

1 Introduction
La vectorisation d’analogues de nucléosides constitue un des défis de la galénique moderne. Parmi les vecteurs pressentis, les cyclodextrines et leurs dérivés constituent une famille très étudiée d’hôtes capables de masquer temporairement les propriétés physico-chimiques d’un invité sans en changer naturellement la structure chimique [1]. Afin de mieux comprendre les interactions qui gouvernent ces édifices supramoléculaires, nous avons entrepris au sein de notre groupe la synthèse de nouveaux composés macrocycliques de la famille des glycophanes associant une partie sucre à des bras espaceurs variés. Après avoir décrit la synthèse de composés cycliques mais pourvus d’une cavité restreinte [2], nous présentons dans ce second article la synthèse de cages moléculaires de grande taille, toujours à partir du d-glucal, mais par une autre réaction de Ferrier.
2 Stratégies de synthèse
Les synthèses mises en œuvre ont toutes pour point de départ un double réarrangement de Ferrier [3] sur le d-glucal par un diol symétrique en présence d’un acide de Lewis, réaction qui fournit un accès facile à des glycosides symétriques, dont nous différencions les deux fonctions alcools primaires par réaction avec le chlorure de trityle (Fig. 1).

Synthèse de glycophanes chiraux à 22 ou 23 chaînons.
Cette première approche ayant livré des glycophanes rigides, mais de taille réduite [2], ou plus grands mais instables [4], nous avons voulu utiliser un autre diol ou spacer (le 1,4-diméthanol-benzène), l’insaturation du sucre étant alors supprimée avant de réaliser une alkylation par le bromure de propargyle, celle-ci pouvant être simple ou qualifiée de « pas à pas » (cf. voie a) ou double pour l’autre voie (cf. voie b) en vue d’un couplage direct. Dans la voie a, après un premier couplage oxydant [5], la seconde fonction alcool secondaire sera alors alkylée par le même réactif, pour permettre la cyclisation de la molécule (Fig. 2).

Deux voies convergentes pour la préparation de tétrasaccharides semi-rigides.
3 Résultats et discussion
3.1 Préparation de précurseurs acétyléniques
La première étape commune à ces deux voies est un réarrangement allylique, rapporté pour la première fois par Ferrier et Prasad [3]. Wieczorek et Thiem [6] ont montré que la présence de certains groupements protecteurs (tel un tert-butyldiméthylsilyl) sur l’alcool en C-3 conduisait à des rendements médiocres, alors qu’un bon groupe partant (généralement un acétyl comme c’est le cas ici) favorisait la migration de la double liaison (Fig. 3).

Réaction du 1,4-diméthanol-benzène sur le d-glucal.
Le sous-produit 2 est très aisément séparable par chromatographie et peut être remis en réaction. Le glycoside symétrique 1, très majoritairement α, est purifiable par recristallisation préparative. La suite de la synthèse est conforme au schéma général, présenté sur la Fig. 2 (Fig. 4).

Obtention et propargylation sélective du tétrol 3.
Afin de réduire l’insaturation C-2/C-3 du sucre, présumée responsable de l’instabilité de certains composés déjà préparés [4], il était indispensable de mettre en œuvre un catalyseur chimiosélectif, le risque d’hydrogénolyser la liaison O-benzyl avec la copule aromatique, voire même de détrityler [7], n’étant pas nul. Après de multiples essais, nous avons retenu le palladium déposé sur carbonate de calcium à 5% : en présence d’hydrogène à la pression atmosphérique, ce catalyseur mène à l’intermédiaire 5, qui est ensuite traité par le bromure de propargyle en quantité stœchiométrique dans les conditions du transfert de phase [8]. Les deux précurseurs 6 et 7 ont donc été obtenus à partir du tri-O-acétyl-d-glucal en cinq étapes, avec un rendement global respectif supérieur à 20%.
3.2 Cyclisation par la méthode « pas à pas » (voie a)
Nous avons réalisé le couplage de l’éther 7 dans les conditions de Hay [9], c’est-à-dire dans la pyridine et sous un courant d’oxygène pour régénérer des ions Cu2+. Dans ce cas, la concentration initiale ne joue guère, puisque ce substrat est monofonctionnalisé, ce qui exclut toute possibilité de polymérisation et explique certainement le très bon rendement en 8 (Fig. 5).

Couplage oxydant de 7 et di-O-alkylation du diol 8.
À ce stade de la synthèse, une seule étape nous séparait formellement du macrocycle que nous nous proposions de synthétiser. Avant de pouvoir cycliser le tétrasaccharide 9, il est apparu rapidement indispensable de le débarrasser de ses groupements protecteurs, qui gênaient le couplage oxydant intramoléculaire (répulsion stérique ?). Ainsi, le traitement du tétrasaccharide 9 en présence d’une quantité catalytique d’acide fort dans le méthanol nous a permis d’isoler, avec un rendement acceptable, le tétrol 10, qu’il n’a pas été possible de cycliser dans les conditions adoptées par Vasella et Bürli [10]. C’est pourquoi nous avons dû acétyler ce précurseur à nouveau pour pouvoir réaliser ce dernier couplage oxydant (Fig. 6).

Déprotection et peracétylation du précurseur 9.
La mise en présence du précurseur 11 avec un excès d’ions cuivriques dans un mélange de pyridine et d’acétonitrile à moyenne dilution (5 × 10–3 M) vers 0 °C conduit à deux macrocycles aisément séparables par chromatographie sur silice : le tétrasaccharide 12 et son cyclodimère, l’octasaccharide 13, très minoritaire (Fig. 7).

Cyclisation par couplage oxydant du précurseur 11.
Nous avons relevé dans la littérature que ce type de couplage conduisait majoritairement au produit issu de la réaction intramoléculaire, avec, il est vrai, toujours un peu du dimère et du trimère correspondant [11].
3.3 Cyclisation par la méthode directe (voie b)
Tenant compte des difficultés rencontrées pour la cyclisation du précurseur 9, nous avons résolu de détrityler puis d’acétyler préalablement le précurseur 6 avant de le soumettre à un quelconque couplage oxydant (Fig. 8).

Détritylation et acétylation du précurseur 6.
Pour tenter de tirer profit de l’effet de matrice des ions cuivre, qui favoriserait la formation du cycle à quatre motifs, nous avons réalisé le couplage oxydant du bis-acétylénique 14 dans les mêmes conditions que celles utilisées pour 11 (température de réaction proche de 0 °C, mêmes concentration et durée). Nous isolons alors directement, avec un très bon rendement, le même glycophane 12 (Fig. 9).

Macrocyclisation par dimérisation directe du précurseur 14.
Dans ces conditions, nous ne décelons pas de trace de l’octasaccharide 13 par chromatographie sur couche mince (ccm), ce qui permet de supposer qu’à faible température et dans ce système solvant, les effets de matrice dus au cuivre orientent quasi-exclusivement le second couplage en faveur du tétrasaccharide 12, au détriment d’autres oligomères ouverts ou cycliques.
En conclusion, l’objectif de ce travail, qui était la synthèse par deux voies d’une cage moléculaire de la famille des glycophanes, a été atteint. Nous sommes parvenus à synthétiser préférentiellement le tétrasaccharide cyclique 12 par la méthode baptisée « pas à pas » au dépend de son dimère, l’octasaccharide cyclique 13, a priori moins intéressant. De plus, la méthode de couplage directe nous a permis d’accéder au même tétrasaccharide 12 avec un meilleur rendement global, soit 40% à partir du d-glucal, en sept étapes seulement. L’étude du pouvoir complexant de ces glycophanes déprotégés et de leurs dérivés hydrosolubles [12], mimes potentiels de certaines cyclodextrines [13], est notre prochain objectif.
4 Partie expérimentale
4.1 Généralités
Les solvants ont été distillés avant usage : le chlorure de méthylène (DCM) sur P2O5, le méthanol et l’éthanol sur magnésium. Sauf indication contraire, les spectres RMN 1H ont été enregistrés sur un appareil Bruker Avance DRX 400 à 400 MHz et Bruker AC 250 à 250 MHz pour le proton, à respectivement 100,6 et 62,9 MHz pour le 13C. Les déplacements chimiques δ sont exprimés en ppm, les spectres 1H et 13C réalisés dans le chloroforme deutéré et calibrés respectivement à 7,26 et 77,16 ppm. L’attribution des pics a été faite par RMN à deux dimensions ou par irradiation sélective, la description des spectres RMN se référant à la numérotation simplifiée proposée pour chacune des molécules (à cause des symétries qui existent dans cette famille de molécules, les spectres RMN 1H et 13C ne montrent généralement qu’une fraction des signaux attendus). Les spectres infrarouge ont été enregistrés sur un spectromètre PerkinElmer FT-IR spectrum 1000 sur pastille NaCl. Les spectres de masse ont été enregistrés sur un appareil Fisons Trio-1000 à l’UHP–Nancy-1 ou sur spectromètre Bruker reflex IV (laboratoire de spectrométrie de masse et de chimie laser du Prof. J.-F. Muller à Metz) pour les spectres MALDI avec l’acide 2,5-dihydroxybenzoïque (DHBA) comme matrice. Les pouvoirs rotatoires ont été mesurés à 25 °C à l’aide d’un appareil PerkinElmer automatique modèle 141, dans une cuve de 10 cm de longueur, et les points de fusion en capillaire sur un appareil Tottoli (températures non corrigées). Les mesures par chromatographie sur couche mince (ccm) ont été réalisées sur des plaques de silice Kiesegel 60F 254 (Merck), la visualisation des plaques sous lumière UV et/ou par pulvérisation de H2SO4 18 N dans MeOH, suivie d’un chauffage vers 270°C, ou d’une solution de KMnO4 à 0,5% dans H2SO4 1 N. Les éluants de chromatographie sont des mélanges d’hexane (H) et d’acétate d’éthyle (AE), ou de dichlorométhane (DCM) et de méthanol (MeOH), dans les proportions indiquées dans le texte. Les chromatographies sur colonne ouverte de silice ont été réalisées sur gel de silice SI 60 (63–200 μM) de Merck. Toutes les réactions, à l’exception de celles réalisées par transfert de phase ou des réactions de couplage en présence de sels de cuivre, l’ont été sous atmosphère contrôlée (Ar ou à défaut N2).
4.1.1 1,4-Bis-[1-(4,6-di-O-acétyl-2,3-didésoxy-α-d-érythro-hex-2-énopyranosyloxy)-méthyl]-benzène 1
La structure du composé 1 est représentée sur la Fig. 10.

Structure du composé 1.
15,13 g (55,0 mmol) de tri-O-acétyl-d-glucal commercial sont dissous dans 150 ml de DCM anhydre, auquel on ajoute en une fois 3,85 g (soit 0,5 équiv) d’alcool 4-hydroxyméthylbenzylique et 2,00 g de tamis moléculaire 4 Å broyé. À cette suspension, préalablement refroidie à l’aide d’un bain de glace vers +4 °C et vigoureusement agitée magnétiquement, est additionné goutte à goutte 0,9 ml (0,25 équiv) du complexe BF3·Et2O. Le milieu réactionnel fonce progressivement et la réaction, suivie par chromatographie sur couche mince (H/A, 2:1), est achevée après 90 min d’agitation à la même température. Le traitement consiste à neutraliser, par ajout de NaHCO3 (3,0 g), à filtrer sur papier filtre pour éliminer les solides, à extraire au DCM (3 × 50 ml), à rassembler les phases organiques, à laver à l’eau, à sécher sur MgSO4 et à concentrer finalement sous vide. Une chromatographie préparative sur silice (H/A, 2:1) suivie d’une cristallisation lente (i-Pr2O/i-PrOH, 1:2) fournit le glycoside 1 (12,38 g) sous la forme d’un solide blanc homogène en ccm. Rdt ~ 80%. F : 102–103°C ; spectre IR : 1743 cm–1 (νC=O) ; [α]D : +77,0 (c = 4 ; CHCl3) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 2,06 (s, 6 H, 2 OAc) ; 2,08 (s, 6 H, 2 OAc) ; 4,08–4,28 (massif, 6 H, 2 H-6, 2 H-6′, 2 H-5, Jgem = 12,5 Hz, J5–6 = 6 Hz, J5–6' = 3 Hz) ; 4,48 (d, 2 H, 2 H-7, Jgem = 11,5 Hz) ; 4,73 (d, 2 H, 2 H-7′) ; 5,08 (sl, 2 H, 2 H-1) ; 5,28 (m, 2 H, 2 H-4, J4–5 = 9 Hz, J3–4 = 2,1 Hz, J2–4 = 1,6 Hz) ; 5,80–5,91 (massif, 4 H, 2 H-2, 2 H-3) ; 7,38 (sl, 4 H, Ar) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 20,5 (CH3) ; 20,6 (CH3) ; 62,6 (C-7) ; 64,9 (C-4) ; 66,8 (C-5) ; 69,5 (C-6) ; 93,0 (C-1) ; 126,7 (C-3) ; 127,8 (C-2) ; 128,0 & 129,0 (C-2 Ar) ; 136,9 (C-1 Ar) ; 169,9 (C=O) ; 170,4 (C=O) ; analyse élémentaire calculée pour C28H34O12 : C% 59,79 ; H% 6,11 ; trouvée : C% 59,9 ; H% 6,1.
4.1.2 (4-Hydroxyméthyl-benzyl) 4,6-di-O-acétyl-2,3-dideoxy-α-d-érythro-hex-2-énopyranoside 2
La structure du composé 2 est représentée sur la Fig. 11.

Structure du composé 2.
Par chromatographie isocratique sur silice (H/A, 2:1), à la suite de 1 sont isolés 3,13 g (17%) de l’alcool 2 sous la forme d’une gomme incolore ; [α]D : +53,0 (c = 1 ; CHCl3) ; spectre IR : 1742 cm–1 (νC=O) ; spectre de RMN 1H 400 MHz (CDCl3) δ : 2,03 (s, 3 H, OAc) ; 2,08 (s, 3 H, OAc) ; 2,28 (sl, 1 H, OH) ; 3,98–4,28 (massif, 3 H, H-6, H-6′, H-5) ; 4,60–4,80 (massif, 4 H, H-7, H-7′, H-8, H-8') ; 5,10 (sl, 1 H, H-1) ; 5,30 (d, 1 H, H-4, J = 9,4 Hz) ; 5,80–5,91 (massif, 2 H, H-2, H-3) ; 7,35 (sl, 4 H, Ar) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 20,7 (2 × CH3) ; 62,7 (C-7) ; 64,6 (C-8) ; 65,0 (C-4) ; 66,8 (C-5) ; 69,5 (C-6) ; 93,6 (C-1) ; 126,7 (C-3) ; 126,9 (C-2) ; 127,6 (C-2 Ar) ; 128,1 (C-3 Ar) ; 136,5 (C-1 Ar) ; 142,0 (C-4 Ar) ; 169,9 (C=O) ; 170,4 (C=O).
4.1.3 1,4-Bis-[1-(2,3-didésoxy-α-d-érythro-hex-2-énopyranosyloxy)-méthyl]-benzène 3
La structure du composé 3 est représentée sur la Fig. 12.

Structure du composé 3.
Une quantité catalytique (~30 mg) de sodium métallique fraîchement préparé est ajoutée à une solution de 10,06 g du disaccharide 2 dans le MeOH abs. (75 ml) ; la réaction suivie par ccm (DCM/MeOH, 7:1) est achevée après 2 h d’agitation à température ambiante. Le traitement consiste à neutraliser la solution avec de la résine Dowex H+, à filtrer et à concentrer le filtrat sous vide jusqu’à prise en masse. Une recristallisation dans l’i-PrOH livre le tétrol 3 (~6,35 g) avec un rendement d’environ 90%. Solide blanc ; F : 180–181°C ; [α]D : +39,0 (c = 1 ; Pyr) ; spectre de RMN 1H (400 MHz, DMSO-d6) δ : 3,45–3,57 (massif, 4 H, 2 H-6, 2 H-6') ; 3,66 (m, 2 H, 2 H-5) ; 3,87 (m, 2 H, 2 H-4) ; 4,48 (d, 2 H, 2 H-7, Jgem = 11,6 Hz) ; 4,65 (t, 2 H, 2 OH, JOH-6 = JOH-6′ = 5,8 Hz) ; 4,73 (d, 2 H, 2 H-7′) ; 5,01 (sl, 2 H, 2 H-1) ; 5,05 (d, 2 H, 2 OH, JOH-4 = 6,8 Hz) ; 5,68 (m, 2 H, 2 H-3, J2–3 = 10,1 Hz) ; 5,85 (d, 2 H, 2 H-2) ; 7,30 (s, 4 H, Ar) ; analyse élémentaire calculée pour C20H26O8 : C% 60,90 ; H% 6,64 ; trouvée C% 61,0 ; H% 6,6.
4.1.4 1,4-Bis-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hex-2-énopyranosyloxy)-méthyl]-benzène 4
La structure du composé 4 est représentée sur la Fig. 13.
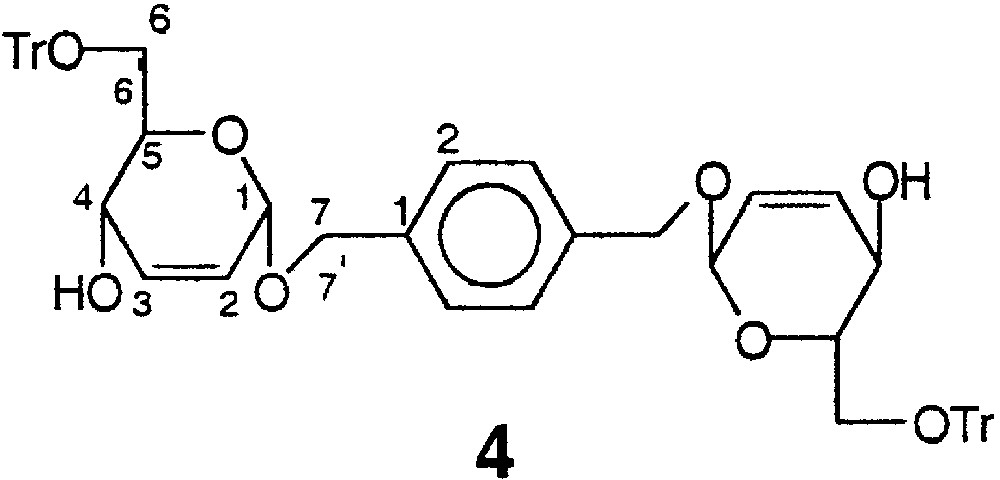
Structure du composé 4.
À une solution de 6,90 g (17,5 mmol) du tétrol 3 dans 25 ml de pyridine sont ajoutés 19,50 g (4 équiv) de TrCl en une fois et le mélange réactionnel est maintenu sous agitation à température ambiante pendant 48 h. Après évaporation partielle de la pyridine en dessous de 30 °C, le sirop est repris dans du DCM (200 ml), puis lavé avec une solution de NaHCO3 sat. (2 × 40 ml), puis à l’eau (2 × 40 ml). La phase organique est séchée sur MgSO4, puis concentrée sous pression réduite. Une purification de la gomme obtenue sur colonne de silice préalablement désactivée par la triéthylamine (éluant H/A, 3:1) permet d’isoler 12,73 g (83%) du diol symétrique 4 sous la forme d’une mousse blanche ; F : 90–92°C ; [α]D : +87,0 (c = 1 ; CHCl3) ; spectre de RMN 1H 400 MHz (CDCl3) δ : 2,68 (sl, 2 H, 2 OH) ; 3,28–3,48 (massif, 4 H, 2 H-6, 2 H-6') ; 3,93 (ddd, 2 H, 2 H-5, J5–6 = 5 Hz, J5–6’ = 3 Hz, J4–5 = 10 Hz) ; 4,13 (dd, 2 H, 2 H-4, J3–4 = 2 Hz) ; 4,58 (d, 2 H, 2 H-7, Jgem = 11,7 Hz) ; 4,85 (d, 2 H, 2 H-7’) ; 5,03 (sl, 2 H, 2 H-1) ; 5,74 (dd, 2 H, 2 H-2, J2–3 = 10 Hz, J2–4 < 2 Hz) ; 5,93 (m, 2 H, 2 H-3) ; 7,20–7,60 (m, 34 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 62,7 (C-6) ; 64,9 (C-7) ; 70,0 (C-4) ; 72,0 (C-5) ; 87,5 (CPh3) ; 94,0 (C-1) ; 126,0 (C-4 Tr) ; 127,4 (C-2 Ar) ; 128,3 (C-2) ; 128,7 (C-2 Tr) ; 133,4 (C-3 Tr) ; 133,7 (C-3) ; 137,5 (C-1 Ar) ; 143,8 (C-1 Tr) ; analyse élémentaire calculée pour C58H54O8 : C% 79,24 ; H% 6,19 ; trouvée C% 78,9 ; H% 6,3.
4.1.5 1,4-Bis-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-benzène 5
La structure du composé 5 est représentée sur la Fig. 14.

Structure du composé 5.
11,19 g (12,7 mmol) de disaccharide tritylé 4 sont dissous dans 400 ml d’AE et la solution purgée par bullage à l’Ar pendant 5 min. 0,50 g de Pd sur CaCO3 à 5% sont ajoutés en une fois, mis en suspension par agitation magnétique et le mélange réactionnel est saturé en H2 à la pression atmosphérique. L’avancement de la réaction est suivie par ccm (H/Tol/AE, 1:2:2). Après 17 h d’agitation, la suspension est filtrée sur célite, qui est rincée au DCM puis à l’AcOEt pur. L’évaporation du filtrat sous pression réduite livre le diol 5 sous la forme d’un solide blanc amorphe (10,10 g, ~90%), utilisable directement pour l’étape suivante ; F : 92–94°C ; [α]D : +16,0 (c = 0,6 ; CHCl3) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,68–1,93 (massif, 8 H, 4 H-2, 4 H-3) ; 2,64 (d, 2 H, 2 OH, J4-OH ~ 3 Hz) ; 3,35 (dd, 2 H, 2 H-6, Jgem = 12 Hz, J5–6 = 5 Hz) ; 3,43 (dd, 2 H, 2 H-6’, J5–6′ = 3 Hz) ; 3,58 (m, 2 H, 2 H-5) ; 3,73 (m, 2 H, 2 H-4) ; 4,43 (d, 2 H, 2 H-7, Jgem = 10,0 Hz) ; 4,70 (d, 2 H, 2 H-7′) ; 4,85 (sl, 2 H, 2 H-1) ; 7,18–7,50 (massif, 34 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 27,5 (C-2) ; 29,5 (C-3) ; 66,2 (C-6) ; 68,6 (C-7) ; 69,2 (C-4) ; 72,3 (C-5) ; 87,8 (CPh3) ; 95,5 (C-1) ; 127,7 (C-4 Tr) ; 128,5 (C-2 Ar) ; 128,7 (C-2 Tr) ; 129,2 (C-3 Tr) ; 137,9 (C-1 Ar) ; 144,2 (C-1 Tr) ; analyse élémentaire calculée pour C58H58O8 : C% 78,89 ; H% 6,62 ; trouvée C% 78,6 ; H% 6,7.
4.1.6 1,4-Bis-[1-(2,3-didésoxy-4-O-propargyl-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-benzène 6
La structure du composé 6 est représentée sur la Fig. 15.

Structure du composé 6.
9,90 g (11,2 mmol) de diol 5 sont mis en solution dans 20 ml de xylène, auxquels on ajoute successivement 30 ml de NaOH aqueux à 50%, 7,23 g (2 équiv) de Bu4NBr et 3,0 ml de bromure de propargyle, en maintenant la température en dessous de 15 °C ; la réaction, suivie par ccm (H/A, 1:1), est achevée après 4 h d’agitation vigoureuse en dessous de 20 °C. Le traitement consiste à diluer le mélange réactionnel avec de l’eau glacée (50 ml), à isoler la phase organique par décantation, à extraire la phase aqueuse au DCM (3 × 50 ml). Les phases organiques sont rassemblées, lavées avec une solution saturée de NH4Cl jusqu’à neutralisation, puis finalement à l’eau, séchées sur MgSO4 et finalement concentrées sous pression réduite. Une purification sur colonne de silice désactivée à la triéthylamine (H/A, 6:1) permet d’isoler 4,62 g (43%) de l’éther symétrique 6 sous la forme d’une mousse blanche homogène en ccm ; F : 71–73°C ; [α]D : +69,5 (c : 0,4 ; CHCl3) ; spectre IR : 3303 et 2247 cm–1 (νC≡CH) ; spectre de RMN 1H (400 MHz, CDCl3) δ: 1,78–2,13 (m, 8 H, 4 H-2, 4 H-3) ; 2,33 (sl, 2 H, 2 H-10) ; 3,13 (dd, 2 H, 2 H-6, Jgem = 10 Hz, J5–6 = 5 Hz) ; 3,46 (dd, 2 H, 2 H-6′, J5–6′ = 3 Hz) ; 3,62 (m, 2 H, 2 H-5, J4–5 ~ 10 Hz) ; 3,83 (dd, 2 H, 2 H-4, J3–4 = 4 Hz) ; 4,08 (m, 4 H, 4 H-8) ; 4,53 (d, 2 H, 2 H-7, Jgem = 12,0 Hz) ; 4,86 (d, 2 H, 2 H-7′) ; 4,93 (sl, 2 H, 2 H-1) ; 7,18–7,63 (massif, 34 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 24,4 (C-2) ; 28,9 (C-3) ; 56,2 (C-8) ; 63,4 (C-6) ; 68,0 (C-7) ; 71,6 (C-4) ; 73,0 (C-5 & C-10) ; 74,0 (C-9) ; 86,3 (CPh3) ; 94,9 (C-1) ; 126,9 (C-4 Tr) ; 127,7 (C-2 Ar) ; 128,2 (C-2 Tr) ; 128, 8 (C-3 Tr) ; 137,4 (C-1 Ar) ; 143,3 (C-1 Tr) ; analyse élémentaire calculée pour C64H62O8 : C% 80,14 ; H% 6,52 ; trouvée : C% 80,1 ; H% 6,5.
4.1.7 1-[1-(2,3-Didésoxy-4-O-propargyl-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-4-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-benzène 7
La structure du composé 7 est représentée sur la Fig. 16.

Structure du composé 7.
À la suite de 6, de la même colonne en isocratique sont isolés 4,30 g (42%) de l’alcool 7, sous la forme d’une mousse blanche ; F : 80–81°C ; [α]D : +35,6 (c = 1 ; CHCl3) ; spectre IR : 3470 cm–1 (νC≡CH) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,72–2,07 (massif, 8 H, 2 H-2, 2 H-3, 2 H-2', 2 H-3') ; 2,25 (sl, 1 H, H-10) ; 2,64 (d, 1 H, OH, JOH-4′ = 3,0 Hz) ; 3,18 (dd, 1 H, H-6, Jgem = 10,2 Hz, J5–6 = 5,1 Hz) ; 3,28–3,53 (massif, 3 H, H-6′, H-6′′, H-6′′′) ; 3,58–3,66 (massif, 2 H, H-4, H-4′) ; 3,68–3,88 (massif, 2 H, H-5′, H-5) ; 4,03 (m, 2 H, 2 H-8) ; 4,38–4,80 (massif, 4 H, H-7, H-7′, H-7′′, H-7′′′) ; 4,83 (sl, 1 H, H-1) ; 4,93 (sl, 1 H, H-1′), 7,18–7,48 (massif, 34 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 24,4 (C-2′) ; 26,7 (C-2) ; 28,9 (C-3′′) ; 29,0 (C-3) ; 56,2 (C-8) ; 63,4 (C-6′′) ; 66,1(C-6) ; 68,1 (C-7′′) ; 68,2 (C-7) ; 69,1 (C-4) ; 71,1 (C-4′) ; 71,7 (C-5 & C-10) ; 73,1 (C-5′) ; 74,0 (C-9) ; 86,3 (CPh3) ; 87,6 (CPh3) ; 95,1 (C-1) ; 95,1 (C-1′) ; 126,9 (C-4 Tr) ; 127,3 (Ar) ; 127,7 (Ar) ; 127,9 (Ar) ; 128,0 (C-2 Tr) ; 128,2 (C-2′ Tr) ; 128,7 (C-3 Tr) ; 128,9 (C-3′ Tr) ; 137,4 (C-1 & C-4 Ar) ; 143,7 (C-1 Tr) ; 144,2 (C-1′ Tr) ; SM–MALDI : m/z = 943 [M + Na]+ (100%), m/z = 959 [M + K]+ (70%) ; analyse élémentaire calculée pour C61H60O8 : C% 79,53 ; H% 6,57 ; trouvée : C% 79,3 ; H% 6,7.
4.1.8 1,6-Bis-{1-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-4-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-phényl}-hex-2,4-diyne 8
La structure du composé 8 est représentée sur la Fig. 17.

Structure du composé 8.
À une solution de 1,30 g (1,40 mmol) du précurseur 7 dans 20 ml de pyridine à 20 °C sont additionnés 1,27 g de Cu(OAc)2 anh. (5 équiv). La suspension est agitée pendant 18 h à 20 °C, puis 1 h à 50 °C. Après complet refroidissement, le mélange réactionnel est évaporé sous vide, puis repris dans un mélange de DCM (150 ml) et d’eau (100 ml). La phase organique est isolée par décantation, la phase aqueuse épuisée au DCM (3 × 50 ml). Les phases organiques sont rassemblées, lavées avec NH4Cl sat. (2 × 15 ml) et à l’eau (2 × 50 ml), puis séchées sur MgSO4 et enfin concentrées à sec. Une purification sur colonne de silice désactivée à la triéthylamine (H/A, 2:1 → 1:1) permet d’isoler 965 mg (75%) du produit de couplage 8 sous la forme d’un solide blanc ; F : 99–100°C ; [α]D : +75,6 (c = 0,3 ; CHCl3) ; spectre IR : 3450 cm–1 (νOH), 2500 cm–1 (νC≡C) ; spectre de RMN 1H (250 MHz, CDCl3) δ : 1,66–2,20 (massif, 16 H, 4 H-2, 4 H-2′, 4 H-3, 4 H-3′) ; 2,90 (sl, 2 H, 2 OH) ; 3,35–3,75 (massif, 12 H, 2 H-5, 2 H-5′, 2 H-6, 2 H-6′, 2 H-6′′, 2 H-6′′′) ; 3,79–3,95 (m, 4 H, 2 H-4, 2 H-4′) ; 4,10 (sl, 4 H, H-8) ; 4,55 (d, 2 H, H-7, J7–7′ = 12,4 Hz) ; 4,62 (d, 2 H, H-7′′, J7′′'-7′′′ ~ 12 Hz) ; 4,82 (d, 2 H, H-7′) ; 4,88 (d, 2 H, H-7′′′) ; 4,91 (sl, 2 H, H-1) ; 5,01 (sl, 2 H, H-1′) ; 7,15–7,65 (massif, 68 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 26,8 (C-2 & C-2′) ; 28,9 (C-3 & C-3′) ; 56,6 (C-8) ; 65,0 (C-6 & C-6′′) ; 67,9 (C-7 & C-7′′) ; 68,6 (C-4′) ; 69,9 (C-10) ; 71,5 (C-4) ; 71,7 (C-5′) ; 73,3 (C-5) ; 75,6 (C-9) ; 86,3 (CPh3) ; 87,2 (C'-Ph3) ; 94,9 (C-1 & C-1′) ; 126,8 (C-4′ Tr) ; 127,1 (C-4 Tr) ; 127,8 (C-2 & C-3 Ar) ; 128,1 (C-2 & C-2′Tr) ; 128,6 (C-3 Tr) ; 128,7 (C-3′ Tr) ; 137,3 (C-1 & C-4 Ar) ; 143,7 (C-1 Tr) ; 144,2 (C-1′ Tr) ; SM–MALDI : m/z = 1861,3 : [M + Na]+ (70%) ; analyse élémentaire calculée pour C122H118O16 : C% 79,63 ; H% 6,46 ; trouvée : C% 78,9 ; H% 6,4.
4.1.9 1,6-Bis-{1-[1-(2,3-didésoxy-4-O-propargyl-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-4-[1-(2,3-didésoxy-6-O-trityl-α-d-érythro-hexopyranosyloxy)-méthyl]-phényl}-hex-2,4-diyne 9
La structure du composé 9 est représentée sur la Fig. 18.

Structure du composé 9.
3,91 g (2,13 mmol) de diol 8 sont mis en solution dans 15 ml de benzène, auxquels on ajoute successivement 30 ml de NaOH aq. à 50%, 1,37 g (2 équiv) de Bu4NHSO4 et 3 ml de bromure de propargyl vers 4 °C ; la réaction suivie par ccm (H/A, 1:1) est arrêtée après 36 h d’agitation vigoureuse à température ambiante. Le traitement consiste à diluer avec de l’eau glacée (50 ml), à isoler la phase organique par décantation et à extraire la phase aqueuse au DCM (3 × 30 ml), à rassembler les phases organiques, à laver avec une solution aqueuse saturée de NH4Cl (30 ml), puis avec H2O (30 ml), à sécher sur MgSO4, puis à concentrer sous pression réduite ; une purification sur colonne de silice permet d’isoler 3,18 g, soit 78% du produit dialkylé 9 sous la forme d’un solide blanc ; F : 98–100°C ; [α]D : +55,0 (c = 0,5 ; CHCl3) ; spectre IR : 3304 cm–1 (νC≡CH) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,80–2,26 (massif, 16 H, 4 H-2, 4 H-2′, 4 H-3, 4 H-3) ; 2,43 (sl, 2 H, 2 H-10) ; 3,50 (m, 4 H, 2 H-4, 2 H-4′) ; 3,80 (m, 4 H, 2 H-5, 2 H-5′) ; 4,18–4,34 (massif, 16 H, 4 H-8, 4 H-11, H-6, H-6′, H-6′′, H-6′′′) ; 4,42 (d, 4 H, H-7, H-7′′, J7–7′ = J7′′-7′′′ = 11,7 Hz) ; 4,63 (d, 4 H, H-7′, H-7′′′) ; 4,83 (sl, 4 H, 2 H-1, 2 H-1′) ; 7,18–7,63 (massif, 68 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 24,4 (C-2 & C-2′) ; 28,9 (C-3 & C-3′) ; 56,02 (C-8 & C-11) ; 65,7 (C-6 & C-6′′) ; 67,9 (C-7 & C-7′′) ; 69,9 (C-13) ; 71,6 (C-4 & C-4′) ; 72,9 (C-5 & C-5′) ; 73,3 (C-10) ; 74,03 (C-9) ; 75,6 (C-12) ; 86,2 (CPh3) ; 94,9 (C-1 & C-1′), 126,8 (C-4 Tr) ; 127,7 (C-2 & C-3 Ar) ; 128,1 (C-2 Tr) ; 128,7 (C-3 Tr) ; 137,3 (C-1 & C-4 Ar) ; 144,1 (C-1 Tr) ; SM–MALDI : m/z = 1938 [M + Na]+ (75%) ; m/z = 1954 [M + K]+ (35%) ; analyse élémentaire calculée pour C128H122O16 : C% 79,63 ; H% 6,46 ; trouvée : C% 79,7 ; H% 6,5.
4.1.10 1,6-Bis-{1-[1-(2,3-didésoxy-4-O-propargyl-α-d-érythro-hexopyranosyloxy)-méthyl]-4-[1-(2,3-didésoxy-α-d-érythro-hexopyranosyloxy)-méthyl]-phényl}-hex-2,4-diyne 10
La structure du composé 10 est représentée sur la Fig. 19.
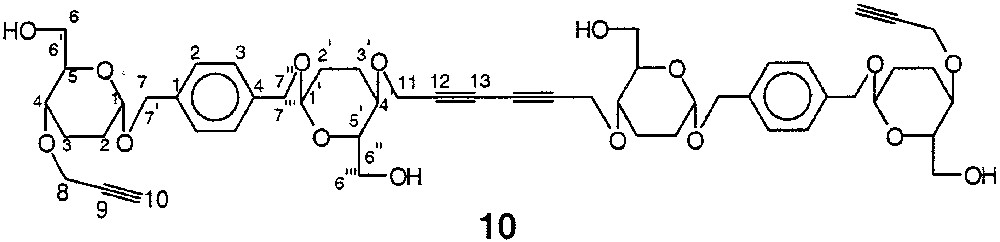
Structure du composé 10.
3,18 g de tétrasaccharide dialkylé 9 (1,66 mmol) sont dissous dans un mélange de 70 ml de MeOH, 70 ml de DCM et de 30 mg d’APTS. L’agitation est maintenue pendant 4 h à température ambiante, l’avancement de la réaction étant suivie par ccm (DCM/MeOH, 9:1). 100 mg de bicarbonate de sodium sont ajoutés au mélange réactionnel, qui est agité pendant 15 min supplémentaires. Les solvants chassés sous vide, le résidu obtenu est purifié sur une colonne ouverte de silice (H/A, 1:1), qui livre 3,67 g (~80%) du produit détritylé 10 sous la forme d’une gomme incolore ; [α]D : +4,0 (c = 3 ; CHCl3) ; spectre IR : 3400 cm–1 (νOH) ; spectre de RMN 1H 400 MHz (CDCl3) δ : 1,36 (sl, 4 H, 4 OH) ; 1,58–2,08 (massif, 16 H, 4 H-2, 4 H2′, 4 H-3, 4 H-3′) ; 2,43 (sl, 2 H, 2 H-10) ; 3,53 (m, 4 H, 2 H-6, 2 H-6′), 3,58–3,88 (massif, 12 H, 2 H-4, 2 H-4′, 2 H-5, 2 H-5′, 2 H-6′′, 2 H-6′′′) ; 4,18 (sl, 8 H, 4 H-8, 4 H-11) ; 4,43 (d, 4 H, 2 H-7, 2 H-7′′, J7–7′ = J7′′-7′′′ ~ 10 Hz) ; 4,63 (d, 4 H, 2 H-7′, 2 H-7′′′) ; 4,88 (sl, 4 H, 2 H-1, 2 H-1′) ; 7,38 (sl, 8 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 23,7 (C-2 & C-2′) ; 29,6 (C-3 & C-3′) ; 56,2 (C-8 & C-11) ; 63,1 (C-6 & C-6′′) ; 68,9 (C-7 & C-7′′) ; 72,8 (C-4 & C-4′) ; 73,5 (C-5 & C-5′ + C-10) ; 74,7 (C-9) ; 96,0 (C-1 & C-1′) ; 129,1 (C-2 & C-3 Ar) ; 137,7 (C-1 & C-4 Ar) ; SM-MALDI : m/z = 969,4 [M + Na]+ (100%), m/z = 985,4 [M + K]+ (20%) ; analyse élémentaire calculée pour C52H66O16 : C% 65,94 ; H% 7,02 ; trouvée : C% 65,7 ; H% 7,1.
4.1.11 1,6-Bis-{1-[1-(6-O-acétyl-2,3-didésoxy-4-O-propargyl-α-d-érythro-hexopyranosyloxy)-méthyl]-4-[1-(6-O-acétyl-2,3-didésoxy-α-d-érythro-hexopyranosyloxy)-méthyl]-phényl}-hex-2,4-diyne 11
La structure du composé 11 est représentée sur la Fig. 20.

Structure du composé 11.
À une solution de 3,70 g (3,9 mmoles) du tétrol 10 dans 20 ml de pyridine sont ajoutés 5 ml d'anhydride acétique et 1 ml de triéthylamine. Le mélange est agité pendant 24 h à température ambiante, les solvants évaporés sous vide et le brut repris dans du DCM (50 ml), lavé avec une solution saturée de NH4Cl puis à l’eau jusqu’à pH 7. La phase organique est séchée sur MgSO4, filtrée, concentrée et le brut purifié par simple élution sur une tulipe de silice (AE) pour donner 3,89 g (90%) du peracétate 11 sous la forme d’une gomme jaunâtre ; [α]D : +128,0 (c : 1 ; CHCl3) ; spectre IR : 1740 cm–1 (νC=O), 3270 cm–1 (νC≡CH) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,63–1,88 (massif, 12 H, 4 H-2, 4 H-2′, 4 H-3) ; 1,98 (m, 4 H, 4 H-3′) ; 2,03 (s, 6 H, 2 OAc) ; 2,06 (s, 6 H, 2 OAc) ; 2,38 (sl, 2 H, 2 H-10) ; 3,48 (m, 4 H, 2 H-4, 2 H-4′) ; 3,78 (m, 4 H, 2 H-5, 2 H-5′) ; 4,18 (m, 4 H, 2 H-6, 2 H-6′) ; 4,28 (s, 8 H, 4 H-8, 4 H-11) ; 4,33 (m, 4 H, 2 H-6′′, 2 H-6′′′) ; 4,48 (d, 4 H, 2 H-7, 2 H-7′′, J7–7′ = J7′′-7′′′ = 12,4 Hz) ; 4,68 (dd, 4 H, 2 H-7′, 2 H-7′′′) ; 4,88 (sl, 4 H, 4 H-1) ; 7,38 (sl, 8 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 20,7 (CH3) ; 23,1 (C-2 & C-2′) ; 29,4 (C-3 & C-3′) ; 55,3 (C-8) ; 55,7 (C-11) ; 63,5 (C-6) ; 63,7 (C-6′′) ; 68,1 (C-7 & C-7′′) ; 69,5 (C-5 & C-5′) ; 72,1 (C-4 & C-4′) ; 72,2 (C-10) ; 74,2 (C-9) ; 95,1 (C-1 & C-1′) ; 127,7 (C-2 & C-3 Ar) ; 136,9 (C-1 & C-4 Ar) ; 170,6 (C=O) ; SM-MALDI : m/z = 1137,3 [M + Na]+ (76%), m/z = 1307,5 [M + Na + DHBA]+ (100%) ; analyse élémentaire calculée pour C60H74O20 : C% 64,61 ; H% 6,69 ; trouvée : C% 64,5; H% 6,8%.
4.1.12 Synthèse du glycophane 12
La structure du composé 12 est représentée sur la Fig. 21.

Structure du composé 12.
4.1.12.1 Par cyclisation du précurseur 11
À un mélange de 88 ml de pyridine et de 220 ml de MeCN, préalablement refroidi à 0 °C, sont ajoutés 0,85 g du précurseur 11 et 8,31 g de Cu(OAc)2 (30 équiv). La suspension est agitée pendant 22 h, alors que la température est maintenue en dessous de +5°C avec de la glace fondante. Le mélange réactionnel est concentré sous vide puis repris dans du DCM (100 ml), lavé avec NH4Cl sat. (2 × 10 ml) et enfin à l’eau (2 × 50 ml) ; la phase organique est isolée par décantation, séchée sur MgSO4 et finalement concentrée à sec. Une purification sur colonne de silice (H/A, 3:2) permet d’isoler 0,50 g (~ 60%) d’une gomme translucide, dont les caractéristiques correspondent au tétrasaccharide 12 (vide infra).
4.1.12.2 Par cyclisation directe du précurseur 14
À un mélange de 80 ml de pyridine, de 200 ml de MeCN et de 1,54 g du précurseur 14 (vide infra), refroidi vers 0 °C sont additionnés 7,52 g de Cu(OAc)2 (30 équiv). La suspension est agitée pendant 22 h et la température maintenue en dessous de +5°C. Le mélange réactionnel est évaporé sous vide puis repris dans du DCM (150 ml), lavé avec NH4Cl (2 ×15 ml) et à l’eau (2 × 50 ml) ; la phase organique est isolée par décantation, séchée sur MgSO4 et concentrée à sec. Une purification sur colonne de silice, désactivée par la triéthylamine (H/A, 3:2), permet d’isoler 1,37 g (~ 90%) d’un composé organique unique en tout point identique au produit préparé à partir de 11 ; [α]D : +127,6 (c : 0,8 ; CHCl3) ; spectre IR : 1739 cm–1 (νC=O), 2360 cm–1 (νC≡C) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,70–1,93 (massif, 12 H, 8 H-2, 4 H-3) ; 2,10 (m, 4 H, 4 H-3) ; 2,12 (s, 12 H, 4 OAc) ; 3,28 (m, 4 H, 4 H-4) ; 3,84 (m, 4 H, 4 H-5) ; 4,22–4,31 (massif, 16 H, 4 H-6, 4 H-6′, 8 H-8) ; 4,48 (d, 4 H, 4 H-7, Jgem = 12,4 Hz) ; 4,69 (d, 4 H, 4 H-7′) ; 4,86 (d, 4 H, 4 H-1, J1–2 = 2,5 Hz) ; 7,33 (sl, 8 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 20,9 (CH3) ; 23,7 (C-2) ; 28,7 (C-3) ; 57,0 (C-8) ; 63,7 (C-6) ; 68,2 (C-7) ; 69,7 (C-5) ; 73,4 (C-4) ; 75,6 (C-10) ; 76,5 (C-9) ; 95,1 (C-1) ; 128,2 (C-2 Ar) ; 137,2 (C-1 Ar) ; 170,9 (C=O) ; SM-MALDI : m/z = 1136,0 : [M + Na]+ (100%), m/z = 1151,9 : [M + K]+ (40%).'
4.1.13 Synthèse du glycophane 13
La structure du composé 13 est représentée sur la Fig. 22.

Structure du composé 13.
Par chromatographie isocratique sur silice (H/A, 3:2), à la suite de 12 sont isolés 100 mg (~ 12%) du macrocycle 13 sous la forme d’une gomme jaunâtre ; [α]D = +64,0 (c = 1 ; CHCl3) ; spectre IR : 1739 cm–1 (νC=O), 2360 cm–1 (νC≡C) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,68–1,93 (massif, 24 H, 16 H-2, 8 H-3) ; 2,03 (m, 8 H, 8 H-3) ; 2,08 (s, 24 H, 8 OAc) ; 3,48 (m, 8 H, 8 H-4) ; 3,83 (m, 8 H, 8 H-5) ; 4,18–4,38 (massif, 32 H, 8 H-6, 8 H-6′, 16 H-8) ; 4,48 (d, 8 H, 8 H-7, Jgem = 12,4 Hz) ; 4,68 (d, 8 H, 8 H-7′) ; 4,88 (sl, 8 H, 8 H-1) ; 7,33 (s, 16 H, Ar) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 22,0 (CH3) ; 24,0 (C-2) ; 29,8 (C-3) ; 57,0 (C-8) ; 65,0 (C-6) ; 68,0 (C-7) ; 72,0 (C-5) ; 73,0 (C-4) ; 74,8 (C-10) ; 77,5 (C-9) ; 97,1 (C-1) ; 130,2 (C-2 Ar) ; 139,3 (C-1 Ar) ; 172,0 (C=O) ; SM-MALDI : m/z = 2248,7 : [M + Na]+ (100%).
4.1.14 1,4-Bis-[1-(2,3-didésoxy-4-O-propargyl-6-O-acétyl-α-d-érythro-hexopyranosyloxy)-méthyl]-benzène 14
La structure du composé 14 est représentée sur la Fig. 23.

Structure du composé 14.
Le mode opératoire pour la détritylation est identique à celui utilisé pour la préparation du composé 10. À partir de 4,35 g de 6 sont isolés 3,81 g (~ 80%) du diol correspondant, immédiatement remis en réaction dans les mêmes conditions que pour l’acétylation du tétrol 10. Par chromatographie isocratique sur silice (H/A, 4:1) ont été obtenu 1,82 g (72%) du bis-acétate 14 sous la forme d’une gomme incolore ; [α]D : +86,0 (c = 1 ; CHCl3) ; spectre IR : 3470 cm–1 (νC≡CH) ; spectre de RMN 1H (400 MHz, CDCl3) δ : 1,60–1,95 (massif, 8 H, 4 H-2, 4 H-3) ; 2,11 (s, 6 H, CH3) ; 2,41 (sl, 2 H, H-10) ; 3,54 (m, 2 H, 2 H-5) ; 3,87 (m, 2 H, 2 H-4) ; 4,15 (dd, 2 H, 2 H-6, J5–6 = 2,3 Hz, Jgem = 15,8 Hz) ; 4,21 (dd, 2 H, 2 H-6', J5–6′ < 2 Hz) ; 4,30 (sl, 4 H, 4 H-8) ; 4,46 (d, 2 H, 2 H-7, Jgem = 11,7 Hz) ; 4,70 (d, 2 H, 2 H-7′) ; 4,90 (sl, 2 H, H-1) ; 7,33 (s, 4 H, ArH) ; spectre de RMN 13C (62,9 MHz, CDCl3) δ : 20,85 (CH3) ; 23,3 (C-2) ; 28,6 (C-3) ; 55,5 (C-8) ; 63,9 (C-6) ; 68,3 (C-7) ; 69,8 (C-4) ; 72,1 (C-5 & C-10) ; 74,4 (C-9) ; 95,3 (C-1) ; 128,0 (C-2 Ar) ; 137,2 (C-1 Ar) , 170,8 (C=O) ; analyse élémentaire calculée pour C30H38O10 : C% 64,50 ; H% 6,86 ; trouvée : C% 64,4 ; H% 7,0.