

1 Introduction
Molecular networks are hyper molecules generated upon self-assembly of molecular building blocks or tectons bearing within their structure interactions sites. The self-assembly [1] process takes place through molecular recognition phenomena between complementary tectons [2, 3] or through coordination events if a combination of metallic and organic tectons is used [4, 5]. The generation of new molecular networks requires the design and synthesis of new tectons. In this context, calixarene derivatives [6] are rather interesting scaffolds. In particular, calix[4]arene backbone, a cyclic tetramer of phenol, offers many possibilities that may be explored for the design of new coordinating tectons [7]. Indeed, taking advantage of the existence of four non inter convertible conformations (cone, partial cone, 1,2-alternate and 1,3-alternate) that may be generated upon functionalisation of the OH moieties by bulky groups, one may design a series of tectons bearing a variety of coordination sites. For example, if the calix[4]arene core is functionalised at the lower rim by four monodentate units, four blocked conformers, defined by the relative orientation of the monodentate ligands with respect to each other, may be formed in principle (Fig. 1). Each conformer, when coupled with metal centres adopting a specific coordination geometry, should lead to coordination networks or coordination polymer. The same type of principle has been employed using different atropoisomers of substituted porphyrin derivatives [8–10].
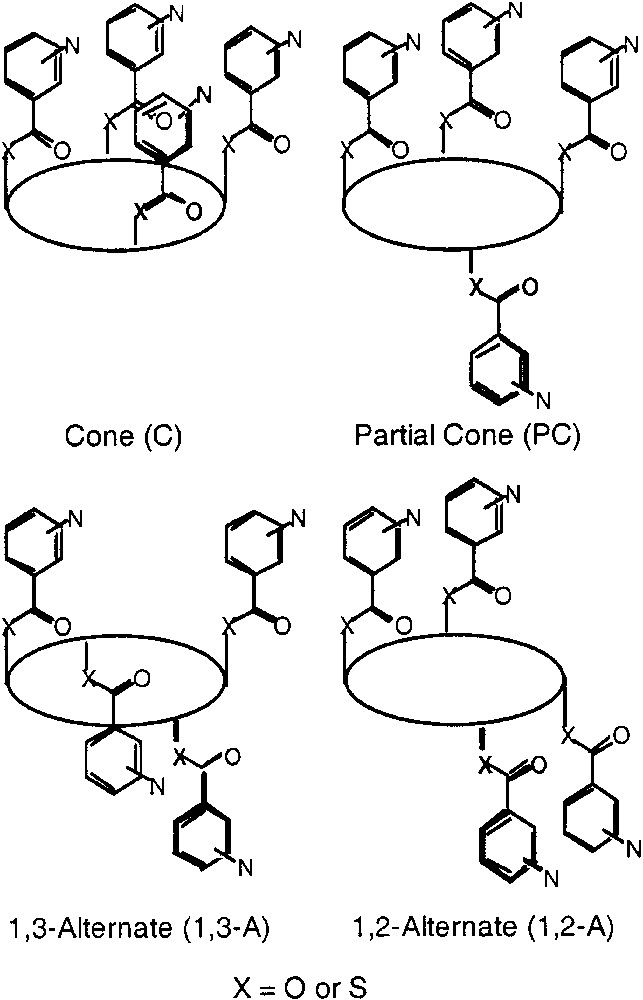
Schematic representations of all four conformational isomers of the substituted tetrathia- and tetramercapto-tetrathia-calix [4]arene derivatives. In principle, all four isomers (cone (C), partial cone (PC), 1,2-alternate (1,2-A) and 1,3-alternate (1,3-A) may be formed during the coupling reaction. The ellipsoid represents the calixarene core.
Here we report the design, synthesis and structural analysis of a series of new tectons based on the tetrathiacalix[4]arene scaffold bearing four pyridine units.
2 Results and discussion
2.1 Design and synthesis of tectons
Tetrathiacalix[4]arenes such as 1 and 2 are analogues of the classical calix[4]arene for which the methylene junctions between the phenolic moieties are replaced by sulphur atoms [11, 12] (Fig. 2). Based on the same backbone, new derivatives resulting from partial oxidation of the thio junctions affording the tetrasulfoxide derivatives [13, 14] or complete oxidation of the sulphur atoms leading to the tetrasulfone derivatives [15] have been also reported. The synthesis of the tetramercaptotetrathiacalix[4]arene 3, for which the methylene junctions between the aromatic moieties are replaced by S atoms and all four OH groups are substituted by SH moiety has been also reported [16]. Dealing with other mercaptocalixarene derivatives, the synthesis of di- [17–18] and tetra- [19–20] mercaptocalix[4]arenes for which two or four OH groups are replaced by SH groups was also reported. Furthermore, the remarkable binding abilities towards transition metals of thiacalixarenes were investigated [21]. In particular, recently, using compounds 1 and 3 and mercury cation, the formation of tetra- and hexanuclear mercury complexes based on two or three 1 or 3 units respectively was reported [22].
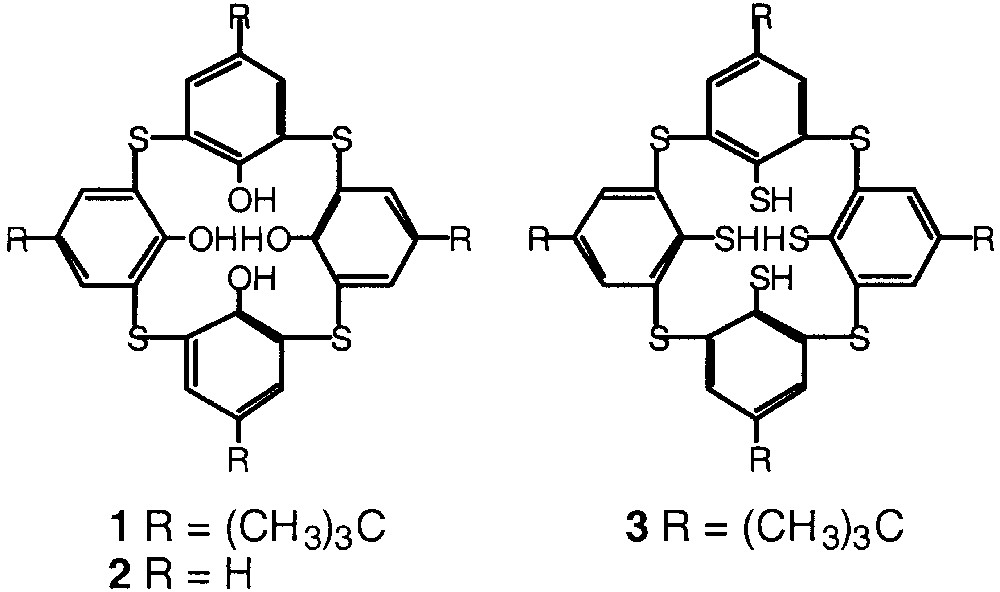
Tetrathiacalix[4]arene derivatives 1, 2, and 3.
As in the case of calix[4]arene, tetrathiacalix[4]arene derivatives are subject to conformational mobility and may adopt four limit conformations. These conformers differ from each other by the positions of the aromatic rings with respect to the main plane of the macrocycle defined by the sulphur atoms. For that reason, the design of new coordinating tectons based on different conformers of tetrathiacalix[4]arene appeared as interesting to us. In particular, we considered the two derivatives 1 (R = t-butyl) and 2 (R = H) of tetrathiacalix[4]arene bearing four thioether and four OH groups and the tetramercaptotetrathiacalix[4]arene 3 (Fig. 2) bearing four thioether and four SH groups for the design of tectons 4–8 (Fig. 3).
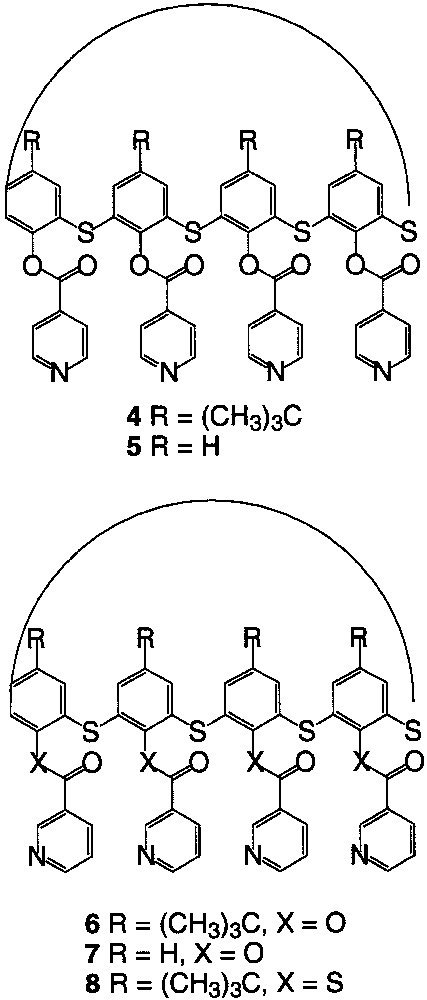
Coordinating tectons 4–8.
Calix[4]arene [23, 24] and thiacalix[4]arene [25] derivatives functionalised at the lower rim by pyridine units through ether junctions have been reported.
The starting material, the tetrathiacalix[4]arene derivatives 1 [11], 2 [26] and 3 [16] (Fig. 2) were prepared according to published procedures and used, as stated above, as scaffolds for the design of new coordinating tectons 4–8 (Fig. 3). Dealing with the pendent monodentate-coordinating moiety, pyridine was chosen. The junction between the pyridine and the calix backbone was achieved through an ester group in the case of compounds 4–7 using calix derivatives 1 and 2 and through a thioester functionality in the case of compound 8 when starting with compound 3. Dealing with the orientation of the N atom on the pyridine ring with respect to the axis perpendicular to the calix plane, the connection was either achieved at the position 3 (compounds 6–8) or position 4 (4–5) of the pyridine (Fig. 3).
All the condensation reactions between the calix and pyridine derivatives were performed in pyridine under reflux over night. The condensation of 1 or 2 with iso-nicotinoyl chloride afforded different conformers of compounds 4 and 5 in ca 66% overall yield. On the other hand, compounds 6–8 were obtained in 82, 81 and 57% overall yields as mixtures of conformers upon treatment of calix derivatives 1–3 respectively with nicotinoyl chloride.
2.2 Conformation analysis of tectons
Upon condensation of the calix derivatives 1–3 with either nicotinoyl chloride or isonicotinoyl chloride, four blocked conformers may be expected (Fig. 1). For the condensation of 1 with iso-nicotinoyl chloride, although all four conformers (1,2-A, 1,3-A, PC and C) of the tetra-substituted compound 4 were formed, however only the 1,2-A (60%) and the PC (7%) conformers could be isolated and purified. The other two conformers (C and 1,3-A) were present in less that 5%.
The same reaction with 2 afforded almost exclusively the 1,2-A conformer of 5. The latter appeared to be rather insoluble in usual solvents. Concerning the condensation of nicotinoyl chloride with compounds 1 and 2, the 1,3-A conformers of both 6 and 7 were found to be the most abundant species (86 and 89% respectively). However, the 1,2-A conformer of 6 and 7 were also isolated in 5 and 9% yields respectively. In the case of 1, 6% of the PC conformer of 6 was also isolated. In marked contrast, in the case of the condensation of nicotinoyl chloride with the mercaptotetrathiacalixarene 3, only the cone conformer of 8 could be isolated in 57% yield. We have no obvious explanation for the distribution of different conformers.
Due to the blocked conformation of the substituted calix unit, all conformers reported were identified, based on their symmetry, by 1H and 13C NMR spectroscopy and further confirmed in the solid state by X-ray diffraction on single crystal. This was particularly important for differentiating the 1,3-A with and the cone conformation.
2.3 Solid-state structural studies
The structure of 4(1,2-A), 5(1,2-A), 6(1,2-A), 7(1,3-A) and 8(C) in the solid state was investigated by X-ray diffraction on single crystals obtained from a CHCl3/MeOH solution (see table).
For both derivatives 4(1,2-A) and 6(1,2-A), in contrast with the parent compound 1 which was shown to adopt the cone conformation [26], the calix unit adopts the 1,2-alternate conformation positioning thus the adjacent pyridine units on the same face of the calix, which appears to be almost parallel to each other (Fig. 4). For the S atoms connecting the aromatic rings, an average CS bond distance of ca 1.80 Å is observed. With respect to the ester junction, the average C=O and C–O distances are 1.19 Å and 1.36 Å. All four C=O groups are outwardly oriented.
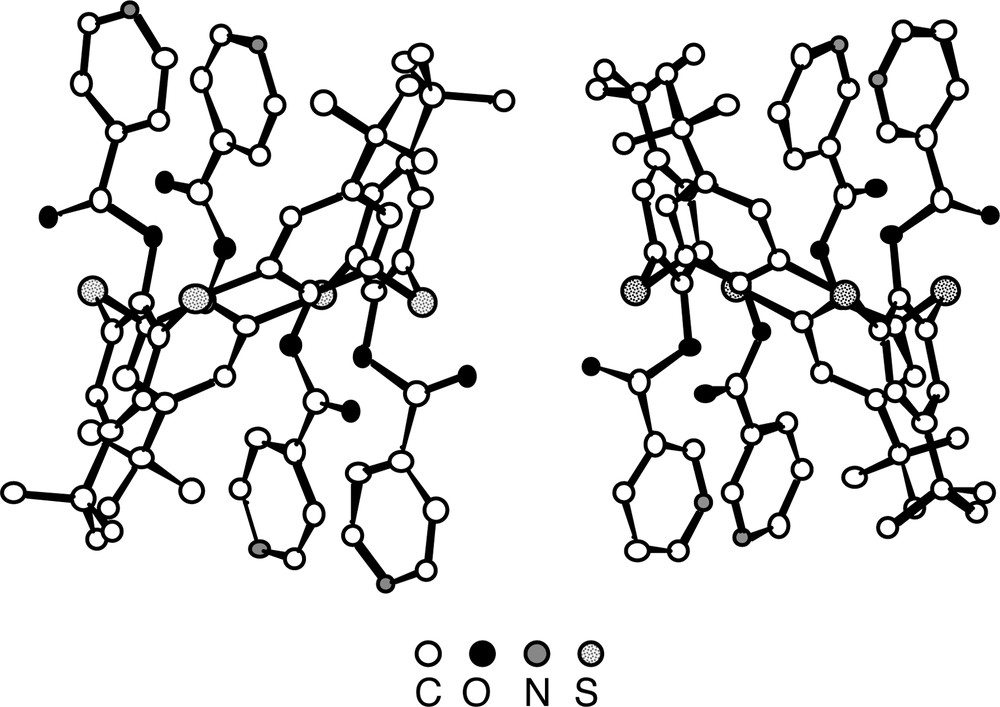
Solid-state structures of compounds 4(1,2-A) (left) and 6(1,2-A) (right). The calix unit adopts in both cases the 1,2-alternate conformation positioning thus two adjacent pyridine units on the same face of the calix unit. Solvent molecules and H atoms are not represented for clarity. For bond distances and angles, see text.
For both derivatives 5(1,2-A) and 7(1,3-A), generated from the tetrathiacalix [4]arene 2, again in contrast with the parent compound 2, which was shown to adopt the cone conformation [26], the calix unit adopts the 1,2-alternate conformation in the case of the condensation with iso-nicotinoyl chloride and the 1,3-alternate conformation for the reaction with nicotinoyl chloride (Fig. 5). In the latter case, the four pyridine units are located in an alternate fashion below and above the main plane of the calix. For the S atoms connecting the aromatic rings, an average CS bond distance of ca 1.78 Å is observed. With respect to the ester junction the average C=O and C–O distances are 1.20 Å and 1.37 Å. All four C=O groups are again outwardly oriented.
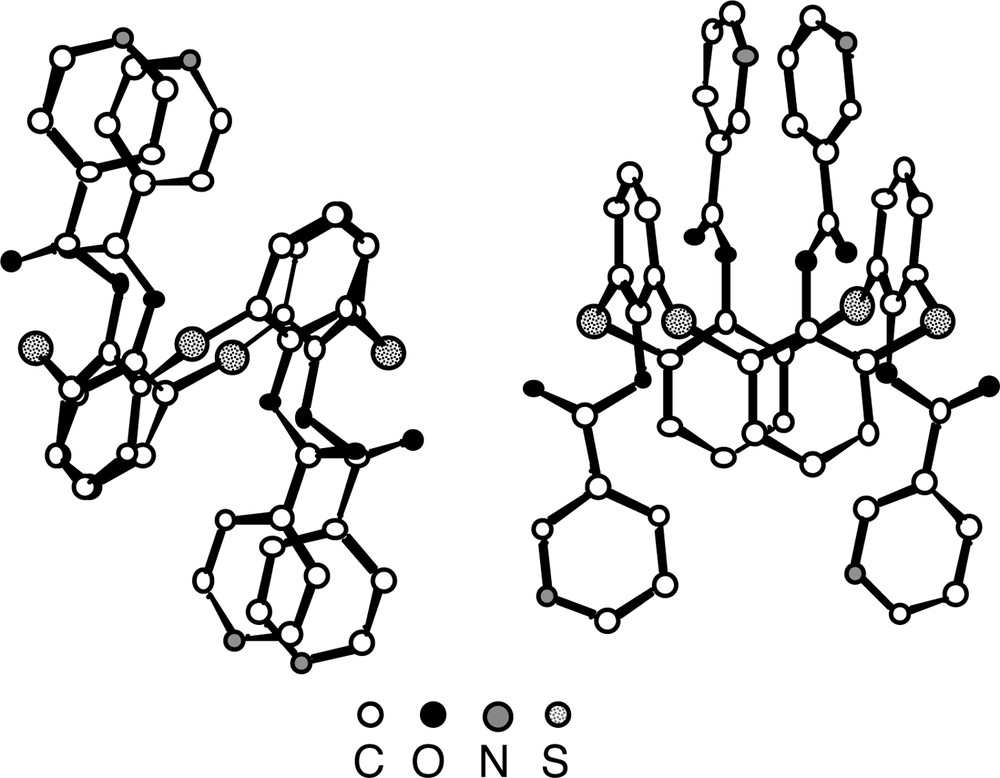
Solid-state structures of compounds 5(1,2-A) (left) and 7(1,3-A) (right). The calix unit adopts, in the case of 5, the 1,2-alternate conformation positioning thus two adjacent pyridine units on the same face of the calix unit and, in the case of 7, the 1,3-alternate conformation positioning in an alternating fashion the pyridine units above and below the main plane of the calix unit. Solvent molecules and H atoms are not represented for clarity. For bond distances and angles, see text.
The structure analysis of 8(C) (Fig. 6) showed the following features. In contrast with the parent compound 3, which was shown to adopt the 1,3-alternate conformation [16], the calix unit in 8 adopts the cone conformation allowing thus to position the four pyridine units on the same face of the calix. For the S atoms connecting the aromatic rings, an average CS bond distance of ca 1.79 Å is observed. With respect to the thioester junction the average C=O and C–S distances are 1.20 Å and 1.80 Å. All four C=O groups are outwardly oriented.
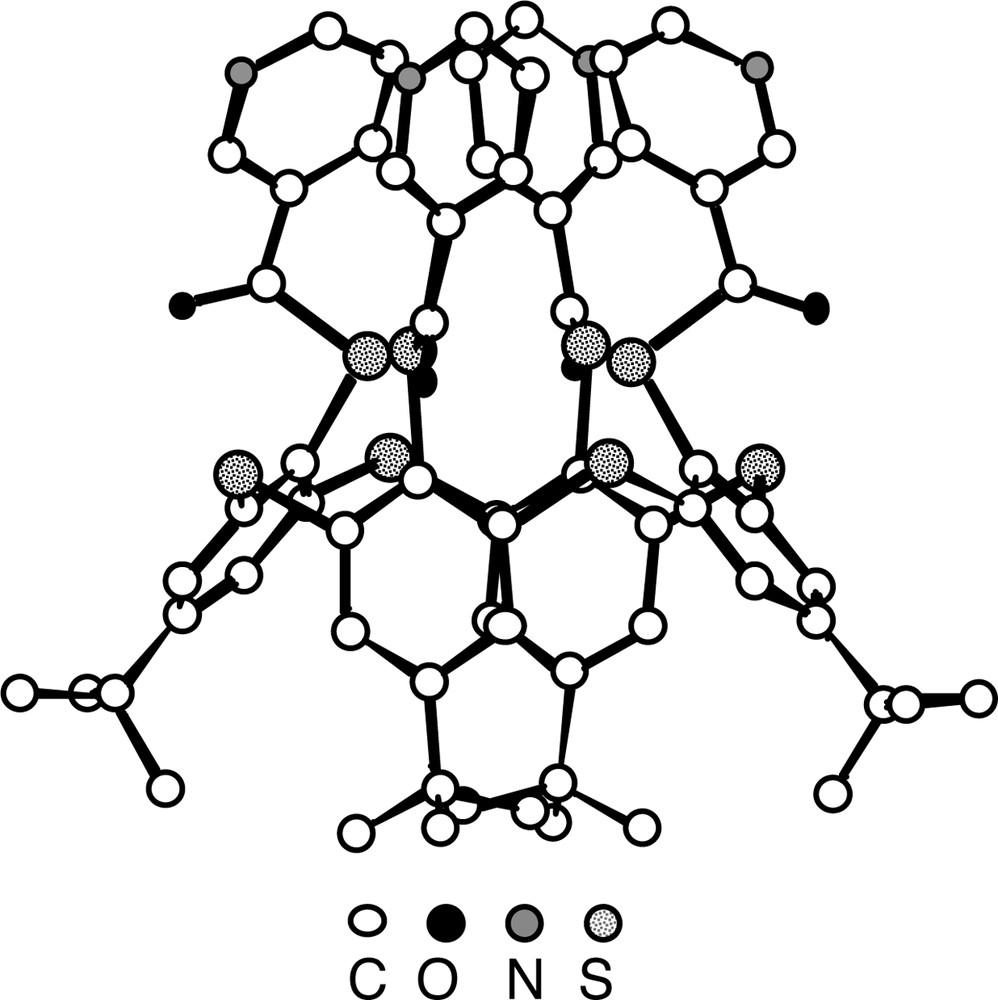
Solid-state structure of the compound 8(C) (lateral view). The calix unit adopts the cone conformation positioning thus all four pyridine units connected to the calix backbone at the 3-position on the same face of the calix unit. H atoms are not represented for clarity. For bond distances and angles, see text.
2.4 Conclusion
Upon treatment of the thiacalix derivatives 1-3 by iso-nicotinoyl or nicotinoyl chloride, five new derivatives bearing four pyridine units were prepared. From each condensation, a series of tetrasubstituted conformers were isolated. In the case of 4 and 5, the most abundant conformer was found to be the 1,2-alternate, whereas in the case of 6 and 7, the 1,3-alternate conformation was the major component of the mixture. Finally, in the case of 8, the cone conformer was exclusively isolated. Thus overall nine new conformers bearing four pyridines were isolated and characterised. In the case of 4(1,2-A), 5(1,2-A), 6(1,2-A), 7(1,3-A) and 8(C), their structures in the solid state were investigated by X-ray diffraction on single crystals. The use of all reported compounds in the formation of coordination networks using metal centres is currently under investigation.
3 Experimental section
3.1 Synthesis
4(1,2-A) and 4(PC). To a suspension of p-tert-butylthiacalix[4]arene 1 (1 g, 1.3 mmol) in 50 ml of pyridine, iso-nicotinoyl chloride (1.9 g, 10 mmol) was added and the mixture refluxed overnight. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated and the residue was extracted with CH2Cl2 (100 ml), washed with dilute aqueous HCl (30 ml, 2 N) and aqueous solution of K2CO3 (30 ml, 2.5 N). The organic layer was dried over MgSO4 before the solvent was removed under reduced pressure leaving a mixture that was purified by fractional crystallisation (CHCl3–MeOH) affording 950 mg of 4(1,2-A) and 110 mg of 4(PC). The overall yield of the reaction was 67%. Anal. calcd for C64H60N4O8S4: (1141.45 g mol–1): C 67.35,% H 5.30%, N 4.91%, S 11.23%.
4(1,2-A). fr = 0.5 (SiO2, CH2Cl2/MeOH, 95/5), mp > 350 °C, anal. found: C 67.35%; H 5.32%, N 4.90%, S 11.54%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 1.02 (s, 36H, C(CH3)3), 6.77 (d, J = 5.8 Hz, 8H, Ar pyr), 7.19 (d, J = 2.3 Hz, 4H, Ar calix), 7.85 (d, J = 2.3 Hz, 4H, Ar calix), 8.10 (d, J = 5.8 Hz, 8H, Ar pyr); 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 31.0 (CH3), 34.9 C(CH3)3), 122.5, 128.6, 130.7, 131.1, 134.6, 135.1, 149.5, 150.5, 150.6, 162.7 (Ar).
4(PC). fr = 0.4 (SiO2, CH2Cl2/MeOH, 95/5), mp = 290°C (decomp.), anal. found: C 67.65%; H 5.07%, N 4.44%, S 10.52%; 1H-NMR (300 MHz, CDCl3, 25°C): δ (ppm): 0.79 (s, 18H, C(CH3)3), 1.24 (s, 9H, C(CH3)3), 1.51 (s, 9H, C(CH3)3), 6.39 (m, 2H, Ar pyr), 7.03 (d, J = 3 Hz, 2H, Ar pyr), 7.39 (s, 2H, Ar calix), 7.48 (m, 2H, Ar pyr), 7.62 (m, 4H, Ar calix), 7.69 (m, 2H, Ar pyr), 7.89 (sl, 2H, Ar pyr), 8.07 (s, 2H, Ar calix) 8.10 (d, J = 3 Hz, 2H, Ar pyr), 8.58 (d, J = 3Hz, 4H, Ar pyr), 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm) 30.4; 31.0; 31.3 (CH3); 34.2; 34.7; 35.1 (C(CH3)3; 122.4; 122.6; 122.7; 122.8; 126.2; 127.3; 129.8; 130.5; 131.3; 134.6; 135.6; 135.9; 135.9; 148.3; 149.6; 150.0; 150.3; 150.4; 150.5; 150.6; 150.6; 151.3; 159.7; 163.5; 163.8.
5(1,2-A). To a suspension of thiacalix[4]arene 2 (0.5 g, 1.1 mmol) in 25 ml of pyridine, iso-nicotinoyl chloride (0.9 g, 5 mmol) was added and the mixture refluxed overnight. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated and the residue was extracted with CH2Cl2 (50 ml), washed with dilute aqueous HCl (20 ml, 2.5 N) and aqueous solution of K2CO3 (20 ml, 2 N). The organic layer was dried over MgSO4 before the solvent was removed under reduced pressure leaving a mixture that was purified by fractional crystallisation (CHCl3–MeOH) affording 610 mg of 5(1,2-A) in 66% yield. mp 220 °C (decomp.); fr = 0.5 (SiO2, CH2Cl2/MeOH, 95/5), Anal. calcd for C48H28N4O8S4·2 MeOH: (981.10 g mol–1): C 61.21,% H 3.70%, N 5.71%, S 13.07%, found: C 61.25%; H 3.09%, N 5.81%, S 13.75%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm):6.86 (dd, J1 = 1.5 Hz, J2 = 3 Hz, 8H, Ar pyr); 7.17 (m, 8H, Ar Calix); 8.05 (dd, J1 = 1.9 Hz, J2 = 5.6 Hz, 4H, Ar calix), 8.40 (dd, J1 = 1.6 Hz, J2 = 2.7 Hz, 8H, Ar pyr); 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 122.7, 127.1, 129.2, 131.4, 134.5, 134.8, 137.2, 150.5, 151.2, 162.2 (Ar).
6(1,3-A), 6(1,2-A), and 6(PC). To a suspension of p-tert-butylthiacalix[4]arene 1 (0.5 g, 0.69 mmol) in 25 ml of pyridine, nicotinoyl chloride (0.9 g, 5 mmol) was added and the mixture refluxed overnight. After allowing the reaction mixture to cool to rt., the solvent was evaporated and the residue was extracted into 50 ml of CH2Cl2, washed with dilute aqueous HCl (20 ml, 2.5 N), aqueous solution of K2CO3 (20 ml, 2 N) and dried over MgSO4. After removing the volatiles, upon crystallisation from CHCl3–MeOH mixture 560 mg of 6(1,3-A) could be isolated. The other two conformers 6(PC) (50 mg) and 6(1,2-A) (40 mg) have been isolated by preparative thin layer chromatography. The overall yield was 82%. Anal. calcd for C64H60N4O8S4: (1141.45 g mol–1): C 67.35,% H 5.30%, N 4.91%, S 11.23%: (overall yield of 82%).
6(1,3-A). fr = 0.5 (SiO2, CH2Cl2/MeOH, 95/5), mp 320 °C (decomp), anal. found: C 67.33%; H 5.34%, N 4.80%, S 11.51%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 0.7 (s, 36H, CH3), 7.07 (s, 8H, Ar calix), 7.76 (dd, J1 = 5.0 Hz, J2 = 2.8 Hz, 4H, Ar pyr), 8.07 (m, 4H, Ar pyr), 8.7 (s large, 4H, Ar pyr), 8.86 (dd, J1 = 1.6 Hz, J2 = 3.2 Hz, 4H, Ar pyr); 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 30.6 (CH3), 34.3 (C(CH3)3), 124.4, 124.8, 130.2, 130.8, 137.9, 149.8, 150.6, 151.4, 153.8, 162.3 (Ar).
6(PC): fr = 0.6 (SiO2, CH2Cl2/MeOH, 95/5), mp 330 °C (decomp.), anal. found: C 67.08%; H 5.31%, N 4.91%, S 11.63%; 1H-NMR (300 MHz, CDCl3, 25°C): δ (ppm): 0.81 (s, 18H, C(CH3)3), 1.17 (s, 9H, C(CH3)3), 1.48 (s, 9H, C(CH3)3), 6.11–6.15 (m, 1H, Ar pyr), 6.91–6.95 (m, 1H, Ar pyr), 7.02 (d, J = 2.5 Hz, 2H, Ar calix), 7.29–7.36 (m, 3H, Ar pyr, Ar calix), 7.41–7.46 (m, 3H, Ar pyr), 7.70 (d, J = 2.5 Hz, 2H, Ar calix), 7.91–7.95 (m, 1H, Ar pyr), 8.02 (m, 2H, Ar calix), 8.20–8.28 (m, 3H, Ar pyr), 8.51–8.53 (m, 1H, Ar pyr), 8.73–8.80 (m, 3H, Ar pyr), 9.01 (m, 2H, Ar pyr), 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 30.6, 31.0, 31.2 (CH3), 34.2, 34.8, 35.0 (C(CH3)3, 122.7; 123.3, 124.1, 124.5, 126.3, 127.8, 129.9, 130.3, 131.0, 132.3, 135.5, 135.7, 136.6; 137.0, 137.7, 149.7, 150.1, 150.3, 150.5, 150.8, 151.2, 152.7, 153.3, 153.6, 163.6.
6(1,2-A). fr = 0.9 (SiO2, CH2Cl2/MeOH, 95/5), mp 305 °C (decomp.), anal. found: C 67.29%; H 5.50%, N 4.85%, S 11.47%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 1.19 (s large, 36H, CH3), 6.89 (s, 4H, Ar pyr), 7.36 (bs, 4H, Ar calix), 7.61 (m, 4H, Ar pyr), 8.04 (bs, 4H, Ar pyr), 8.20 (bs, 4H, Ar calix), 8.53 (s large, 4H, Ar pyr), 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 30.9 (CH3), 34.7 (C(CH3)3, 123.0; 124.0, 130.5, 130.8, 134.9, 137.0, 149.1, 150.3, 150.7, 153.4, 163.0.
7(1,3-A) and 7(1,2-A): To a suspension of thiacalix[4]arene 2 (0.5 g, 1 mmol) in 25 ml of pyridine, nicotinoyl chloride (0.9 g, 5 mmol) was added and the mixture refluxed overnight. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated and the residue was extracted in 50 ml of CH2Cl2, washed with dilute aqueous HCl (20 ml, 2.5 N), aqueous solution of K2CO3 (20 ml, 2 N) and dried over MgSO4. After removing the volatiles, upon crystallisation from CHCl3–MeOH mixture 670 mg of 7(1,3-A) was isolated. The other conformer 7(1,2-A) (80 mg) was obtained by preparative thin layer chromatography. The overall yield was 81%. Anal. calcd for C48H28N4O8S4: (1141.45 g mol–1): C 62.87,% H 3.08%, N 6.11%, S 13.98%.
7(1,3-A): fr = 0.4 (SiO2, CH2Cl2/MeOH, 95/5), mp 270 °C (decomp), anal. found: C 62.42%; H 3.07%, N 5.95%, S 14.09%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 6.73 (t, J = 7.7 Hz, 4H, Ar calix), 7.10 (d, J = 7.7 Hz, 8H, Ar calix), 7.95 (m, 4H, Ar pyr), 8.09 (m, 4H, Ar pyr), 8.92 (d, J = 1.6, 4H, Ar pyr), 9.06 (m, 4H, Ar pyr); 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 123.6, 124.1, 126.3, 130.0, 136.0, 138.6, 152.1, 153.1, 154.8, 161.4 (Ar).
7(1,2-A): fr = 0.3 (SiO2, CH2Cl2/MeOH, 95/5), mp 305 °C (decomp.), anal. found: C 62.54%; H 3.24%, N 5.90%, S 14.15%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 7.04 (dd, J1 = 4.8 Hz, J2 = 3.0, 4H, Ar pyr), 7.21 (m, 8H, Ar calix), 7.54 (dt, J1 = 1.9 Hz, J2 = 8.0 Hz, 4H, Ar calix), 8.11 (dd, J1 = 1.9 Hz, J2 = 5.6 Hz, 4H, Ar pyr), 8.21 (bs, 4H, Ar pyr), 8.76 (dd, J1 = 1.3 Hz, J2 = 3.4 Hz, 4H, Ar pyr), 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 122.9, 123.5, 127.0, 128.9, 131.2, 134.5, 137.2, 137.5, 150.8, 150.9, 154.1, 162.5 (Ar).
8(C): To a suspension of the mercapto-p-tert-butylthiacalix[4]arene 3 (0.3 g, 0.38 mmol) in 25 ml of pyridine, nicotinoyl chloride (0.9 g, 5 mmol) was added and the mixture refluxed overnight. After allowing the reaction mixture to cool to room temperature, the solvent was evaporated and the residue was extracted in 50 ml of CH2Cl2, washed with dilute aqueous HCl (20 ml, 2.5 N), aqueous solution of K2CO3 (20 ml, 2 N) and dried over MgSO4. After removing the volatiles the crude product was purified by fractional crystallisation that afforded 260 mg (57% yield) of the compound 8(C). mp 280 °C (decomp.); fr = 0.6 (SiO2, CH2Cl2/MeOH, 95/5), anal. calcd for C64H60N4O4S8 (1205.69 g mol–1): C 63.76,% H 5.02%, N 4.65%, S 21.27%, found: C 63.45%; H 5.03%, N 4.69%, S 21.22%; 1H-NMR (200 MHz, CDCl3, 25°C): δ (ppm): 1.17 (s, 36H, CH3), 7.35 (m, 4H, Ar pyr), 7.63 (s, 8H, Ar calix), 8.24 (m, 4H, Ar pyr), 8.81 (m, 4H, Ar pyr), 9.17 (m, 4H, Ar pyr), 13C-NMR (75 MHz, CDCl3, 25°C): δ (ppm): 31.0 (CH3), 34.9 (C(CH3)3), 123.7, 134.3, 134.4, 135.1, 143.4, 148.6, 153.7, 154.0, 188.4 (Ar).
3.2 X-ray structure analysis
For all structures reported (see Table 1), datasets were measured at 173 K on a kappa CCD diffractometer with graphite monochromated Mo Kα radiation. All structures were solved by direct methods using OpenMoleN 2.2 [27] and refined anisotropically using absorption corrected data. Atomic coordinates, bond lengths and angles, and thermal parameters have been deposited at the Cambridge Crystallographic Data Centre.
Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic data Centre, 12 Union Road, Cambridge CB2 1EZ, UK as cif files No. CCDC 197905-197909, and can be obtained by contacting the CCDC.
Acknowledgements
We would like to acknowledge the ‘Université Louis-Pasteur’and the French ‘Ministère de la Recherche et de la Technologie’for financial support.