Our experimental and theoretical study showed that the NCSe− ligands induce a stronger ligand field than the NCS− one, and thus a higher SCO transition temperature, T1/2 [24]. Comparisons between the transition temperatures of similar complexes are also easier to rationalize in solution studies, as the influence of the intermolecular interaction vanishes. For example, we have shown that it was possible to rationalize the transition temperature in the family [Fe(Rbik)3]2+ and [Fe(bim)3]2+ complexes (R are methyl, ethyl, or vinyl groups grafted on the imidazolyl donors of the bik ligand; bim is similar to bik but the ketone is replaced by a methane group). Our experimental multitechnique approach led to results that are coherent with the theoretical prediction. In addition, we showed in this study that among the different techniques used to follow the SCO equilibrium (UV–vis, Evans NMR, magnetic measurements in solution), the monitoring of an adequately chosen 1H NMR signal could be a very straightforward and accurate approach.
More interestingly, we have systematically studied all of these [Fe(bik)2(N–)2] systems by comparing the photomagnetic effect at 20 K by using different wavelengths in the visible and near-infrared regions (from 405 to 1313 nm). When replacing the [FeIII(Tp)(CN)3]− metalloligand with [MoV(CN)8]3−, we obtained another square complex of formula {[Mo(CN)8]2 [Fe(bik)2]2}(HMeIm)2·5H2O·CH3CN, which exhibits similar [FeII(bik)2(N)2] chomophores [23]. Here the thermally induced spin transition is very gradual. It occurs near room temperature and is accompanied by a solvent loss that makes the spin-state change irreversible. The dehydrated compound is still photomagnetic with a close relaxation temperature (Trelax ca. 48 K). However, the most efficient irradiation wavelength is located at 405 nm, which strikingly contrasts with the situation observed in the previously reported {FeIII2FeII2} square, whose photomagnetic effect is higher near 750 nm. These results are to be compared with the model SCO compound [FeII(bik)3](BF4)2, which shows an SCO centered at T1/2 = 316 K and a photomagnetic effect, which is more efficient (but incomplete) at 635 nm. Finally, in the [FeII(bik)2(NCX)2] complexes, the photomagnetic effect is still observed although it is incomplete. The most efficient wavelength to promote an LIESST effect is 900 nm (whereas the induced magnetization is lower at 808 and 635 nm). However these systems relax at 20 K (the temperature set for all our photomagnetic measurements) when switching off the light, indicating thus the existence of an efficient relaxation pathway. It is worth noticing that no significant differences are observed in the visible absorption spectra of the all these SCO compounds. They all exhibit a broad absorption centered at ca. 630 nm and ascribe to a metal–ligand charge transfer. No metal–metal charge transfer (MMCT) are observed for the polymetallic square complexes. Continuing these experimental investigations, we looked here at the properties of three novel {Fe2Fe2}2+ square complexes {[FeIII(Tp*)(CN)3]2[FeII(bik)2]}2(ClO4)2·2H2O (2), {[FeIII(Tp)(CN)3]2[FeII(bik)2]2}(ClO4)2·2CH3CN (3), and {[FeIII(Tp)(CN)3]2[FeII(bik)2]}2(BF4)2·2CH3OH (4) (Tp = hydrotris (pyrazol-1-yl)borate, Tp* = hydrotris (3,5-dimethyl-pyrazol-1-yl)borate, bik = bis(1-methylimidazol-2-yl)ketone). On the one hand, two different [FeIII(L)(CN)3]− metalloligands were used, and on the other hand, the counterions are different so as the intermolecular interactions. Our purpose was to check to which extent the photomagnetic properties can be influenced by the metalloligand, the solid state structure, and intermolecular interactions.
2 Results and discussion
2.1 Syntheses and general characterization
The PPh4[FeIII(Tp)(CN)3]·H2O complex was prepared in three steps by following our previously reported procedure [30]. First, we isolated a [FeII(Tp)2] complex by direct reaction of the scorpionate ligand, KTp, and a FeII salt. In a second step, the reaction of three equivalents of KCN with the recrystallized dried [FeII(Tp)2] allows the formation of the tricyanido complex [FeII(Tp)(CN)3]2−. This one is then carefully oxidized by hydrogen peroxide and recrystallized from acetonitrile solution after a salt metathesis reaction. The ammonium salt of the [FeIII(Tp*)(CN)3]− building block can be obtained similarly in three steps, as reported by Holmes et al. [31], however, the result is more uncertain. The instability of the first intermediate product, [FeII(Tp*)2], which needs to be immediately used, the potential hazards related to the partial solvent removal from a hydrogen peroxide–contaminated acetonitrile solution, and its layering by diethyl ether are major drawbacks. We thus developed a new one-step synthesis whose yield is lower (ca. 32%) but allows a straightforward access to single crystals of PPh4[FeIII(Tp*)(CN)3]·CH3CN (crystallographic data and a view of the crystal structure, Fig. S1, are given in the Supplementary material). In this synthesis, the degassed solution containing equivalent amount of FeII salt and KTp* is to be added to the cyanide solution shortly after its preparation as [FeII(Tp*)2] tends to precipitate over time. Then the oxidation is carried out by opening the flask to the air. As shown in the Supplementary material (Fig. S2), the redox potential of the obtained PPh4[FeIII(Tp*)(CN)3]·CH3CN complex (−1.00 V to be compared with −0.82 V for [FeIII(Tp)(CN)3]−.versus [FeIII/II(Cp)2], 10−3 M in CH3CN) allows a clean oxidation by air's oxygen in acetonitrile, avoiding the critical oxidation step by hydrogen peroxide.
The square complexes were obtained by self-assembling the [FeIII(Tp)(CN)3]− or [FeIII(Tp*)(CN)3]− building blocks with the in situ formed [FeII(bik)2(S)2]2+ complexes in alcoholic or acetonitrile solutions. Although the use of FeCl2 salt leads to the crystallization of the previously reported {FeIII2FeII2}2+ square complex with the [FeIII(Tp)(CN)3]− complex acting as counterion [22], the presence of fluoroborate or perchlorate anions in the solution (coming from the use of FeII(BF4)2 or FeII(ClO4)2 salts) leads to the fast crystallization of the dicationic {FeIII2FeII2}X2 square complexes (X = ClO4, BF4) in a relatively good yield (2, 40%; 3, 60%; and 4, 60%).
The compounds were all characterized by FT-IR spectroscopy. This technique is very useful to characterize the electronic states of cyanido-bridged complexes as the stretching cyanide vibration are sensitive to the oxidation state of the coordinated metal ions and to the bridging mode of the cyanide (bridging and nonbridging). Typically, nonbridging FeIII–CN moieties are observed in the range 2130–2110 cm−1, as observed for the precursors: 2123 cm−1 for PPh4 [FeIII(Tp*)(CN)3]·CH3CN and 2121 cm−1 for PPh4 [FeIII(Tp)(CN)3]·H2O. In contrast, stretching vibrations due to bridging cyanide moieties are expected at higher wavenumber (ca. > 2145 cm−1). Here the observed cyanide stretching vibrations of 2–4 are reported in Table 1 together with other characteristic FT-IR peaks. They are very similar for all compounds and reveal the presence of one nonbridging FeIII–CN unit and two different FeIII–CN–FeII bridges in agreement with the crystal structure of the compounds (see subsequently). The elemental analyses carried out on microcrystalline samples agree with the above formula for 4, whereas a higher amount of water molecules (5H2O) is found in 2 in comparison to the crystal structure (2H2O), and a lower amount of CH3CN is found for 3, in comparison to the crystal structure. This can be associated with the presence of disordered water molecules in the crystal lattice of 2 and a partial loss of solvent molecule in 3.
Analytical and selected infrared data of 2, 3 and 4.
| Compounds | Selected FT-IR vibration (cm−1) | Analyses (%)a | ||||||
| νCN | Counterions | νBH | νCO | C | H | N | ||
| Bridging | Terminal | |||||||
| 2 | 2160 2147 | 2130 | 1093, 1064, 1054 (ClO4−) | 2538 | 1634 | 42.88 (42.73) | 4.43 (4.68) | 23.49 (23.53) |
| 3 | 2160 2148 | 2113 | 1076, 1064, 1045 (ClO4−) | 2503 | 1632 | 41.45 (41.23) | 3.60 (3.52) | 27.39 (27.14) |
| 4 | 2161 2148 | 2113 | 1109, 1094, 1045, 995 (BF4−) | 2510 | 1633 | 39.47 (41.27) | 3.80 (3.80) | 26.78 (26.40) |
a Calculated data are given in parentheses for the formula {[FeIII(Tp*)(CN)3]2[FeII(bik)2]}2(ClO4)2·5H2O (2), {[FeIII(Tp)(CN)3]2[FeII(bik)2]2}(ClO4)2·1CH3CN (3), and {[FeIII(Tp)(CN)3]2[FeII(bik)2]}2(BF4)2·2CH3OH (4).
2.2 Description of the structures
Red prismatic crystals of 1 and orange-red square-like crystals of 3–4 suitable for single-crystal X-ray diffraction experiment were obtained by slow evaporation of the mother solutions. The crystal data were all collected at low temperature (200 or 140 K). Compounds 2 and 3–4 crystallize in the P-1 and P21/c space groups, respectively (see Table 2). The structures of 2–4 consist of centrosymmetric dicationic cyanide-bridged tetrametallic units {FeIII2FeII2}, perchlorate (2–3), or tetrafluoroborate (4) counterions and lattice solvent molecules. The mixed valence tetrametallic units are made of [FeIII(L)(CN)3]− complex units (L = Tp* for 2 and L = Tp for 1, 3 and 4) acting as metalloligand (through cis-coordinated cyanide) toward divalent FeII ions (see Fig. 1). The coordination sphere of the FeII ions is completed by two bik α-ketone-β-diimine ligands. The tetrametallic core of 2–4 exhibits a [2 + 2]-type diamond-like geometry whose main angles are reported in Table 3. The edge distances in 2 are slightly longer than those found in 3–4, in agreement with the slightly longer FeII-ligand bond distances. This is likely because of the spin-state of the FeII ions, which is expected to be purely LS at 200 K in 3–4 although it may contain some HS components in 2, as shown by the magnetic measurements (see subsequently). In all complexes, only moderate deviations from linearity are observed on both sides of the cyanide bridges as shown in Table 3.
Crystal data and structure refinement parameters for 2–4.
| Compounds | 2 | 3 | 4 |
| Formula | C72H88B2Fe4N34O14Cl2 | C64H66B2Fe4N36O12Cl2 | C62H68B4Fe4N34O6F8 |
| Fw (g/mol) | 1969.62 | 1847.34 | 1804.04 |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| a (Å) | 13.430 (3) | 15.2373 (4) | 14.9451 (5) |
| b (Å) | 13.470 (3) | 18.5182 (4) | 18.3777 (6) |
| c (Å) | 13.907 (3) | 15.1215 (4) | 15.5415 (5) |
| α (°) | 102.34 (3) | 90 | 90 |
| β (°) | 108.67 (3) | 110.4755 (3) | 101.927 (2) |
| γ (°) | 107.01 (3) | 90 | 90 |
| V (Å3) | 2143.9 (12) | 3997.24 (17) | 4176.4 (2) |
| Z | 1 | 2 | 2 |
| Space group | P | P21/c | P21/c |
| μ (cm−1) | 0.808 | 0.861 | 0.771 |
| ρ (g cm3) | 1.525 | 1.535 | 1.485 |
| Merging R | 0.241 | 0.0925 | 0.038 |
| Ra | 0.0877 | 0.0507 | 0.0601 |
| Rwb | 0.1582 (0.1819)c | 0.1196 (0.1324)c | 0.1789 (0.2049)c |
| GOF | 0.997 | 0.963 | 0.988 |
| Δρmin (e Å−3) | −2.82 | −0.574 | −1.53 |
| Δρmax (e Å−3) | 1.42 | 0.657 | 1.88 |
| T (K) | 200 | 140 | 200 |
a R = Σ||Fo| − |Fc||/Σ|Fo|.
b [Σw (||Fo| − |Fc||)2/ΣwFo2]1/2.
c For I > 2σ (for all data in parentheses).
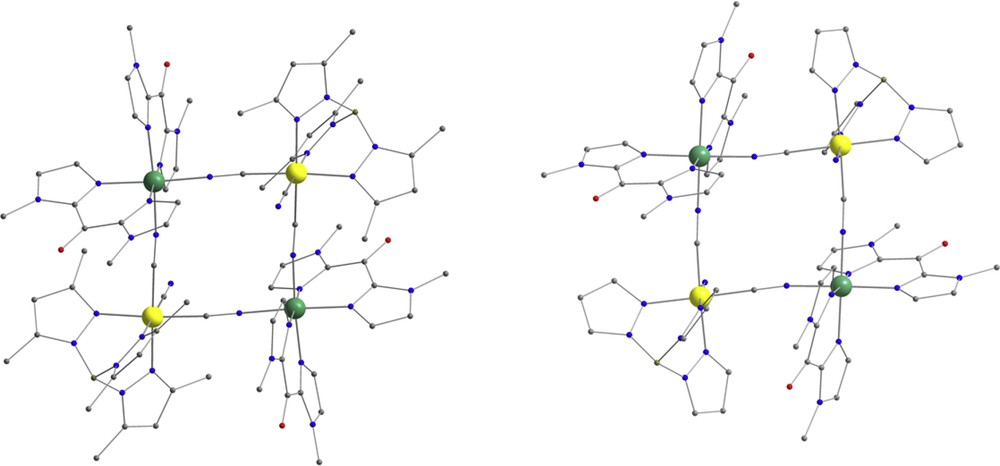
Perspective views of the {FeIII2FeII2} square complexes 2 (left) and 3 (right). C, gray; N, blue; O, red; B, pink-gray; FeIII, yellow; FeII, green. Hydrogen atoms are omitted for clarity.
Selected M–M distances,a bond lengths (Å), and angles (degrees) for 2–4.
| Compounds | 2 | 3 | 4 |
| FeIII–FeII–FeIII | 96.0° | 94.5° | 91.9° |
| FeII–FeIII–FeII | 84.0° | 85.4° | 88.1° |
| FeIII–CN–FeII | 5.038–5.038 | 4.954–4.964 | 4.947–4.971 |
| FeII–N(CN) | 1.961(6)–1.976(7) | 1.918(3)–1.924(3) | 1.918(2)–1.928(2) |
| FeIINbik | 1.973(7)–1.996(6) | 1.966(3)–1.986(3) | 1.964(2)–1.982(2) |
| FeIII–C(CN) (average) | 1.910(9)–1.930(8) (1.920) | 1.901(4)–1.930(5) (1.911) | 1.897(3)–1.934(3) (1.912) |
| FeIII–N(Tp/Tp*) (average) | 1.985(6)–2.024(6) (2.007) | 1.963(3)–1.986(3) (1.977) | 1.972(2)–1.978(2) (1.975) |
| FeIINC | 174.3–179.1° | 171.4–175.9° | 173.3–171.7° |
| FeIII–CN | 174.4(6)–177.6(7) | 174.2–178.2° | 177.4–178.4° |
| ΣFeII | 23.2° | 20.8° | 25.0° |
| ΣFeIII | 26.8° | 22.4° | 29.9° |
| Fe⋯Fe (shortest)b | 9.46 | 7.617 | 7.540 |
a All distances are given with an error of 6–9 on the last digit.
b Shortest intermolecular Fe–Fe distance.
Both FeII and FeIII ions exhibit slightly distorted octahedral coordination sphere. The FeII ions are in a N6 coordination sphere formed by four N atoms of the bik ligands and two N atoms of the cyanide located at cis position. The distortion of the coordination sphere can be evaluated by the octahedral distortion, Σ, defined as the sum of the deviation to 90° of the 12 angles around the metal atom. The octahedral distortion, Σ, measured at 200 K amount to 23.2°, 20.8°, and 25.0° for 2–4, respectively. These moderate values agree with the essentially LS states of the complexes 2–4 at 200 K. This is confirmed by the averaged FeII–N bond lengths (1.981, 1.952, 1953 Å for 1, 2, and 3), which compare well with those found in similar FeII LS complexes [22,32,33]
The FeIII coordination environment is formed by three imine moieties of the pyrazolyl groups and by the carbon atoms of three cyanides. The Fe–CCN bond lengths (average of 1.920, 1.911, 1.912 Å for 2–4) are in agreement with those previously reported for similar FeIII–LS complexes [31,34]. The FeIII–N(Tp/Tp*) bond lengths are longer with mean values of 2.007, 1.977, and 1.975 Å for 2–4. The octahedral distortion, Σ, increases upon coordination of the N-donor atom. They amount to 26.8°, 22.4°, and 29.9° in 2–4, respectively, to be compared with 17.9° and 18.5° in the PPh4 [FeIII(Tp*)(CN)3]·CH3CN and PPh4 [FeIII(Tp)(CN)3]·H2O precursors [31,34].
Finally, the square units are well separated from each other by the counterions and the solvent lattice molecules. The shortest metal–metal distances are over 7.5 Å in 3–4 (see Table 3). In the case of 2, a water molecule establishes a hydrogen bridge between the nonbridging cyanide and an oxygen atom of the perchlorate counterion, a feature commonly observed in these materials.
2.3 Magnetic properties
The magnetic properties of 2–4 are shown in Fig. 2 as the χMT products versus temperature (χM is the molar magnetic susceptibility per {Fe2IIFeIII2} formula unit). The χMT product of compound 2 shows a sigmoidal curve whose shape is typical of SCO complexes. The χMT variation between low and high temperatures, ca. 7.1 cm3 mol−1 K, suggests an almost complete LS ⇔ HS conversion for the two FeII ions, the expected value being 2 × 3.6 = 7.2 cm3 mol−1 K, with S = 2 and g ≈ 2.2. The transition temperature, T1/2 = 227 K, is lower than those measured in our previous studies on SCO molecule based on the {Fe(bik)2(N–)2} unit. The χMT value measured at low temperature, ca. 1.4 cm3 mol−1 K at 20 K, is ascribed to the presence of two FeIII LS ions (χMT ≈ 0.4 cm3 mol−1 K at low temperature for each FeIII ion) and possibly to a small amount of residual FeII HS. Once heated at 400 K in the magnetometer, the spin transition is only slightly shifted toward lower temperature with T1/2 ≈ 223 K (Fig. S3). This shift is likely because of the loss of crystallization solvent. To check this hypothesis, an ex situ desolvated sample of 2 was studied. The sample was prepared by heating the crystals in a TGA under a N2 gas flow up to 80 °C. The resulting χMT curve matches well with that obtained for the sample desolvated inside the magnetometer (Fig. S3).
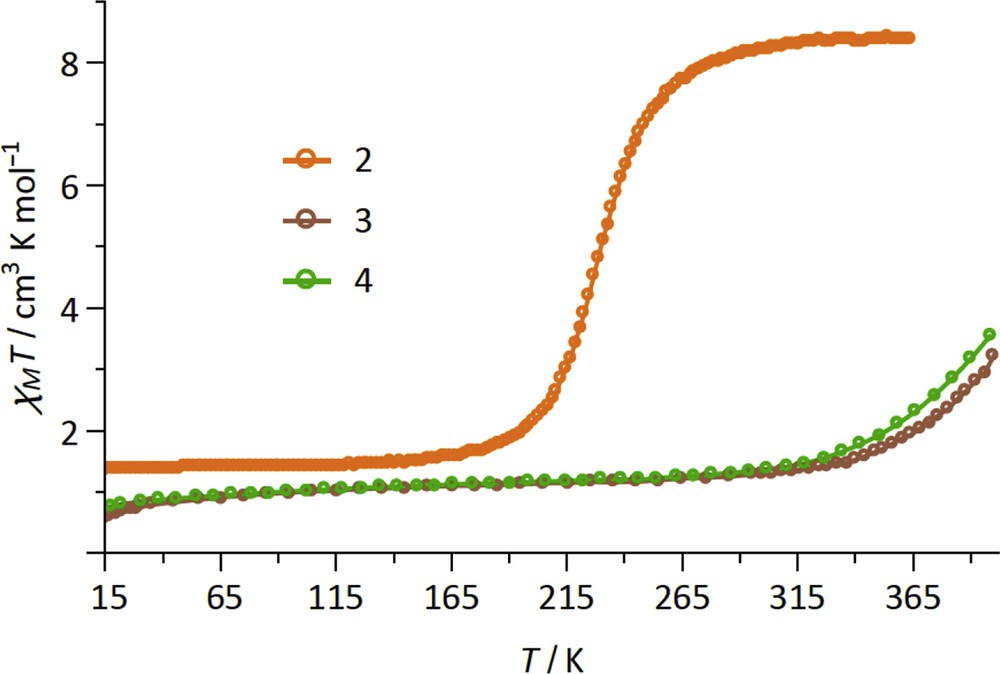
Magnetic properties of 2–4 depicted as the χMT versus T curve.
In contrast with 2, the compounds 3 and 4 do not show a full sigmoidal curve in the 10–400 K temperature range, but only an increase in the χMT products above room temperature, which suggests the occurrence of gradual spin-state conversion at high temperatures. The χMT values measured at 400 K, ca. 3.2 and 3.7 cm3 mol−1 K for 3 and 4, account for roughly 25% and 32% of HS FeII and indicate transition temperatures that are above 400 K. The χMT products measured at low temperature for 3 and 4, χMT≈ 1.10 cm3 mol−1 K at 20 K, are also coherent with the occurrence of two LS FeII and two LS FeIII ions. Here the non-Curie behavior observed between 20 and 350 K (a slight slope is observed on the χMT vs T curves) is due to the spin–orbit coupling occurring in the 3T1g ground state term of the LS [FeIII(Tp)(CN)3]− subunit [35]. It is also worth noticing that the spin transition in the compounds 3 and 4 are reversible. Upon cooling from 400 K, the χMT curves superimpose on that of the fresh compounds (see Fig. S4).
The magnetic curve of 2, which shows a complete and gradual SCO, can be simulated using the Slichter–Drickamer mean-field model (Eq 1) to extract the thermodynamic parameters associated with the spin-state equilibrium and to evaluate the cooperativity [36].
| (1) |
Finally, it is interesting to compare the magnetic behavior of these three compounds with that of the previously published square complex 1, {[FeIII(Tp)(CN)3]2 [FeII(bik)2]2}·[FeIII(Tp)(CN)3]2·18H2O·4CH3OH. Although the {[FeIII(Tp)(CN)3]2[FeII(bik)2]2}X2 square complexes (X = [Fe (Tp)(CN)3]− (1), ClO4−, (3), and BF4− (4)) exhibit spin-transition temperature above room temperature (330 K for 1, and over 400 K for 3 and 4), the T1/2 value of 2 is much lower, near 230 K. This suggests at first glance that [FeIII(Tp)(CN)3]− metalloligand is a better donor inducing a stronger ligand field on the [Fe (bik)2(N–)2] SCO units, as compared with the [FeIII(Tp*)(CN)3]−. However the situation is more complicated as the desolvation of 1 leads to a T1/2 of ca. 240 K, a temperature close to the transition temperature of 2. Actually intermolecular interactions can play a drastic role on the spin transition behavior. In 1, a complex hydrogen-bond network is observed, including a quite strong hydrogen bond between the nonbridging cyanide and a water molecule (N⋯O approx. 2.8 Å). The impact of the desolvation is much lower for 3 and 4. This could be correlated with the nature of the intermolecular interactions. In 3 and 4, only weak intermolecular interactions with CH3CN (3) and MeOH (4) are observed, and the molecular squares remain well isolated from each other. The impact of the metalloligand could be compared by using solution studies, as we previously did for the [Fe (bik)3]2+ complexes; however, we observed that the square complexes partially disassembled in CH3CN or MeOH solution preventing such a study.
The LIESST was probed on fresh samples of 2–4 by measuring the magnetization versus time upon laser light irradiation at low temperature. The experimental conditions for 2–4 are similar to those previously used for the {FeIII2FeII2} (1) and {MoV2FeII2} squares complexes, which include the {Fe (bik)2(NC–)2} subunits [22,23]. Among the three new compounds, compound 2 shows the most important photomagnetic effect. In fact compound 3 does not show any photomagnetic effect whereas only a weak increase in the magnetization is observed on 4 upon irradiation with a visible light (see Fig. S6). As shown in Fig. 3, all of the irradiations lead to a rapid and efficient increase in the χMT product of 2 for the six wavelengths that were probed. As for 1, a plateau is reached after only ca. 20 min [22]. The irradiations at 635, 808, and 900 nm are the most efficient ones. These wavelengths correspond to metal–ligand charge transfer band of the [FeII(bik)2(N–)2] units [22]. The χMT saturation value obtained in 2, ca. 12 cm3 mol−1 K, is notably higher than that found in 1. It is also higher than that obtained at room temperature. This suggests the occurrence of ferromagnetic interactions in the photoexcited state. Actually both ferromagnetic and antiferromagnetic exchange pathways can be expected in a {FeIIILS–CN–FeIIHS} bridge. However, it has already been observed that the ferromagnetic interaction can dominate in similar low-dimensional systems [40,41]. The drop of the χMT product at low temperature (less than 10 K) is likely because of the occurrence of antiferromagnetic intermolecular interaction or/and magnetic anisotropy effects. The stability of the metastable state of 2 was then probed by measuring the χMT versus T curve after irradiation (at 808 or 900 nm), from 2 to 100 K (Fig. 4). Upon heating, the magnetization relaxes back to the diamagnetic state and becomes close to initial value at ca. 35 K. This behavior is similar to that observed for 1. Overall, the LIESST effect is much more efficient in 2 than in 3 and 4. This is coherent with previous studies, which reported that the higher the thermal transition is (the higher the energy difference between the LS ground state and the HS state is), the smaller the stabilization of the metastable state is, and the lower the relaxation temperature is [42–44].
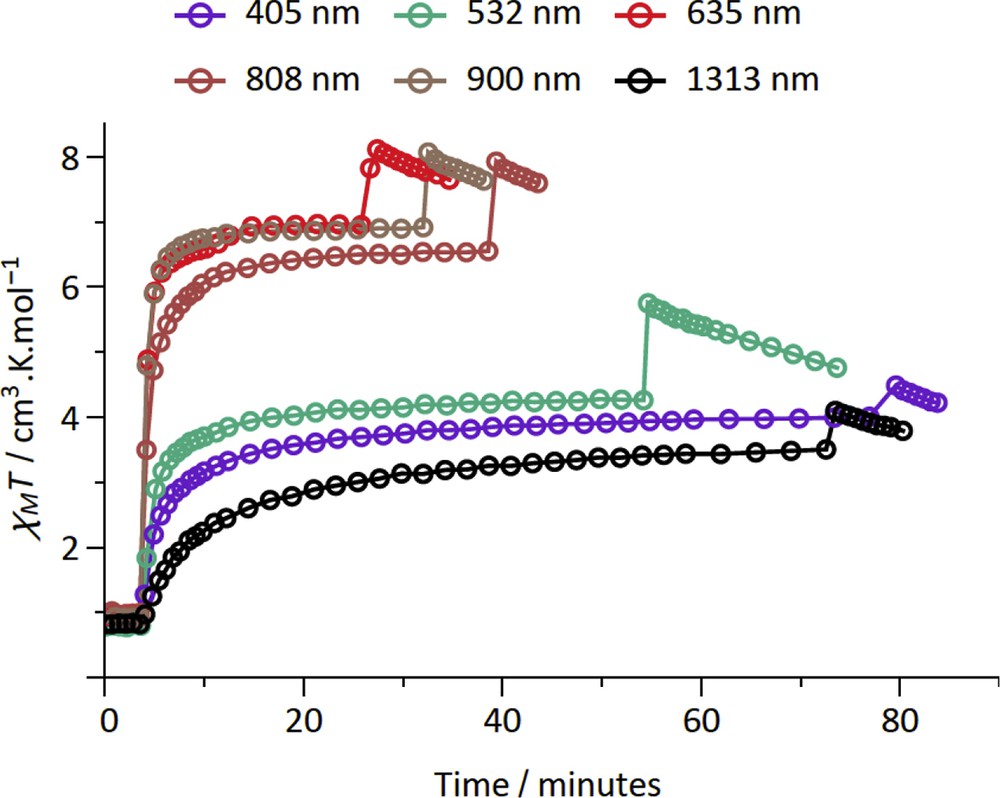
Measurement of the χMT product versus time under laser light irradiation (ca. 5 mW/cm2) at 20 K for the compound 2. The first jump corresponds to the moment the light is switched on (photomagnetic effect), whereas the second jump corresponds to the moment the light is switched off and the temperature of the sample decreases.
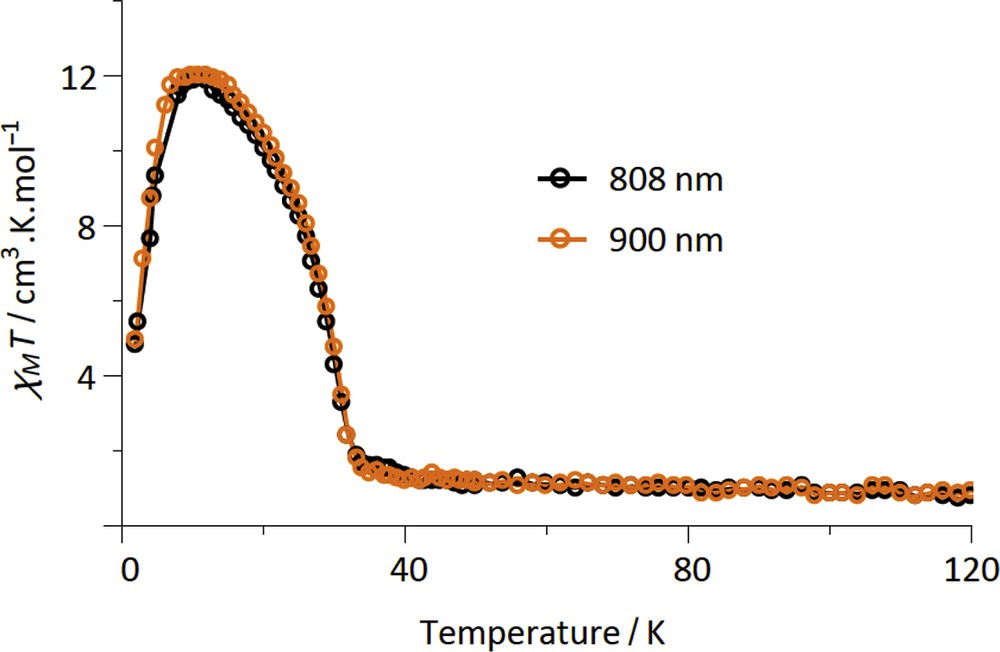
Measurement of the χMT product for 2 after laser light irradiation at 20 K (H = 1 T). The sample is measured from 2 to 100 K at 0.4 K/min.
3 Conclusions
Continuing our investigation on the [FeII(bik)2(N–)2] SCO units, we reported here three novel mixed valence cyanide-bridged {FeIII2FeII2}X2 square complexes (X = ClO4, BF4), which are reminiscent of our previously published {[FeIII(Tp)(CN)3]2[FeII(bik)2]2} [FeIII(Tp)(CN)3]2 square complex. These squares were obtained by reacting [FeII(bik)2(S)2]2+ with the [FeIII(Tp)(CN)3]− or [FeIII(Tp*)(CN)3]− N-donor metalloligands. The compounds 3 and 4 based on the [FeIII(Tp)(CN)3]−metalloligand exhibit SCO at high temperature (T1/2 > 400 K) and no (3) or moderate (4) photomagnetic effect. In contrast, the use of the [FeIII(Tp*)(CN)3]− as N-donor metalloligand leads to a shift in the SCO of the [FeII(bik)2(N-)2] units toward lower temperature, with T1/2 = 233 K. The spin-state change in 2 is gradual and the thermodynamic parameter extracted from the magnetic measurements leads to weak value of cooperative factor (Γ < 2RT1/2) in agreement with the absence of significant intermolecular interactions in 2 The decrease in the T1/2 temperature is associated with a stabilization of the HS metastable state and thus a more efficient LIESST effect. Remarkably, the irradiation with different laser lights covering the visible and near-infrared range leads to a significant photomagnetic effect. However, the strongest increase in magnetization is observed when irradiating the sample at 635, 808, or 900 nm, in the region corresponding to ligand–metal cherge transfer (LMCT) band of the [FeII(bik)2(N–)2] units. The resulting photoinduced χMT values are high and indicate the occurrence of ferromagnetic interactions in the {FeIIILS–CN–FeIIHS} metastable state.
Finally, this experimental study nicely complete our previous investigations on the photomagnetic properties of the [FeII(bik)2(N-)2] SCO units. When confronting all of these studies, it appears that the nature of the N-donor metalloligand strongly influences the energy of the most efficient laser light inducing the LIESST effect. While the most efficient irradiation is located at 405 nm when using the [Mo(CN)8]3− as N-donor ligands, it becomes 635 nm for the bik N-donor ligand, 750 nm for the [FeIII(Tp)(CN)3]− metalloligand, and 900 nm for the NCS− N-donor ligand. The present study leads to the best results as a remarkable magnetization increase is observed with the same efficiency at 635, 808, and 900 nm when using the [Fe (Tp*)(CN)3]− N-donor metalloligand. The correlation with the absorption spectra of these compounds do not lead to clear conclusion as all of them are dominated by an intense LMCT bands of the [FeII(bik)2(N–)2] units. We believe that the rationalization of this behavior would require advanced physical measurements to better investigate the excitation states involved in the LIESST effect.
4 Experimental section
4.1 General
All chemicals used were of reagent grade quality. They were purchased from commercial sources and used as received. The bik ligand was prepared as reported by Braussaud et al. [45]. The PPh4 [FeIII(Tp)(CN)3]·H2O complex was prepared in three steps by following our previously reported procedure [30].
4.2 Preparations
4.2.1 PPh4[FeIII(Tp*)(CN)3]·CH3CN
A degassed solution of KTp* (319 mg, 1.0 mmol) in 5 mL methanol was added dropwise to a methanolic solution of FeCl2·4H2O (199 mg, 1.0 mmol in 15 mL). The violet suspension was stirred for 1 h, then added dropwise to a methanolic solution of KCN (214 mg, 3.3 mmol). The resulting red suspension was stirred at room temperature overnight and the methanol was evacuated to dryness. The red resulting solid was redissolved in acetonitrile and filtered. Crystals of K[FeIII(Tp*)(CN)3] were produced by slow evaporation of the acetonitrile solution. Crystals of PPh4[FeIII(Tp*)(CN)3]·H2O suitable for X-ray diffraction analysis were produced in 1–2 weeks by slow evaporation of an acetonitrile solution of K[FeIII(Tp*)(CN)3] in which was added 1 equiv of tetraphenylphosphonium chloride and a small amount of water. Yield: 32%. Elemental analysis (%): calculated for C42H42BFeN9P·CH3CN·0.5H2O: C 64.41, H 5.65, N 17.07; found: C 64.71, H 5.43, N 16.79. ESI-MS m/z (%): 431.14 (100) [FeIII(Tp*)(CN)3]−, 339.13 (100), [PPh4]+.
4.2.2 {[FeIII(Tp*)(CN)3]2[FeII(bik)2]}2(ClO4)2·2H2O (2)
FeII(ClO4)2·6H2O (19 mg, 0.05 mmol) and bik ligand (19 mg, 0.1 mmol) were dissolved in 15 mL of a methanol/water (5/1) mixture. The resulting deep dark blue solution was added to a solution of K[Fe (Tp*)(CN)3] (25 mg) in 15 mL of the same mixture of solvents. The purple solution was further stirred for about 10 min before being filtered. Slow evaporation of the reaction mixture produced carmine red crystals suitable for X-ray diffraction analysis. Yield 40%.
4.2.3 {[FeIII(Tp)(CN)3]2[FeII(bik)2]2}(ClO4)2·2CH3CN (3)
PPh4[FeIII(Tp)(CN)3]·H2O (70.6 mg, 0.01 mmol) was dissolved in ca. 10 mL of acetonitrile and added dropwise to a solution containing [Fe(bik)2(S)2](ClO4)2 (S = solvent). The air stable [FeII(bik)2(S)2]Cl2 complex was prepared in situ by reacting approximately 0.01 mmol (36.2 mg) of Fe(ClO4)2·6H2O and 0.02 mmol (38 mg) of the bik ligand in ca. 5 mL of acetonitrile. The resulting bluish-red solution was stirred at room temperature for 30 min and then filtered. Slow evaporation of the filtrate under ambient conditions afforded orange-red plate-like crystal of {[FeIII(Tp)(CN)3]2 [FeII(bik)2]2}(ClO4)2·2CH3CN (3). Yield 60%.
4.2.4 {[FeIII(Tp)(CN)3]2[FeII(bik)2]}2(BF4)2·2CH3OH (4)
PPh4[FeIII(Tp)(CN)3]·H2O (70.6 mg, 0.01 mmol) was dissolved in ca. 10 mL of a methanolic/acetonitrile mixture and added dropwise to a solution containing [Fe(bik)2(S)2]Cl2. The air stable [Fe(bik)2(S)2]Cl2 complex was prepared in situ by reacting 0.01 mmol (33.8 mg) of Fe(BF4)2·6H2O and 0.02 mmol (38 mg) of the bik ligand in ca. 5 mL of methanol. The resulting bluish-red solution was stirred at room temperature for 30 min and then filtered. Slow evaporation of the filtrate under ambient conditions afforded orange-red plate-like crystal of {[FeIII(Tp)(CN)3]2 [FeII(bik)2]}2(BF4)2·2CH3OH (4). Yield 60%.
4.3 X-ray data collection and refinement
A single crystal of each compound was selected, mounted onto a cryoloop, and transferred in a cold nitrogen gas stream. Intensity data were collected with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using either a Bruker Κ-APEXII or a Rigaku-Oxford Saphire3 Xcalibur diffractometer. Data collection, unit-cell parameters refinement, integration, and data reduction were performed with CrysAlis (Oxford) or APEX2 (Bruker) softwares. Multiscan absorption corrections were applied. The structures were solved with ShelxS86 [46], Superflip [47], or Sir97 [48] program and refined by full-matrix least-squares methods using SHELXL-14 [49] or CRYSTALS [50].
4.4 Physical techniques
Elemental analyses (C, H, N, Co, Mn, and Ni) were performed by the Service Central d'Analyse du CNRS (France). IR spectra were recorded using a Perkin–Elmer 882 spectrophotometer as KBr pellets. Variable-temperature (2.0–400 K) magnetic susceptibility measurements in the direct-current mode were carried out with a superconducting quantum interference device (SQUID) magnetometer (from Quantum Design) using applied magnetic fields of 0.5 T (T > 30 K) and 0.025 T (T < 30 K). The magnetic data were corrected for the diamagnetism of the constituent atoms and the sample holder. The samples of 2–4 were measured as crystalline powders, removed from their mother solution directly before the measurements, and introduced into the magnetometer at 200 K to avoid the loss of solvent crystallization molecules.
Acknowledgments
Financial support was received from the Ministère Français de l'enseignement et de la recherche et du Centre National de la Recherche Scientifique (CNRS) and the DFG-funded transregional collaborative research centre SFB/TRR 88 “Cooperative effects in homo- and heterometallic complexes (3MET)” (projectB4).
Appendix A Supplementary data
The following is the Supplementary data to this article:
X-ray crystallographic files in CIF format (excluding structure factors) for 2–4 and 5 are available from the Cambridge Crystallographic Data Centre, 12 Union road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033; email: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk) on request, on quoting the deposition number CCDC 1891890 (2), 1891892 (3), 1891891 (4), and 1891893 (5).